Abstract
Axenfeld–Rieger syndrome (ARS) encompasses a group of developmental disorders that affect the anterior segment of the eye, as well as systemic developmental defects in some patients. Malformation of the ocular anterior segment often leads to secondary glaucoma, while some patients also present with cardiovascular malformations, craniofacial and dental abnormalities and additional periumbilical skin. Genes that encode two transcription factors, FOXC1 and PITX2, account for almost half of known cases, while the genetic lesions in the remaining cases remain unresolved. Given the genetic similarity between zebrafish and humans, as well as robust antisense inhibition and gene editing technologies available for use in these animals, loss of function zebrafish models for ARS have been created and shed light on the mechanism(s) whereby mutations in these two transcription factors cause such a wide array of developmental phenotypes. This review summarizes the published phenotypes in zebrafish foxc1 and pitx2 loss of function models and discusses possible mechanisms that may be used to target pharmaceutical development and therapeutic interventions.
1. Axenfeld–Rieger Syndrome
Axenfeld–Rieger syndrome (ARS) is a clinically heterogeneous disorder characterized by ocular anomalies with systemic multi-organ system involvement in some patients. This relatively rare disorder, with a prevalence of 1 in 50,000–100,000 live births [1], presents with structural defects in the ocular anterior segment leading to an early onset glaucoma in about 50% of patients. A subset of patients with ARS have developmental anomalies in other tissues and organs that include craniofacial and dental abnormalities [2,3], cerebral vasculature defects that increase in stroke risk [4], hydrocephalus and Dandy-Walker malformation [5,6], and cardiac developmental defects including aberrant formation of valves and the outflow tract [7,8]. Defects in the pituitary gland with secondary endocrinological conditions can also result [9], as can deficits in the auditory system leading to sensorineural hearing loss [10,11,12] and redundant periumbilical skin is also observed in some cases [13,14]. Clinically, ARS is defined by three subtypes assigned by the presence or absence of systemic anomalies in addition to ocular developmental defects, and correlate with mutations in known ARS causing genes. For an in-depth review of phenotypes associated with ARS, please refer to Seifi et al. [1].
ARS is inherited as an autosomal dominant disorder, with the genetic lesion defined in approximately 40% of cases. Mutations or copy number variation (CNVs) in two genes identified through a variety of family-based studies account for ARS with fully penetrant ocular manifestations; FORKHEAD BOX C1 (FOXC1) and PAIRED-LIKE HOMOEDOMAIN (PITX2). DNA lesions involving PITX2 result in ARS type I, in which patients have fully penetrant ocular phenotypes often observed with craniofacial and dental anomalies. Mutations in FOXC1 result in ARS type III, defined by fully penetrant ocular phenotypes often observed with cardiovascular defects and sensorineural hearing loss. While linkage analysis supports an additional gene causing ARS type II on chromosome 13q14 [15], no causative gene has yet been identified. FOXC1 and PITX2 are transcription factors from the forkhead and homeodomain families, respectively, and can regulate gene transcription independently or physically interact on DNA to co-regulate gene transcription [16]. This complex relationship likely accounts for the phenotypic differences and ARS classifications in patients with mutations in one of the two causative genes. Mutations in the CYP1B1 gene have been found in a single family with ARS [17], and while a relatively common cause of congenital glaucoma is some populations [18,19], mutations in CYP1B1 appear to be an extremely rare cause of ARS.
The zebrafish (Danio rerio) has provided valuable mechanistic insights into ARS disease etiology. The zebrafish genome contains two homologues of the FOXC1 gene denoted foxc1a and foxc1b, arising from an ancient duplication in the teleost lineage, and one homologue of PITX2 (pitx2). Gene expression studies using in situ hybridization have highlighted specific cell types that require the expression of foxc1 and pitx2 for normal ocular development. The use of morpholino based antisense inhibition and genome editing have produced zebrafish strains that mimic ARS phenotypes and have provided novel mechanistic data highlighting downstream genes and signaling pathways that are required for ocular and systemic manifestations of the syndrome. This review will focus on the role of foxc1 and pitx2 in the regulation of genes and signaling pathways that regulate formation of the eye, cardiovascular system, and craniofacial skeleton, as defined by loss of function zebrafish mutant strains or antisense inhibition data.
2. Expression of ARS Genes in Zebrafish
At early stages of development, zebrafish express foxc1a and foxc1b in overlapping domains in neural crest cells [4,20] (Figure 1G, (foxc1a)) as well as the lateral plate mesoderm (LPM) [21]. Neural crest cells contribute to the periocular mesenchyme (POM)—a set of cells that are required for closure of the optic fissure and development of the ocular anterior segment [22]. Neural crest cells also populate the central nervous system, craniofacial skeleton, smooth muscle cells of the cerebral vasculature, as well as the cardiac outflow tract and heart valves [23,24,25]. This expression pattern highlights the important role of neural crest cells in development and helps to explain the broad range of phenotypes observed in patients with ARS. The LPM also contributes to the development the heart and cardiovascular system and thus defects in this tissue arising from FOXC1 dysfunction may contribute to cardiovascular anomalies observed in ARS patients [21,26,27]. Although typically known as a developmental disease, foxc1a continues to be expressed in the adult zebrafish eye, including the anterior segment and retinal ganglion cell layer [28], possibly contributing to maintenance of adult ocular tissues.
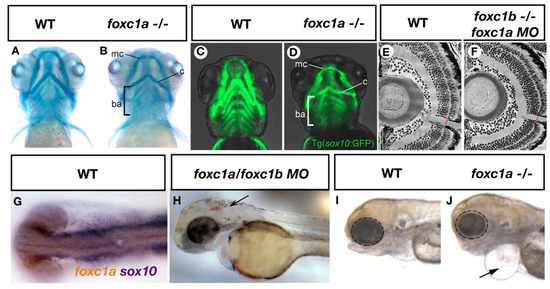
Figure 1.
ARS related phenotypes in zebrafish resulting from loss of foxc1 function. Mutation of foxc1a results in hypoplasia of the Meckel’s cartilage (mc), Ceratohyal cartilages (c) and disorganization of the branchial arches (ba) as assessed by alcian blue (cartilage) staining (A,B) [27]. Such defects may result in part due to decreased sox10 positive neural crest cells observed in these tissues (C,D) [27]. Injection of foxc1a morpholinos into foxc1b homozygous mutants reveals a thinner optic nerve when compared to controls (E,F) [29]. Expression of foxc1a overlaps with sox10 expression in the periocular mesenchyme (G, previously unpublished data from the French lab) while inhibition of both foxc1a and foxc1b paralogs causes cerebral hemorrhages (arrow, H) [4]. Homozygous foxc1a mutants display microphthalmia (dotted circle), cardiac edema (arrow) and jaw hypoplasia in lateral brightfield images (I,J) [27].
The zebrafish genome contains a single pitx2 gene that encodes two isoforms via alternative splicing that correspond to human PITX2A and PITX2C. The expression of pitx2a is found in partially overlapping domains with foxc1a and foxc1b in developing zebrafish embryos. As early as 24 h post fertilization (hpf), pitx2 expression is observed in the periocular mesenchyme [22] (Figure 2A), providing an opportunity for co-regulation of gene expression with Foxc1a and Foxc1b. Like foxc1a and foxc1b, pitx2 is expressed in the neural crest derived tissues of the pharyngeal arches that contribute to the craniofacial skeleton [30]. Additional expression in the dental epithelium and tooth placodes [31,32] likely accounts for dental phenotypes in ARS type 1 patients. Asymmetric expression of the pitx2c isoform in the lateral plate mesoderm [33] (Figure 2B) and expression of pitx2a in the neural crest [34], may contribute to cardiac outflow tract defects and valve incompetence observed in a subset of these ARS patients.
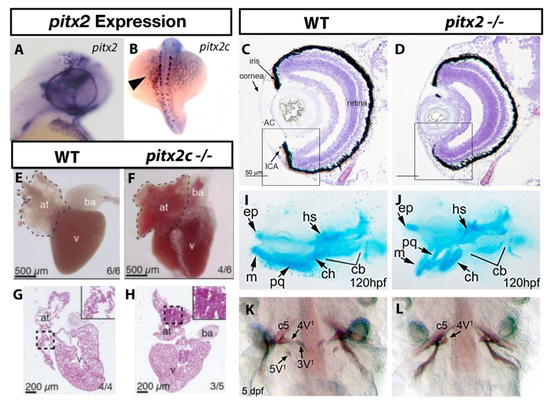
Figure 2.
ARS related phenotypes in zebrafish due to loss of pitx2 function. Expression of pitx2a is found within the brain and periocular mesenchyme (A) [22] while pitx2c is observed in a “left sided” pattern in the lateral plate mesoderm (B) [27]. The anterior ocular chamber is reduced in pitx2 homozygous mutants when compared to wildtype siblings (C,D) [33]. Homozygous pitx2c mutants display hypoplastic cardiac atria and increased fibrosis when compared to wildtype siblings (E–H) [35]. Underdeveloped jaw cartilages are also observed in pitx2 mutants (I,J) [30] as is delayed or absent pharyngeal tooth (3V1 and 5V1) development (K,L) [33]. Abbreviations: atrium (at), bulbus arteriosus (ba), ventricle (v), Anterior, segment (AC), ethmoid plate (ep), hyosympletic (hs), Meckel’s cartilage (mc), palatoquadrate (pq), ceratohyal (ch), ceratobranchial (cb).
3. Ocular Related Phenotypes in foxc1a and foxc1b Mutants
The most distinguishing feature of Axenfeld–Rieger Syndrome is the developmental abnormalities in the ocular anterior segment, leading to an increased risk for early onset glaucoma. Given the ability to create heritable loss of function mutations in zebrafish geusing genome editing techniques, as well as robust antisense (morpholino) inhibition, a number of zebrafish models have been reported that recapitulate the phenotypes of FOXC1-attributable ARS patients. Published alleles of zebrafish foxc1a and foxc1b mutations can be found in Table 1.

Table 1.
Published mutant alleles for zebrafish ARS models.
The zebrafish anterior segment begins to develop as early as 26 hpf when the lens vesicle detaches from the surface ectoderm [45]. By 3 days post fertilization (dpf), rudimentary structures of the anterior segment are present and will continue to develop through the first month of life. Zebrafish with homozygous mutations in foxc1a and foxc1b display underdeveloped or absent anterior segments beginning around 3 dpf [39]. Additional ocular phenotypes, including microphthalmia (Figure 1I,J) and coloboma are often observed in double homozygous foxc1a/foxc1b zebrafish, which are typically not observed in ARS patients. This discrepancy may be the result of complete loss of Foxc1 function in double homozygotes, compared to heterozygous mutations or copy number variations that define ARS. Heterozygous mutations of both foxc1a and foxc1b in zebrafish do not alter early development of the anterior segment, however, published analyses only describe assays performed up to 6 dpf and thus additional analyses on older heterozygous larvae or adults when the anterior segment has matured, could potentially reveal anterior segment anomalies consistent with ARS. Loss of Foxc1 function in zebrafish also leads to endophenotypes of glaucoma, including reduced number of retinal ganglion cells and thinner optic nerves [29] (Figure 1E,F), providing a valuable model to study the increased glaucoma risk associated with ARS.
Given the similarity of phenotypes in zebrafish foxc1 mutants and patients with FOXC1-attributable ARS, mechanistic insights into disease pathology can be gained. The underdeveloped or absent anterior segment in zebrafish foxc1 mutants likely results from the role of foxc1 in regulating the migration and survival of neural crest cells that populate the periocular mesenchyme and subsequently contribute to the iris stroma and cornea [46]. Loss of foxc1 causes migration defects and increased cell death of sox10 positive neural crest cells [4] that contribute to these cell populations.
Certainly, the observed anterior segment defects in foxc1 zebrafish mutants could contribute to mechanism of glaucoma development, given the role of the anterior segment in regulation of intraocular pressure (IOP). However, many ARS patients have normal tension glaucoma (NTG), defined as having normal intraocular pressure, and thus other mechanisms are likely involved. For example, a number of studies have demonstrated that foxc1 regulates the formation of the zebrafish hyaloid and retinal vasculature [39,47]. Defects in embryonic ocular vasculature could reduce trophic support in the eye or allow the build-up of waste products, but how this may specifically affect the RGCs and the optic nerve remains to be determined. Additionally, the reduced numbers of RGCs observed due to loss of Foxc1 function in zebrafish occurred as the result of reduced expression of atonal homolog 7 (atoh7). Atoh7 is required for RCG differentiation during embryonic development [48,49], and mutations in ATOH7 in human populations are associated with persistent hyperplasia of the primary vitreous [50] whereby failure of fetal ocular vascular to regress inhibits the development of adult intrinsic retinal vasculature. Thus, the regulation of atoh7 expression via Foxc1 could account for ocular vascular defects in ARS patients and could influence the development of glaucoma independent of, or in combination with, potential increased IOP caused by the anterior segment dysgenesis. Variants in ATOH7 have also been associated with primary open angle glaucoma [51] and endophenotypes of glaucoma such as optic disc size [52,53,54], indicating its key role in maintaining these structures. This further supports the role for Foxc1 and its transcriptional regulation of atoh7 as a key contributor of RGC and optic nerve health.
In addition to atoh7, other genetic targets regulated by Foxc1a/Foxc1b in zebrafish have been identified that may shed light on potential mechanisms of glaucoma development. Foxc1a/Foxc1b regulates the expression of FOXO1A/foxo1a, a gene expressed in the zebrafish POM that mediates the response to oxidative stress [55]. Manipulation of either foxc1a or foxo1a resulted in aberrant responses to increased oxidative stress and increased cell death in the eye [55], indicating that impaired response to oxidative stress could also be a key facet of glaucoma development due to loss of FOXC1. Foxc1a also regulates the expression of fgf19 [29,56], another gene highly expressed in the zebrafish POM with roles in retinal and lens development [57,58]. Mutations in genes such as foxo1a and fgf19 have not been identified in ARS or glaucoma patients and thus the dysfunction of either gene alone likely cannot cause overt visual defects, however, their reduced expression due to loss of Foxc1 function, as well as other yet to be identified downstream targets, likely contribute to the complex etiology of RGC loss in FOXC1-attributable ARS patients.
4. Craniofacial Defects in Zebrafish foxc1a and foxc1b Mutants
Zebrafish foxc1a mutants, as well as foxc1a/foxc1b double mutants, have craniofacial defects consistent with abnormalities in the facial structure of ARS patients. In these patients, hypertelorism (increased space between the eyes) and a prominent forehead are often described. In zebrafish, foxc1a and foxc1b are expressed in the neural crest cells that populate the first and second pharyngeal arches that give rise to anterior jaw structures. Mutation of foxc1a alone results in craniofacial dysmorphism [27,37] including underdeveloped symplectic cartilage and like many ocular phenotypes observed in these mutants, severity increased in double foxc1a/foxc1b mutants. Phenotypes in double homozygous mutants included under-developed palatoquadrate and hyomandibula cartilages [37], demonstrating that foxc1a and foxc1b play critical roles in the development of anterior facial cartilages (Figure 1A–D, foxc1a). Analysis of genetic targets downstream of foxc1 revealed both foxc1a and foxc1b regulate Sox9-dependent expression of cartilage specification genes, accounting for reduced jaw structures in these animals. While Foxc1 does not directly regulate sox9 expression, loss of foxc1 paralogs in zebrafish causes a decrease in chromatin accessibility for transcription factors such as Sox9 in chondrocytes [38], and thus supports the hypothesis that Foxc1 drives cartilage development through a chromatin remodeling mechanism.
Analysis of heterozygous foxc1a mutants in conjunction with homozygous loss of foxc1b (which survive to adulthood) demonstrates craniofacial abnormalities in adults that include a misshapen head that closely mimics that of ARS patients, as well as mandibular retrognathia and dorsally positioned eyes [39]. Combined with larval-based studies, these data demonstrate that Foxc1 regulates chromatin accessibility in chondrocytes to shape the jaw and head in zebrafish, with such mechanisms likely contributing the craniofacial dysmorphia observed in ARS patients with mutations or CNVs involving FOXC1.
5. Cardiovascular Anomalies in Zebrafish foxc1 Mutants
Cardiac anomalies have been described in ARS patients with FOXC1 mutations or CNVs that include hypoplastic ventricular outflow tract morphology, dysplastic arcade mitral valve, and atrial septal defect [7,59]. Within the brain, cerebral small vessel disease (CSVD) has been described, which includes increased perivascular spaces, subclinical infarcts and white matter hyperintensities [4]—all of which increase stroke risk [60]. These clinical phenotypes clearly indicate that FOXC1 plays an important role in early heart and cerebral vascular development, with such phenotypes recapitulated in zebrafish loss of foxc1 function models.
In zebrafish, mutation of foxc1a alone or in combination with foxc1b leads to cardiac phenotypes that include hypoplastic myocardium and ventricular outflow tract, as well as defects in cardiac valve formation that are similar to that observed in patients with FOXC1-attributible ARS [21,26]. While zebrafish studies generally support a role or foxc1 in heart development, reports differ in their analysis of heart morphology, assay different time points, and study different combinations of mutations. For example, Yue et al. [21] demonstrate a hypoplastic myocardium, shorter outflow tract, defective primitive valve leaflets, and cardiac edema at during early larval development (4-5 dpf) of foxc1a−/− embryos (cardiac edema in foxc1−/− displayed in Figure 1J). Ferre-Fernandez et al. [39] assayed cardiac trabecular zone thickness, outflow tract and AV valve morphology in foxc1a+/−; foxc1b−/− embryos at 6 dpf and found no difference. Compact zone thickness, however, was significantly larger in these fish when compared to wildtype siblings. While these studies support a role for foxc1 in heart development, they indicate that homozygous loss of foxc1a is required to induce heart defects similar to ARS patients, who typically have heterozygous mutations or CNVs involving FOXC1.
Utilizing foxc1 homozygous loss of function embryos, a role for this gene in regulating cardiac progenitor specification and atrioventricular canal (AVC) formation has been proposed. Such studies show that Foxc1a directly binds to the promoter of nkx2.5 [26], a gene required for cardiac progenitor specification in the lateral plate mesoderm (LPM). The reduced expression of nkx2.5 in zebrafish foxc1 homozygous mutants provides mechanistic insight into the hypoplastic myocardium observed in ARS patients. Furthermore, genes expressed specifically in the AVC are downregulated in foxc1 homozygous mutants, including notch1b, tbx2b and bmp4, providing additional genetic targets that downstream of foxc1 that may contribute to cardiac defects. While foxc1a is expressed in LPM, the heart also receives a contribution of cells from the neural crest, and thus foxc1 expression in neural crest cells could also influence the presence and severity of cardiac phenotypes observed in ARS patients and zebrafish foxc1 mutants.
As CSVD in ARS patients increases stroke risk, analysis of cerebral vasculature due to loss of foxc1 function in zebrafish has been undertaken. Combined morpholino inhibition of foxc1a and foxc1b causes cerebral hemorrhages [4] (Figure 1H), a phenotype that is recapitulated in mutant strains [27]. A reduction in neural crest derived pericytes that associate with nascent cerebral vasculature [4,42] is observed, resulting from aberrant neural crest cell migration to the head and increased cell death in sox10 positive neural crest cell populations. Furthermore, defects in cerebral angiogenesis [39] have been reported, including incomplete connections between the earliest developing cerebral arteries that likely contribute to the hemorrhagic phenotype.
6. ARS-Related Defects in Zebrafish pitx2 Mutants
Like FOXC1, mutation of PITX2 results in ARS with early onset glaucoma in many patients. In zebrafish, both morpholino inhibition or mutation of pitx2 results in decreased size of the anterior segment during larval development [30,33,43] (Figure 2C,D). Five-day old pitx2 homozygous mutant larvae have smaller eyes, malformation of the iridocorneal angle, and increased mesenchyme thickness around the cornea with such phenotypes largely phenocopied by morpholino inhibition of pitx2. Interestingly, a four-generation pedigree involving anterior segment dysgenesis (ASD) with early onset glaucoma (a diagnosis similar to ARS) identified a 748 kb deletion containing a conserved PITX2 regulatory element [61]. Removal of this orthologous region using genome editing in zebrafish reduced pitx2 expression and like mutation of the gene itself, resulted in reduced or shallow anterior chambers. These data clearly demonstrate that mutation of pitx2, or genomic modifications that alter pitx2 expression result in anterior segment defects reminiscent of ARS. Although no direct quantification of optic nerve morphology or RGC number has been undertaken, these data demonstrate that mutant pitx2 zebrafish represent an ideal model for subsequent studies that focus on the mechanism by which glaucoma may result in patients with PITX2-attributable ARS.
Analysis of genetic targets in the eye demonstrates that Pitx2 deficiency reduces the expression of the Wnt ligands (wnt3, wnt4a, wnt6b, wnt7aa, wnt9b and wnt10a) [43] as well as the Wnt antagonist dkk2 in the anterior segment of the eye [30]. A role for Wnt signaling in eye development has been hypothesized, and thus a regulation of Wnt signaling via Pitx2 could represent a key facet of the ocular phenotypes observed in zebrafish pitx2 mutants. Wnt signaling plays a key role in the development of neural crest cells in part through regulation of foxd3 and sox10 [25,62] that are required for the earliest events of neural crest cell induction. Given pitx2 expression in early neural crest cells, and its continued expression in the adult anterior segment [28], pitx2 is likely required for development and maintenance of the anterior segment in part through a Wnt dependent mechanism.
Analysis of additional ocular targets due to pitx2 depletion demonstrate normal expression of pax6a that is required for anterior segment development [30]. While no effect on the expression of the anterior segment marker pax6a was observed, it has been shown that pitx2 expression is regulated by Pax6a/b in zebrafish [28] which may contribute to the anterior segment phenotypes (aniridia, Peter’s anomaly) observed in patients and zebrafish with PAX6/pax6 mutations [63]. The expression of crystallins required for lens development were also unchanged due to depletion of Pitx2, however, blood vessel defects are highlighted in the embryonic hyaloid vasculature and thus like foxc1, pitx2 may play an important role in early trophic support of the lens and anterior segment structures early in development.
7. Craniofacial Defects Due to Loss of pitx2
Patients with type I ARS typically have dysmorphic craniofacial features that include maxillary and dental hypoplasia, in addition to the anterior segment ocular phenotypes that define ARS. Similar to foxc1, disruption of pitx2 using antisense morpholinos or with genome editing induced mutations disrupts pharyngeal arch cartilage and jaw formation. Specific defects were observed in the Meckel’s and ceratohyal cartilages, which were under-developed and positioned abnormally [30,43] (Figure 2I,J). Analysis of sox10 positive neural crest cells that line the pharyngeal arches and jaw elements demonstrate reduced cell number [36], indicating craniofacial dysmorphism in foxc1 and pitx2 depleted zebrafish likely involves overlapping mechanisms of decreased neural crest cell migration and survival.
Zebrafish develop pharyngeal teeth that are highly similar to the oral teeth of humans, and mutations that disrupt the DNA binding of Pitx2 in zebrafish cause reduced or absent tooth production [33] (Figure 2K,L)-a common phenotype observed in PITX2-attributable ARS. pitx2 is the earliest expressed transcription factor in tooth bud epithelium and continues to be expressed through tooth development in mice [64] and mammalian cell culture studies demonstrate that Pitx2 acts directly on the promoter of dlx2 [65,66], a gene required for tooth development. In zebrafish, morpholino inhibition of pitx2 reduces dlx2a expression in the posterior pharyngeal arches [30], demonstrating the conservation of transcription factors that are required for tooth development between fish and mammals and that zebrafish can serve as a useful model to understand defects in tooth development due to loss of PITX2 in ARS patients.
8. Cardiovascular Defects Due to Depletion of pitx2 in Zebrafish
Like humans, the zebrafish pitx2 gene encodes multiple isoforms of the Pitx2 protein produced by the use of different promoters and alternative splicing. At least four transcripts encoding four isoforms are present in humans (PITX2a, PITX2b, PITX2c, PITX2d) [67] with two homologous isoforms currently annotated in the zebrafish genome (pitx2a and pitx2c) [33]. During early development, pitx2a is predominantly expressed in neural crest cells and thus mutations that affect this isoform may result in ocular, craniofacial, and cardiovascular anomalies associated with ARS, while pitx2c is predominately expressed in the lateral plate mesoderm and the heart, which could contribute to cardiac defects observed in some ARS patients. Patients with PITX2-attributable ARS may thus present with or without cardiac defects that may depend in part on the location of the disease-causing mutations [67,68]. Mutant zebrafish have been generated that specifically alter one (pitx2a) or both isoforms (pitx2a and pitx2c) in order to identify similar defects in fish, and to gain mechanistic understanding of such phenotypes in patients and animal models.
Mutations in zebrafish that affect both isoforms (or just pitx2c) did not result in cardiac defects consistent with PITX2-attributable ARS in one study, although asymmetric looping and overall morphology were the only phenotypes tested [33]. A second study analyzed adult heart morphology in a pitx2c specific mutant strain and uncovered cardiomyopathy with fibrosis (Figure 2E–H) and arrythmias, particularly when the fish are stressed [35]. This is consistent with cardiac defects observed in some ARS patients and with the known role of PITX2 as a causative gene for atrial fibrillation [69,70]. While morphological defects (Figure 2E–H) and arrythmias were analyzed in adult hearts, analysis of gene expression in larval hearts revealed dysregulation of genes involved in mitochondrial function and points toward a mechanism whereby altered cellular metabolism in the heart could contribute to later heart dysfunction. Lastly, while foxc1 regulates expression of nxk2.5 in the lateral plate mesoderm, nkx2.5 is required for maintenance of pitx2 expression in this area [71], highlighting a potential overlap in mechanism between foxc1 and pitx2 in regulating heart development.
9. Summary
Analysis of zebrafish foxc1 and pitx2 loss of function models provides understanding of the mechanisms that lead to most ARS related phenotypes. Both genes when mutated in zebrafish result in defects consistent with mammalian ARS models and patient phenotypes. Zebrafish ARS mutants have ocular anterior chamber defects, and foxc1 mutants additionally display endophenotypes of glaucoma. Disruption of foxc1 or pitx2 in zebrafish display craniofacial anomalies consistent with ARS, as well as cardiovascular defects that are often observed in patients. pitx2 mutants additionally display tooth hypoplasia that is often observed in Type 1 ARS. Mechanisms involving downstream gene regulation are beginning to be uncovered using homozygous embryos and larvae, however, analysis of adult phenotypes in heterozygous mutants is somewhat lacking in the literature. The continued analysis of such mutants will reveal novel insights into disease mechanisms, and given the utility of zebrafish for translational research, pharmaceutical approaches using high-throughput drug screening for phenotypic rescue provides a path toward testing therapeutic interventions.
Funding
This work was funded in part by a grant from the Glaucoma Research Society of Canada, and Memorial University of Newfoundland and Labrador.
Institutional Review Board Statement
Work contained in this article that was performed in the French Lab was approved by the Canadian Council of Animal Care and Memorial University’s animal welfare review committee.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I would like to thank Danielle French for critical comments on this manuscript. The French lab is funded in part by the Glaucoma Research Society of Canada which contributed to the open access cost of publishing this review. Permission for data included in Figure 1 and Figure 2 were grated from Elsevier, The Natural Academy of Sciences, or through the Creative Commons open access agreement.
Conflicts of Interest
The author declares no conflict of interest.
References
- Seifi, M.; Walter, M.A. Axenfeld-Rieger syndrome. Clin. Genet. 2018, 93, 1123–1130. [Google Scholar] [CrossRef]
- Waldron, J.M.; Mcnamara, C.; Hewson, A.R.; Mcnamara, C.M. Axenfeld-Rieger syndrome (ARS): A review and case report. Spec. Care Dent. 2010, 30, 218–222. [Google Scholar] [CrossRef]
- Dressler, S.; Meyer-Marcotty, P.; Weisschuh, N.; Jablonski-Momeni, A.; Pieper, K.; Gramer, G.; Gramer, E. Dental and Craniofacial Anomalies Associated with Axenfeld-Rieger Syndrome with PITX2 Mutation. Case Rep. Med. 2010, 2010, 621984. [Google Scholar] [CrossRef]
- French, C.R.; Seshadri, S.; Destefano, A.L.; Fornage, M.; Arnold, C.R.; Gage, P.J.; Skarie, J.M.; Dobyns, W.B.; Millen, K.J.; Liu, T.; et al. Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J. Clin. Investig. 2014, 124, 4877–4881. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Lehmann, O.J.; Hudgins, L.; Chizhikov, V.V.; Bassuk, A.G.; Ades, L.C.; Krantz, I.D.; Dobyns, W.B.; Millen, K.J. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat. Genet. 2009, 41, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Maclean, K.; Smith, J.; St Heaps, L.; Chia, N.; Williams, R.; Peters, G.B.; Onikul, E.; McCrossin, T.; Lehmann, O.J.; Adès, L.C. Axenfeld-Rieger malformation and distinctive facial features: Clues to a recognizable 6p25 microdeletion syndrome. Am. J. Med. Genet. A 2005, 132, 381–385. [Google Scholar] [CrossRef]
- Gripp, K.W.; Hopkins, E.; Jenny, K.; Thacker, D.; Salvin, J. Cardiac anomalies in Axenfeld-Rieger syndrome due to a novel FOXC1 mutation. Am. J. Med. Genet. A 2013, 161, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Mammi, I.; De Giorgio, P.; Clementi, M.; Tenconi, R. Cardiovascular anomaly in Rieger Syndrome: Heterogeneity or contiguity? Acta Ophthalmol. Scand. 1998, 76, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Idrees, F.; Bloch-Zupan, A.; Free, S.L.; Vaideanu, D.; Thompson, P.J.; Ashley, P.; Brice, G.; Rutland, P.; Bitner-Glindzicz, M.; Khaw, P.T.; et al. A novel homeobox mutation in the PITX2 gene in a family with Axenfeld-Rieger syndrome associated with brain, ocular, and dental phenotypes. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Nakamura, T.; Hosono, K.; Yamaguchi, T.; Hiratsuka, Y.; Hotta, Y.; Takahashi, M. Sensorineural hearing loss and hypoplastic cochlea in Axenfeld-Rieger syndrome with FOXC1 mutation. Auris Nasus Larynx 2021, 48, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, A.C.; Wiggs, J.L. Childhood glaucoma genes and phenotypes: Focus on FOXC1 mutations causing anterior segment dysgenesis and hearing loss. Exp. Eye Res. 2020, 190, 107893. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, E.T., Jr.; Eliott, D.; Miller, N.R.; Maumenee, I.H.; Green, W.R. Familial Axenfeld-Rieger anomaly, atrial septal defect, and sensorineural hearing loss: A possible new genetic syndrome. Arch. Ophthalmol. 1998, 116, 78–82. [Google Scholar] [CrossRef]
- Law, S.K.; Sami, M.; Piri, N.; Coleman, A.L.; Caprioli, J. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol. Vis. 2011, 17, 1231–1238. [Google Scholar]
- Zhang, L.; Peng, Y.; Ouyang, P.; Liang, Y.; Zeng, H.; Wang, N.; Duan, X.; Shi, J. A novel frameshift mutation in the PITX2 gene in a family with Axenfeld-Rieger syndrome using targeted exome sequencing. BMC Med. Genet. 2019, 20, 105. [Google Scholar] [CrossRef]
- Phillips, J.C.; del Bono, E.A.; Haines, J.L.; Pralea, A.M.; Cohen, J.S.; Greff, L.J.; Wiggs, J.L. A second locus for Rieger syndrome maps to chromosome 13q14. Am. J. Hum. Genet. 1996, 59, 613–619. [Google Scholar] [PubMed]
- Berry, F.B.; Lines, M.A.; Oas, J.M.; Footz, T.; Underhill, D.A.; Gage, P.J.; Walter, M.A. Functional interactions between FOXC1 and PITX2 underlie the sensitivity to FOXC1 gene dose in Axenfeld-Rieger syndrome and anterior segment dysgenesis. Hum. Mol. Genet. 2006, 15, 905–919. [Google Scholar] [CrossRef]
- Tanwar, M.; Dada, T.; Dada, R. Axenfeld-Rieger Syndrome Associated with Congenital Glaucoma and Cytochrome P4501B1 Gene Mutations. Case Rep. Med. 2010, 2010, 212656. [Google Scholar] [CrossRef]
- Jubair, S.; N Al-Rubae’i, S.H.; M Al-Sharifi, A.N.; Jabbar Suleiman, A.A. Investigation of CYP1B1 Gene Involvement in Primary Congenital Glaucoma in Iraqi Children. Middle East Afr. J. Ophthalmol. 2019, 26, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Tran-Viet, K.N.; Yanovitch, T.L.; Freedman, S.F.; Klemm, T.; Call, W.; Powell, C.; Ravichandran, A.; Metlapally, R.; Nading, E.B. CYP1B1, MYOC, and LTBP2 mutations in primary congenital glaucoma patients in the United States. Am. J. Ophthalmol. 2013, 155, 508–517.e5. [Google Scholar] [CrossRef]
- Topczewska, J.M.; Topczewski, J.; Solnica-Krezel, L.; Hogan, B.L. Sequence and expression of zebrafish foxc1a and foxc1b, encoding conserved forkhead/winged helix transcription factors. Mech. Dev. 2001, 100, 343–347. [Google Scholar] [CrossRef]
- Yue, Y.; Jiang, M.; He, L.; Zhang, Z.; Zhang, Q.; Gu, C.; Liu, M.; Li, N.; Zhao, Q. The transcription factor Foxc1a in zebrafish directly regulates expression of nkx2.5, encoding a transcriptional regulator of cardiac progenitor cells. J. Biol. Chem. 2018, 293, 638–650. [Google Scholar] [CrossRef]
- Van Der Meulen, K.L.; Vöcking, O.; Weaver, M.L.; Meshram, N.N.; Famulski, J.K. Spatiotemporal Characterization of Anterior Segment Mesenchyme Heterogeneity during Zebrafish Ocular Anterior Segment Development. Front. Cell Dev. Biol. 2020, 8, 379. [Google Scholar] [CrossRef]
- Girolamo, F.; de Trizio, I.; Errede, M.; Longo, G.; d’Amati, A.; Virgintino, D. Neural crest cell-derived pericytes act as pro-angiogenic cells in human neocortex development and gliomas. Fluids Barriers CNS 2021, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- George, R.M.; Maldonado-Velez, G.; Firulli, A.B. The heart of the neural crest: Cardiac neural crest cells in development and regeneration. Development 2020, 147, dev188706. [Google Scholar] [CrossRef]
- Rocha, M.; Singh, N.; Ahsan, K.; Beiriger, A.; Prince, V.E. Neural crest development: Insights from the zebrafish. Dev. Dyn. 2020, 249, 88–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liang, D.; Yue, Y.; He, L.; Li, N.; Jiang, D.; Hu, P.; Zhao, Q. Axenfeld-Rieger syndrome-associated mutants of the transcription factor FOXC1 abnormally regulate NKX2-5 in model zebrafish embryos. J. Biol. Chem. 2020, 295, 11902–11913. [Google Scholar] [CrossRef]
- Chrystal, P.W.; French, C.R.; Jean, F.; Havrylov, S.; van Baarle, S.; Peturson, A.M.; Xu, P.; Crump, J.G.; Pilgrim, D.B.; Lehmann, O.J.; et al. The Axenfeld-Rieger Syndrome Gene FOXC1 Contributes to Left-Right Patterning. Genes 2021, 12, 170. [Google Scholar] [CrossRef]
- Takamiya, M.; Weger, B.D.; Schindler, S.; Beil, T.; Yang, L.; Armant, O.; Ferg, M.; Schlunck, G.; Reinhard, T.; Dickmeis, T.; et al. Molecular description of eye defects in the zebrafish Pax6b mutant, sunrise, reveals a Pax6b-dependent genetic network in the developing anterior chamber. PLoS ONE 2015, 10, e0117645. [Google Scholar]
- Umali, J.; Hawkey-Noble, A.; French, C.R. Loss of foxc1 in zebrafish reduces optic nerve size and cell number in the retinal ganglion cell layer. Vis. Res. 2019, 156, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Semina, E.V. pitx2 Deficiency results in abnormal ocular and craniofacial development in zebrafish. PLoS ONE 2012, 7, e30896. [Google Scholar] [CrossRef]
- Aigler, S.R.; Jandzik, D.; Hatta, K.; Uesugi, K.; Stock, D.W. Selection and constraint underlie irreversibility of tooth loss in cypriniform fishes. Proc. Natl. Acad. Sci. USA 2014, 111, 7707–7712. [Google Scholar] [CrossRef] [PubMed]
- Jackman, W.R.; Yoo, J.J.; Stock, D.W. Hedgehog signaling is required at multiple stages of zebrafish tooth development. BMC Dev. Biol. 2010, 10, 119. [Google Scholar] [CrossRef]
- Ji, Y.; Buel, S.M.; Amack, J.D. Mutations in zebrafish pitx2 model congenital malformations in Axenfeld-Rieger syndrome but do not disrupt left-right placement of visceral organs. Dev. Biol. 2016, 416, 69–81. [Google Scholar] [CrossRef]
- Ai, D.; Liu, W.; Ma, L.; Dong, F.; Lu, M.F.; Wang, D.; Verzi, M.P.; Cai, C.; Gage, P.J.; Evans, S.; et al. Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev. Biol. 2006, 296, 437–449. [Google Scholar] [CrossRef]
- Collins, M.M.; Ahlberg, G.; Hansen, C.V.; Guenther, S.; Marín-Juez, R.; Sokol, A.M.; El-Sammak, H.; Piesker, J.; Hellsten, Y.; Olesen, M.S.; et al. Early sarcomere and metabolic defects in a zebrafish pitx2c cardiac arrhythmia model. Proc. Natl. Acad. Sci. USA 2019, 116, 24115–24121. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yue, Y.; Dong, X.; Jia, W.; Li, K.; Liang, D.; Dong, Z.; Wang, X.; Nan, X.; Zhang, Q.; et al. Zebrafish foxc1a plays a crucial role in early somitogenesis by restricting the expression of aldh1a2 directly. J. Biol. Chem. 2015, 290, 10216–10228. [Google Scholar] [CrossRef]
- Xu, P.; Balczerski, B.; Ciozda, A.; Louie, K.; Oralova, V.; Huysseune, A.; Crump, J.G. Fox proteins are modular competency factors for facial cartilage and tooth specification. Development 2018, 145, dev165498. [Google Scholar] [CrossRef]
- Xu, P.; Yu, H.V.; Tseng, K.C.; Flath, M.; Fabian, P.; Segil, N.; Crump, J.G. Foxc1 establishes enhancer accessibility for craniofacial cartilage differentiation. eLife 2021, 10, e63595. [Google Scholar] [CrossRef] [PubMed]
- Ferre-Fernandez, J.J.; Sorokina, E.A.; Thompson, S.; Collery, R.F.; Nordquist, E.; Lincoln, J.; Semina, E.V. Disruption of foxc1 genes in zebrafish results in dosage-dependent phenotypes overlapping Axenfeld-Rieger syndrome. Hum. Mol. Genet. 2020, 29, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Hayer, K.; Hogenesch, J.B.; Granato, M. Zebrafish foxc1a drives appendage-specific neural circuit development. Development 2015, 142, 753–762. [Google Scholar] [CrossRef][Green Version]
- Shin, M.; Nozaki, T.; Idrizi, F.; Isogai, S.; Ogasawara, K.; Ishida, K.; Yuge, S.; Roscoe, B.; Wolfe, S.A.; Fukuhara, S.; et al. Valves Are a Conserved Feature of the Zebrafish Lymphatic System. Dev. Cell 2019, 51, 374–386.e5. [Google Scholar] [CrossRef]
- Whitesell, T.R.; Chrystal, P.W.; Ryu, J.R.; Munsie, N.; Grosse, A.; French, C.R.; Workentine, M.L.; Li, R.; Zhu, L.J.; Waskiewicz, A.; et al. foxc1 is required for embryonic head vascular smooth muscle differentiation in zebrafish. Dev. Biol. 2019, 453, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Hendee, K.E.; Sorokina, E.A.; Muheisen, S.S.; Reis, L.M.; Tyler, R.C.; Markovic, V.; Cuturilo, G.; Link, B.A.; Semina, E.V. PITX2 deficiency and associated human disease: Insights from the zebrafish model. Hum. Mol. Genet. 2018, 27, 1675–1695. [Google Scholar] [CrossRef]
- Collins, M.M.; Maischein, H.M.; Dufourcq, P.; Charpentier, M.; Blader, P.; Stainier, D.Y. Pitx2c orchestrates embryonic axis extension via mesendodermal cell migration. eLife 2018, 7, e34880. [Google Scholar] [CrossRef] [PubMed]
- Soules, K.A.; Link, B.A. Morphogenesis of the anterior segment in the zebrafish eye. BMC Dev. Biol. 2005, 5, 12. [Google Scholar] [CrossRef]
- Akula, M.; Park, J.W.; West-Mays, J.A. Relationship between neural crest cell specification and rare ocular diseases. J. Neurosci. Res. 2019, 97, 7–15. [Google Scholar] [CrossRef]
- Skarie, J.M.; Link, B.A. FoxC1 is essential for vascular basement membrane integrity and hyaloid vessel morphogenesis. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5026–5034. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.N.; Finger-Baier, K.C.; Roeser, T.; Staub, W.; Baier, H. Retinal ganglion cell genesis requires lakritz, a Zebrafish atonal Homolog. Neuron 2001, 30, 725–736. [Google Scholar] [CrossRef]
- Kay, J.N.; Link, B.A.; Baier, H. Staggered cell-intrinsic timing of ath5 expression underlies the wave of ganglion cell neurogenesis in the zebrafish retina. Development 2005, 132, 2573–2585. [Google Scholar] [CrossRef] [PubMed]
- Prasov, L.; Masud, T.; Khaliq, S.; Mehdi, S.Q.; Abid, A.; Oliver, E.R.; Silva, E.D.; Lewanda, A.; Brodsky, M.C.; Borchert, M.; et al. ATOH7 mutations cause autosomal recessive persistent hyperplasia of the primary vitreous. Hum. Mol. Genet. 2012, 21, 3681–3694. [Google Scholar] [CrossRef]
- Chen, J.H.; Wang, D.; Huang, C.; Zheng, Y.; Chen, H.; Pang, C.P.; Zhang, M. Interactive effects of ATOH7 and RFTN1 in association with adult-onset primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 779–785. [Google Scholar] [CrossRef]
- Venturini, C.; Nag, A.; Hysi, P.G.; Wang, J.J.; Wong, T.Y.; Healey, P.R.; Mitchell, P.; Hammond, C.J.; Viswanathan, A.C.; Wellcome Trust Case Control Consortium 2; et al. Clarifying the role of ATOH7 in glaucoma endophenotypes. Br. J. Ophthalmol. 2014, 98, 562–566. [Google Scholar] [CrossRef]
- Macgregor, S.; Hewitt, A.W.; Hysi, P.G.; Ruddle, J.B.; Medland, S.E.; Henders, A.K.; Gordon, S.D.; Andrew, T.; McEvoy, B.; Sanfilippo, P.G.; et al. Genome-wide association identifies ATOH7 as a major gene determining human optic disc size. Hum. Mol. Genet. 2010, 19, 2716–2724. [Google Scholar] [CrossRef]
- Ramdas, W.D.; van Koolwijk, L.M.; Ikram, M.K.; Jansonius, N.M.; de Jong, P.T.; Bergen, A.A.; Isaacs, A.; Amin, N.; Aulchenko, Y.S.; Wolfs, R.C.; et al. A genome-wide association study of optic disc parameters. PLoS Genet. 2010, 6, e1000978. [Google Scholar] [CrossRef] [PubMed]
- Berry, F.B.; Skarie, J.M.; Mirzayans, F.; Fortin, Y.; Hudson, T.J.; Raymond, V.; Link, B.A.; Walter, M.A. FOXC1 is required for cell viability and resistance to oxidative stress in the eye through the transcriptional regulation of FOXO1A. Hum. Mol. Genet. 2008, 17, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, Y.; Skarie, J.M.; Footz, T.; Berry, F.B.; Link, B.A.; Walter, M.A. FGF19 is a target for FOXC1 regulation in ciliary body-derived cells. Hum. Mol. Genet. 2006, 15, 3229–3240. [Google Scholar] [CrossRef]
- Nakayama, Y.; Miyake, A.; Nakagawa, Y.; Mido, T.; Yoshikawa, M.; Konishi, M.; Itoh, N. Fgf19 is required for zebrafish lens and retina development. Dev. Biol. 2008, 313, 752–766. [Google Scholar] [CrossRef] [PubMed]
- Vinothkumar, S.; Rastegar, S.; Takamiya, M.; Ertzer, R.; Strähle, U. Sequential and cooperative action of Fgfs and Shh in the zebrafish retina. Dev. Biol. 2008, 314, 200–214. [Google Scholar] [CrossRef]
- Du, R.F.; Huang, H.; Fan, L.L.; Li, X.P.; Xia, K.; Xiang, R. A Novel Mutation of FOXC1 (R127L) in an Axenfeld-Rieger Syndrome Family with Glaucoma and Multiple Congenital Heart Diseases. Ophthalmic Genet. 2016, 37, 111–115. [Google Scholar] [PubMed]
- Cannistraro, R.J.; Badi, M.; Eidelman, B.H.; Dickson, D.W.; Middlebrooks, E.H.; Meschia, J.F. CNS small vessel disease: A clinical review. Neurology 2019, 92, 1146–1156. [Google Scholar] [CrossRef]
- Volkmann, B.A.; Zinkevich, N.S.; Mustonen, A.; Schilter, K.F.; Bosenko, D.V.; Reis, L.M.; Broeckel, U.; Link, B.A.; Semina, E.V. Potential novel mechanism for Axenfeld-Rieger syndrome: Deletion of a distant region containing regulatory elements of PITX2. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1450–1459. [Google Scholar] [CrossRef][Green Version]
- Lewis, J.L.; Bonner, J.; Modrell, M.; Ragland, J.W.; Moon, R.T.; Dorsky, R.I.; Raible, D.W. Reiterated Wnt signaling during zebrafish neural crest development. Development 2004, 131, 1299–1308. [Google Scholar] [CrossRef]
- Hanson, I.M.; Fletcher, J.M.; Jordan, T.; Brown, A.; Taylor, D.; Adams, R.J.; Punnett, H.H.; van Heyningen, V. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat. Genet. 1994, 6, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Hjalt, T.A.; Semina, E.V.; Amendt, B.A.; Murray, J.C. The Pitx2 protein in mouse development. Dev. Dyn. 2000, 218, 195–200. [Google Scholar] [CrossRef]
- Espinoza, H.M.; Cox, C.J.; Semina, E.V.; Amendt, B.A. A molecular basis for differential developmental anomalies in Axenfeld-Rieger syndrome. Hum. Mol. Genet. 2002, 11, 743–753. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Green, P.D.; Hjalt, T.A.; Kirk, D.E.; Sutherland, L.B.; Thomas, B.L.; Sharpe, P.T.; Snead, M.L.; Murray, J.C.; Russo, A.F.; Amendt, B.A. Antagonistic regulation of Dlx2 expression by PITX2 and Msx2: Implications for tooth development. Gene Expr. 2001, 9, 265–281. [Google Scholar] [CrossRef]
- Zhao, C.M.; Peng, L.Y.; Li, L.; Liu, X.Y.; Wang, J.; Zhang, X.L.; Yuan, F.; Li, R.G.; Qiu, X.B.; Yang, Y.Q. PITX2 Loss-of-Function Mutation Contributes to Congenital Endocardial Cushion Defect and Axenfeld-Rieger Syndrome. PLoS ONE 2015, 10, e0124409. [Google Scholar] [CrossRef] [PubMed]
- Strungaru, M.H.; Dinu, I.; Walter, M.A. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Investig. Ophthalmol. Vis. Sci. 2007, 48, 228–237. [Google Scholar] [CrossRef]
- Zhou, Y.M.; Zheng, P.X.; Yang, Y.Q.; Ge, Z.M.; Kang, W.Q. A novel PITX2c lossoffunction mutation underlies lone atrial fibrillation. Int. J. Mol. Med. 2013, 32, 827–834. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Xu, Y.J.; Li, R.G.; Qu, X.K.; Fang, W.Y.; Liu, X. Prevalence and spectrum of PITX2c mutations associated with familial atrial fibrillation. Int. J. Cardiol. 2013, 168, 2873–2876. [Google Scholar] [CrossRef]
- Shiratori, H.; Sakuma, R.; Watanabe, M.; Hashiguchi, H.; Mochida, K.; Sakai, Y.; Nishino, J.; Saijoh, Y.; Whitman, M.; Hamada, H. Two-step regulation of left-right asymmetric expression of Pitx2: Initiation by nodal signaling and maintenance by Nkx2. Mol. Cell 2001, 7, 137–149. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).