
Neddylation Regulates Class IIa and III Histone Deacetylases to Mediate Myoblast Differentiation




Abstract
:1. Introduction
2. Results
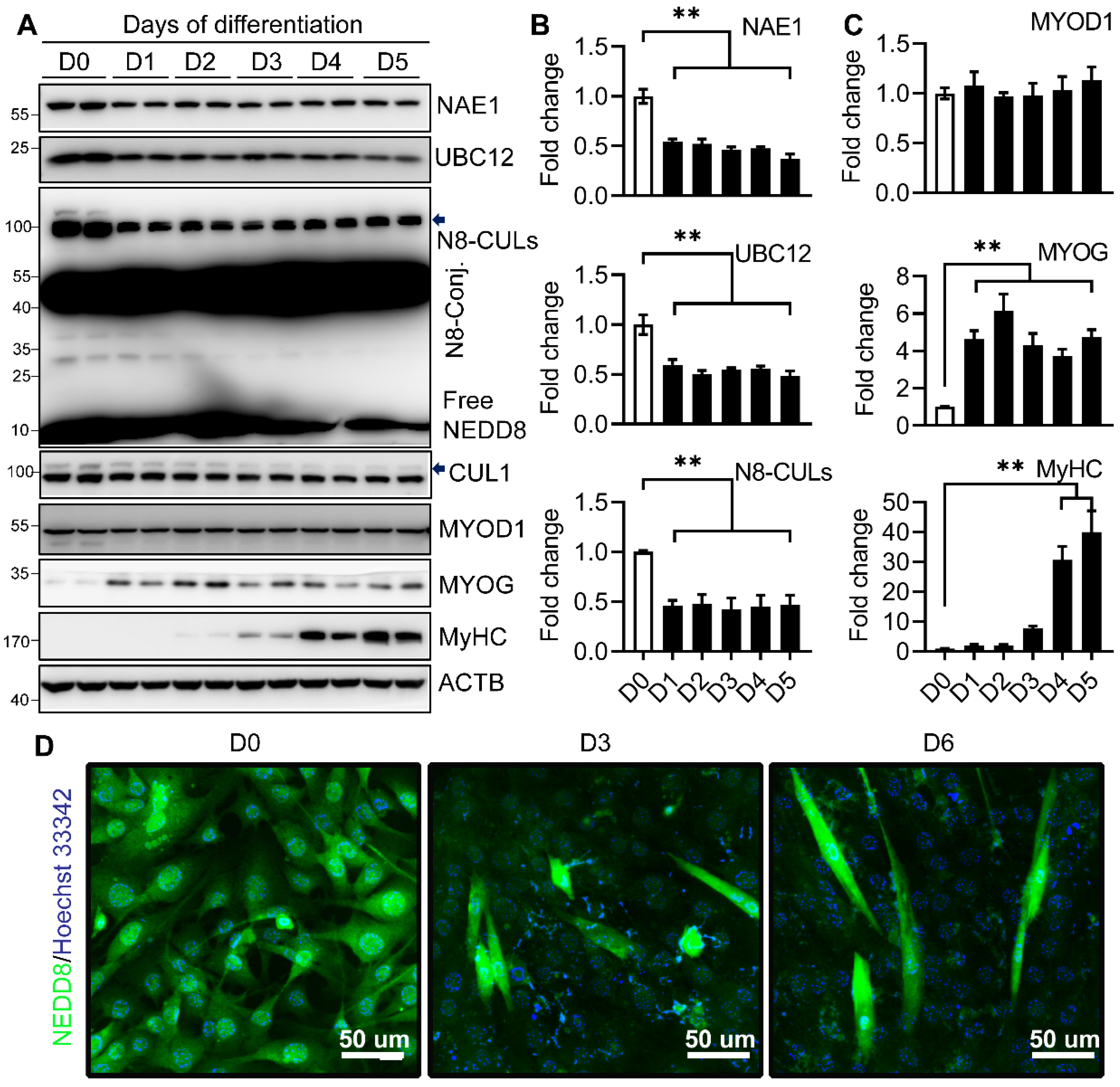
2.1. Neddylation Is Downregulated during Myoblast Differentiation
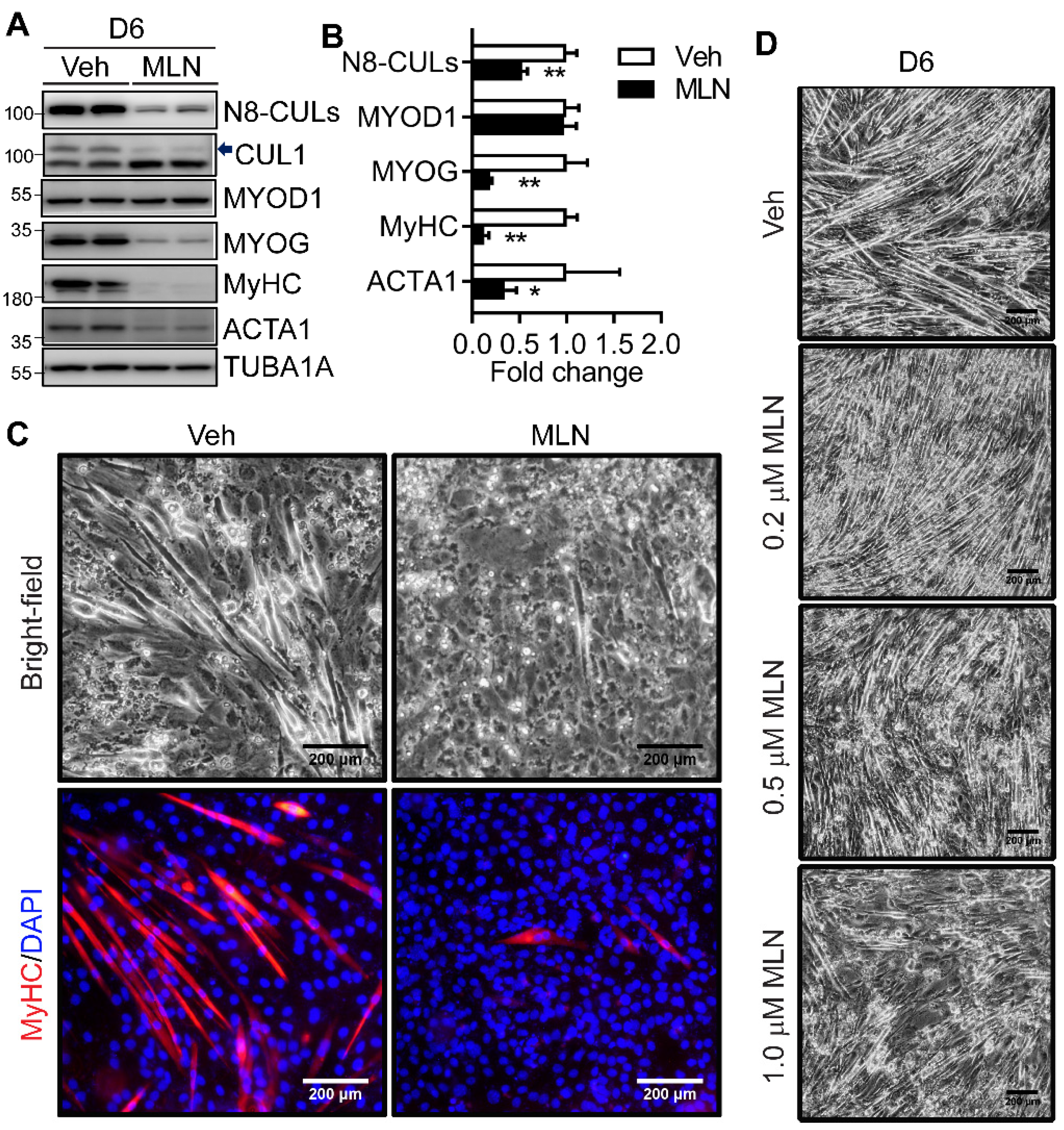
2.2. Neddylation Inhibition Blocks Myotube Formation
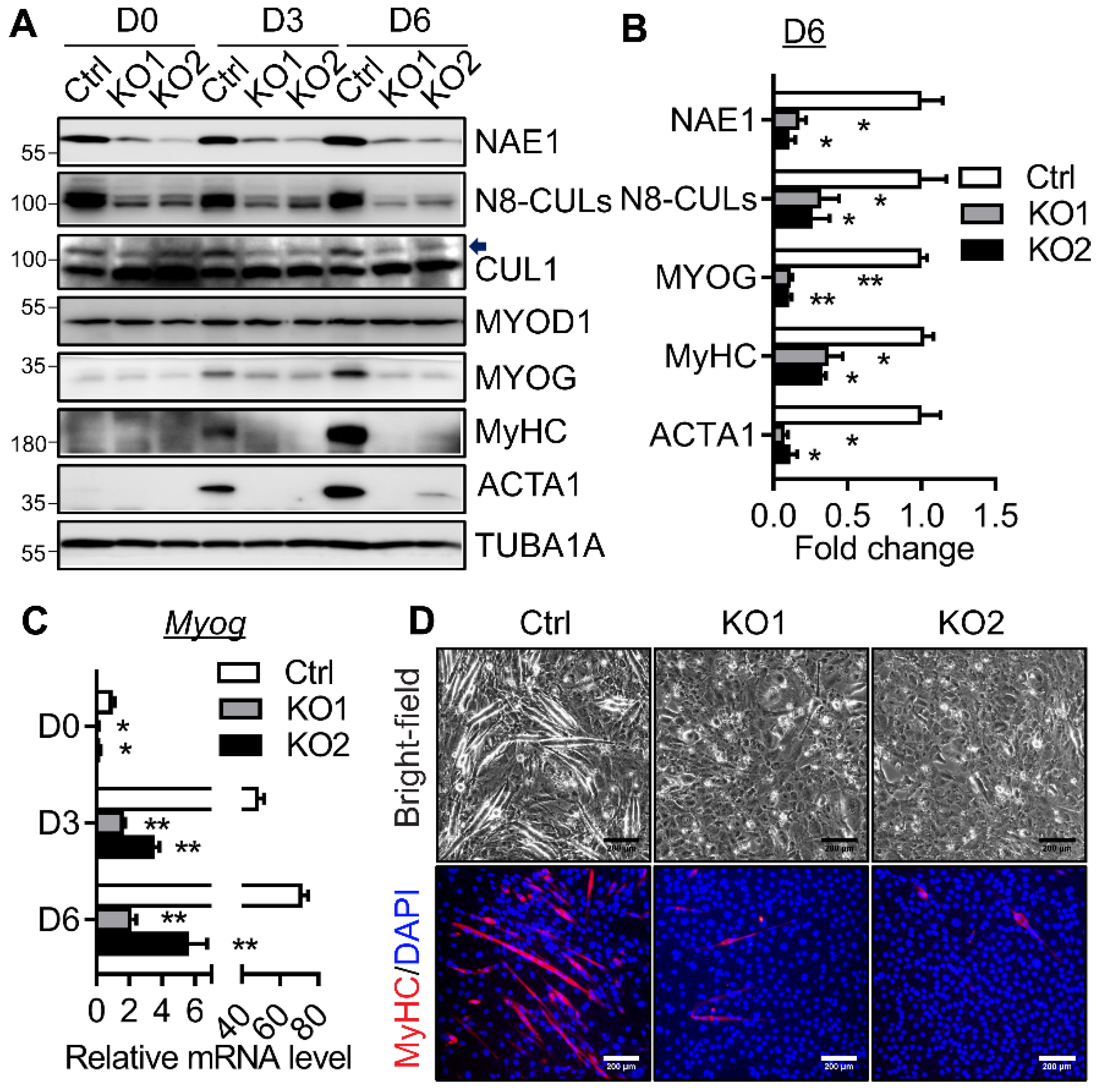
2.3. NAE1 Deletion in C2C12 Cells Prevents Terminal Myoblast Differentiation
2.4. Neddylation Deficiency Blocks Myoblast Differentiation Potentially through Modulating Class II and III HDACs
2.5. Neddylation Deficiency Causes SIRT1 Accumulation in the Nucleus during the Early Stage of Myoblast Differentiation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Myoblast Differentiation and Reagents
4.2. CRISPR/Cas9 Gene Targeting, Lentivirus Production, and Infection of C2C12 Cells
4.3. Quantitative Real-Time PCR (qRT-PCR)
4.4. Cellular Protein Extraction, Subcellular Fractionation, and Western Blot
4.5. Immunofluorescence Microscopy
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abidi, N.; Xirodimas, D.P. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr.-Relat. Cancer 2015, 22, T55–T70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Kandala, S.; Kim, I.M.; Su, H. Neddylation and deneddylation in cardiac biology. Am. J. Cardiovasc. Dis. 2014, 4, 140–158. [Google Scholar] [PubMed]
- Gan-Erdene, T.; Nagamalleswari, K.; Yin, L.; Wu, K.; Pan, Z.Q.; Wilkinson, K.D. Identification and characterization of DEN1, a deneddylase of the ULP family. J. Biol. Chem. 2003, 278, 28892–28900. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Chi, H.; Zhang, H.; Deng, X.W.; Flavell, R.A.; Wei, N. COP9 signalosome subunit 8 is essential for peripheral T cell homeostasis and antigen receptor-induced entry into the cell cycle from quiescence. Nat. Immunol. 2007, 8, 1236–1245. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Omata, M.; Tanaka, K.; Chiba, T. The NEDD8 system is essential for cell cycle progression and morphogenetic pathway in mice. J. Cell Biol. 2001, 155, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Ju, U.I.; Park, J.W.; Song, J.Y.; Shin, D.H.; Lee, K.H.; Jeong, L.S.; Yu, J.; Lee, H.W.; Cho, J.Y.; et al. PPARgamma neddylation essential for adipogenesis is a potential target for treating obesity. Cell Death Differ. 2016, 23, 1296–1311. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Li, J.; Zhang, H.; Ma, W.; Wei, N.; Liu, J.; Wang, X. COP9 signalosome controls the degradation of cytosolic misfolded proteins and protects against cardiac proteotoxicity. Circ. Res. 2015, 117, 956–966. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Ma, W.; Li, J.; Littlejohn, R.; Zhou, H.; Kim, I.M.; Fulton, D.J.R.; Chen, W.; Weintraub, N.L.; Zhou, J.; et al. Neddylation mediates ventricular chamber maturation through repression of Hippo signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E4101–E4110. [Google Scholar] [CrossRef] [Green Version]
- Vogl, A.M.; Brockmann, M.M.; Giusti, S.A.; Maccarrone, G.; Vercelli, C.A.; Bauder, C.A.; Richter, J.S.; Roselli, F.; Hafner, A.S.; Dedic, N.; et al. Neddylation inhibition impairs spine development, destabilizes synapses and deteriorates cognition. Nat. Neurosci. 2015, 18, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cao, Y.; Wu, H.; Ye, X.; Zhu, Z.; Xing, G.; Shen, C.; Barik, A.; Zhang, B.; Xie, X.; et al. Enzymatic Activity of the Scaffold Protein Rapsyn for Synapse Formation. Neuron 2016, 92, 1007–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Pavlick, A.C.; Boasberg, P.; Thompson, J.A.; Mulligan, G.; Pickard, M.D.; Faessel, H.; Dezube, B.J.; Hamid, O. A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Investig. New Drugs 2016, 34, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, J.J.; Jakubowiak, A.J.; O’Connor, O.A.; Orlowski, R.Z.; Harvey, R.D.; Smith, M.R.; Lebovic, D.; Diefenbach, C.; Kelly, K.; Hua, Z.; et al. Phase I Study of the Novel Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (MLN4924) in Patients with Relapsed/Refractory Multiple Myeloma or Lymphoma. Clin. Cancer Res. 2016, 22, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Blondelle, J.; Shapiro, P.; Domenighetti, A.A.; Lange, S. Cullin E3 Ligase Activity Is Required for Myoblast Differentiation. J. Mol. Biol. 2017, 429, 1045–1066. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell 2000, 6, 233–244. [Google Scholar] [CrossRef]
- Sincennes, M.C.; Brun, C.E.; Rudnicki, M.A. Concise Review: Epigenetic Regulation of Myogenesis in Health and Disease. Stem Cells Transl. Med. 2016, 5, 282–290. [Google Scholar] [CrossRef]
- Liu, N.; Nelson, B.R.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 4109–4114. [Google Scholar] [CrossRef] [Green Version]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Verdone, L.; Caserta, M.; Di Mauro, E. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef]
- Mathias, R.A.; Guise, A.J.; Cristea, I.M. Post-translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease. Mol. Cell. Proteom. 2015, 14, 456–470. [Google Scholar] [CrossRef] [Green Version]
- Mal, A.; Sturniolo, M.; Schiltz, R.L.; Ghosh, M.K.; Harter, M.L. A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: Inhibition of the myogenic program. EMBO J. 2001, 20, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.L.; Iezzi, S.; Stiegler, P.; Chen, T.T.; Schiltz, R.L.; Muscat, G.E.; Giordano, A.; Kedes, L.; Wang, J.Y.; Sartorelli, V. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell 2001, 8, 885–897. [Google Scholar] [CrossRef]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070–4075. [Google Scholar] [CrossRef] [Green Version]
- Fulco, M.; Schiltz, R.L.; Iezzi, S.; King, M.T.; Zhao, P.; Kashiwaya, Y.; Hoffman, E.; Veech, R.L.; Sartorelli, V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol. Cell 2003, 12, 51–62. [Google Scholar] [CrossRef]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [Green Version]
- Miska, E.A.; Langley, E.; Wolf, D.; Karlsson, C.; Pines, J.; Kouzarides, T. Differential localization of HDAC4 orchestrates muscle differentiation. Nucleic Acids Res. 2001, 29, 3439–3447. [Google Scholar] [CrossRef] [Green Version]
- Pardo, P.S.; Boriek, A.M. The physiological roles of Sirt1 in skeletal muscle. Aging (Albany NY) 2011, 3, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Rudnicki, M.A.; Schnegelsberg, P.N.; Stead, R.H.; Braun, T.; Arnold, H.H.; Jaenisch, R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 1993, 75, 1351–1359. [Google Scholar] [CrossRef]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef]
- Nabeshima, Y.; Hanaoka, K.; Hayasaka, M.; Esumi, E.; Li, S.; Nonaka, I.; Nabeshima, Y. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 1993, 364, 532–535. [Google Scholar] [CrossRef]
- Shiraishi, S.; Zhou, C.; Aoki, T.; Sato, N.; Chiba, T.; Tanaka, K.; Yoshida, S.; Nabeshima, Y.; Nabeshima, Y.; Tamura, T.A. TBP-interacting protein 120B (TIP120B)/cullin-associated and neddylation-dissociated 2 (CAND2) inhibits SCF-dependent ubiquitination of myogenin and accelerates myogenic differentiation. J. Biol. Chem. 2007, 282, 9017–9028. [Google Scholar] [CrossRef] [Green Version]
- Mal, A.; Harter, M.L. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 1735–1739. [Google Scholar] [CrossRef] [Green Version]
- Ohkawa, Y.; Marfella, C.G.; Imbalzano, A.N. Skeletal muscle specification by myogenin and Mef2D via the SWI/SNF ATPase Brg1. EMBO J. 2006, 25, 490–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacinti, C.; Bagella, L.; Puri, P.L.; Giordano, A.; Simone, C. MyoD recruits the cdk9/cyclin T2 complex on myogenic-genes regulatory regions. J. Cell. Physiol. 2006, 206, 807–813. [Google Scholar] [CrossRef]
- Kwon, D.H.; Eom, G.H.; Ko, J.H.; Shin, S.; Joung, H.; Choe, N.; Nam, Y.S.; Min, H.K.; Kook, T.; Yoon, S.; et al. MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat. Commun. 2016, 7, 10492. [Google Scholar] [CrossRef] [Green Version]
- Lai, Q.Y.; He, Y.Z.; Peng, X.W.; Zhou, X.; Liang, D.; Wang, L. Histone deacetylase 1 induced by neddylation inhibition contributes to drug resistance in acute myelogenous leukemia. Cell Commun. Signal. 2019, 17, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, D.; Hori, D.; Kim, J.H.; Bergman, Y.; Berkowitz, D.E.; Romer, L.H. NEDDylation promotes endothelial dysfunction: A role for HDAC2. J. Mol. Cell. Cardiol. 2015, 81, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef] [Green Version]
- Nomura, Y.; Nakano, M.; Woo Sung, H.; Han, M.; Pandey, D. Inhibition of HDAC6 Activity Protects against Endothelial Dysfunction and Atherogenesis in vivo: A Role for HDAC6 Neddylation. Front. Physiol. 2021, 12, 675724. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Yuan, Z.; Li, Y.; Ling, H.; Izumi, V.; Fang, B.; Fukasawa, K.; Koomen, J.; Chen, J.; Seto, E. Ubiquitinated sirtuin 1 (SIRT1) function is modulated during DNA damage-induced cell death and survival. J. Biol. Chem. 2015, 290, 8904–8912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Xie, L.; Liu, Z.; Li, C.; Liang, Y. MLN4924 Exerts a Neuroprotective Effect against Oxidative Stress via Sirt1 in Spinal Cord Ischemia-Reperfusion Injury. Oxid. Med. Cell. Longev. 2019, 2019, 7283639. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Martin, S.C.; Parkington, J.; Cadena, S.M.; Zhu, J.; Ibebunjo, C.; Summermatter, S.; Londraville, N.; Patora-Komisarska, K.; Widler, L.; et al. HDAC4 Controls Muscle Homeostasis through Deacetylation of Myosin Heavy Chain, PGC-1alpha, and Hsc70. Cell Rep. 2019, 29, 749–763.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward Primers | Backward Primers | |
|---|---|---|
| Hdac4 | CAGGAGATGCTGGCCATGAA | GCACTCTCTTTGCCCTTCTC |
| Hdac5 | GCTTCTTTGGACCAGAGTTCC | CATCTCAGTGGGGATGTTGG |
| Sirt1 | CTTCAGTGTCATGGTTCCTT | ACCGAGGAACTACCTGATTA |
| Myog | GTCCCAACCCAGGAGATCATT | AGTTGGGCATGGTTTCGTCT |
| 36B4 | CGCTTTCTGGAGGGTGTCCGC | TGCCAGGACGCGCTTGTACC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Su, H.; Chen, W. Neddylation Regulates Class IIa and III Histone Deacetylases to Mediate Myoblast Differentiation. Int. J. Mol. Sci. 2021, 22, 9509. https://doi.org/10.3390/ijms22179509
Zhou H, Su H, Chen W. Neddylation Regulates Class IIa and III Histone Deacetylases to Mediate Myoblast Differentiation. International Journal of Molecular Sciences. 2021; 22(17):9509. https://doi.org/10.3390/ijms22179509
Chicago/Turabian StyleZhou, Hongyi, Huabo Su, and Weiqin Chen. 2021. "Neddylation Regulates Class IIa and III Histone Deacetylases to Mediate Myoblast Differentiation" International Journal of Molecular Sciences 22, no. 17: 9509. https://doi.org/10.3390/ijms22179509
APA StyleZhou, H., Su, H., & Chen, W. (2021). Neddylation Regulates Class IIa and III Histone Deacetylases to Mediate Myoblast Differentiation. International Journal of Molecular Sciences, 22(17), 9509. https://doi.org/10.3390/ijms22179509