
Quinolone Resistance of Actinobacillus pleuropneumoniae Revealed through Genome and Transcriptome Analyses

 ,
,
Abstract
:1. Introduction
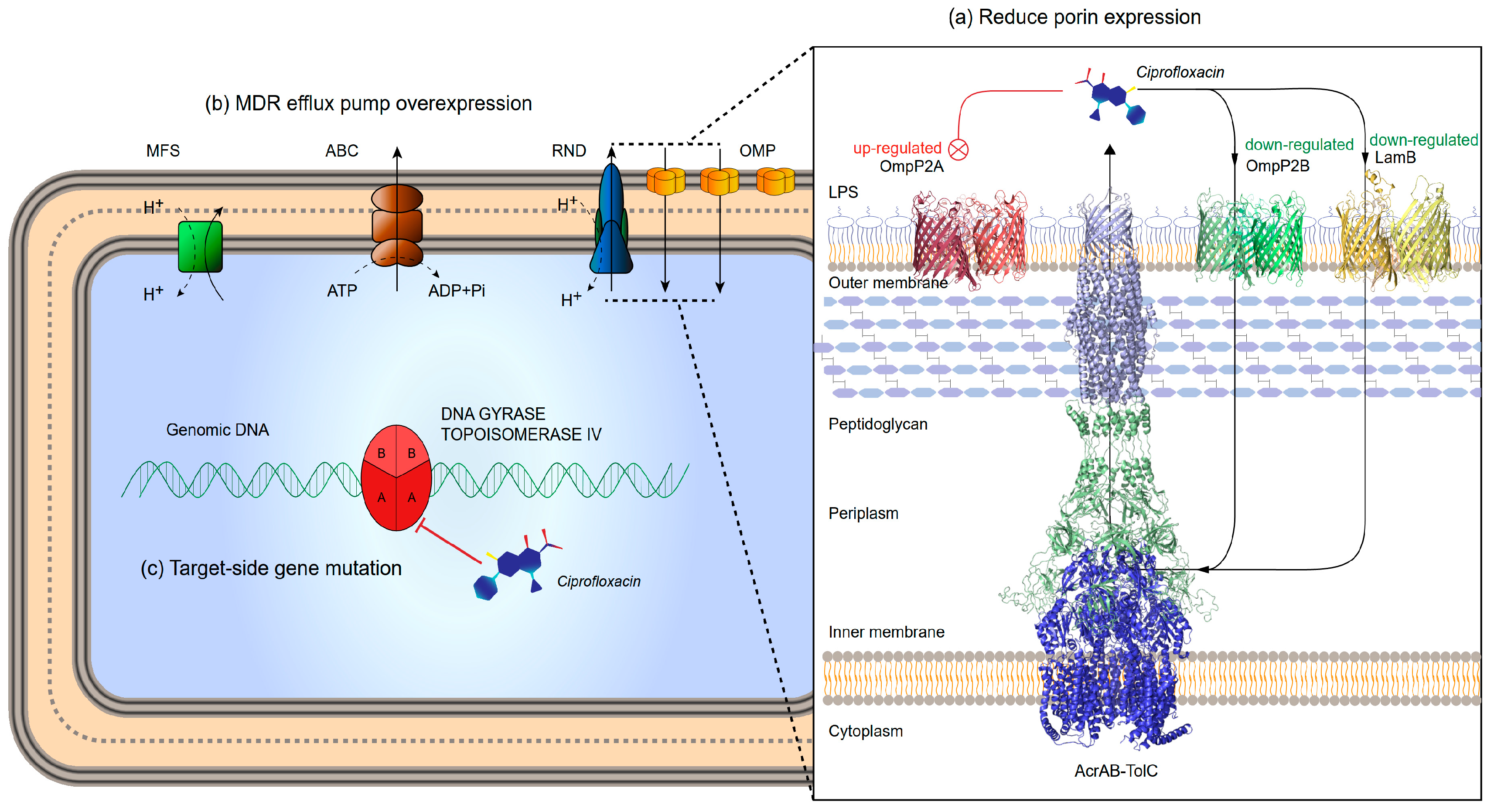
- (i)
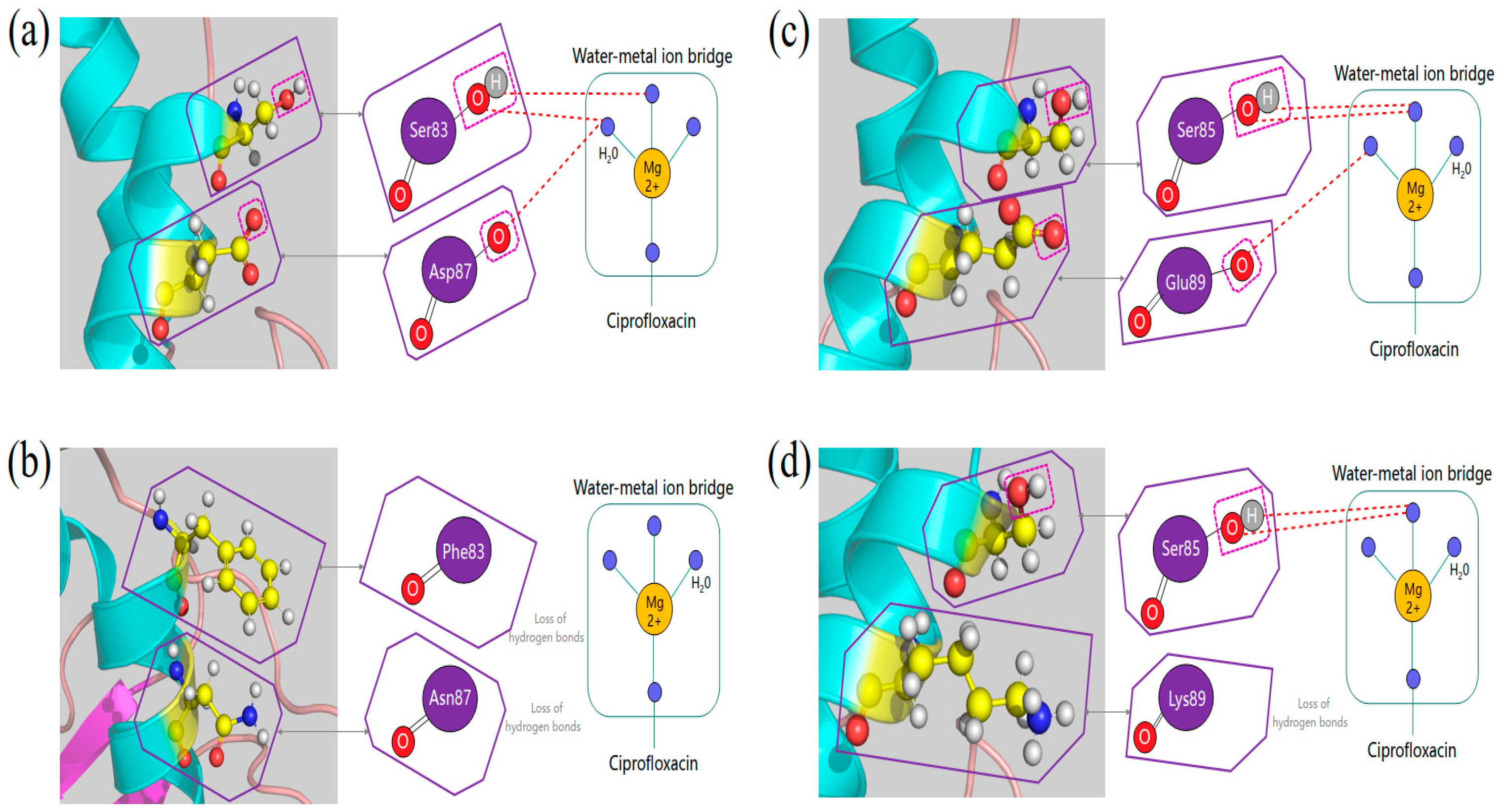
- changes in the affinity of target enzymes and drugs because of chromosomal mutations. Fluoroquinolone resistance is usually associated with mutations in specific regions of gyrase or topoisomerase IV. In general, type II enzyme mutation produces ≤10-fold drug resistance, and higher levels of drug resistance usually appear in strains wherein both enzymes are mutated [7,8]. Quinolones bind to the target enzyme through a water-metal ion bridge formed by a non-catalytic magnesium ion coordinated with four water molecules. Therefore, targeted mutations that lead to drug resistance occur more frequently on amino acid residues that bridge water-metal ions [9,10].
- (ii)
- reduced uptake or increased efflux leading to decreased drug accumulation, mainly related to porin and efflux pumps. Porin is located on the outer membrane of bacteria and is mainly composed of β-sheets. When the mass fraction of the drug is less than its exclusion limit, under the action of free diffusion, porin can become the internal flow channel of the drug [11]. Down-regulation, deletion or narrowing of the channel can lead to low-level resistance to quinolones [12,13,14]. Drug efflux is another important means for development of fluoroquinolone resistance. Resistance nodulation division family (RND) is one of the causes for quinolone resistance in gram-negative bacteria [15].
- (iii)
2. Results
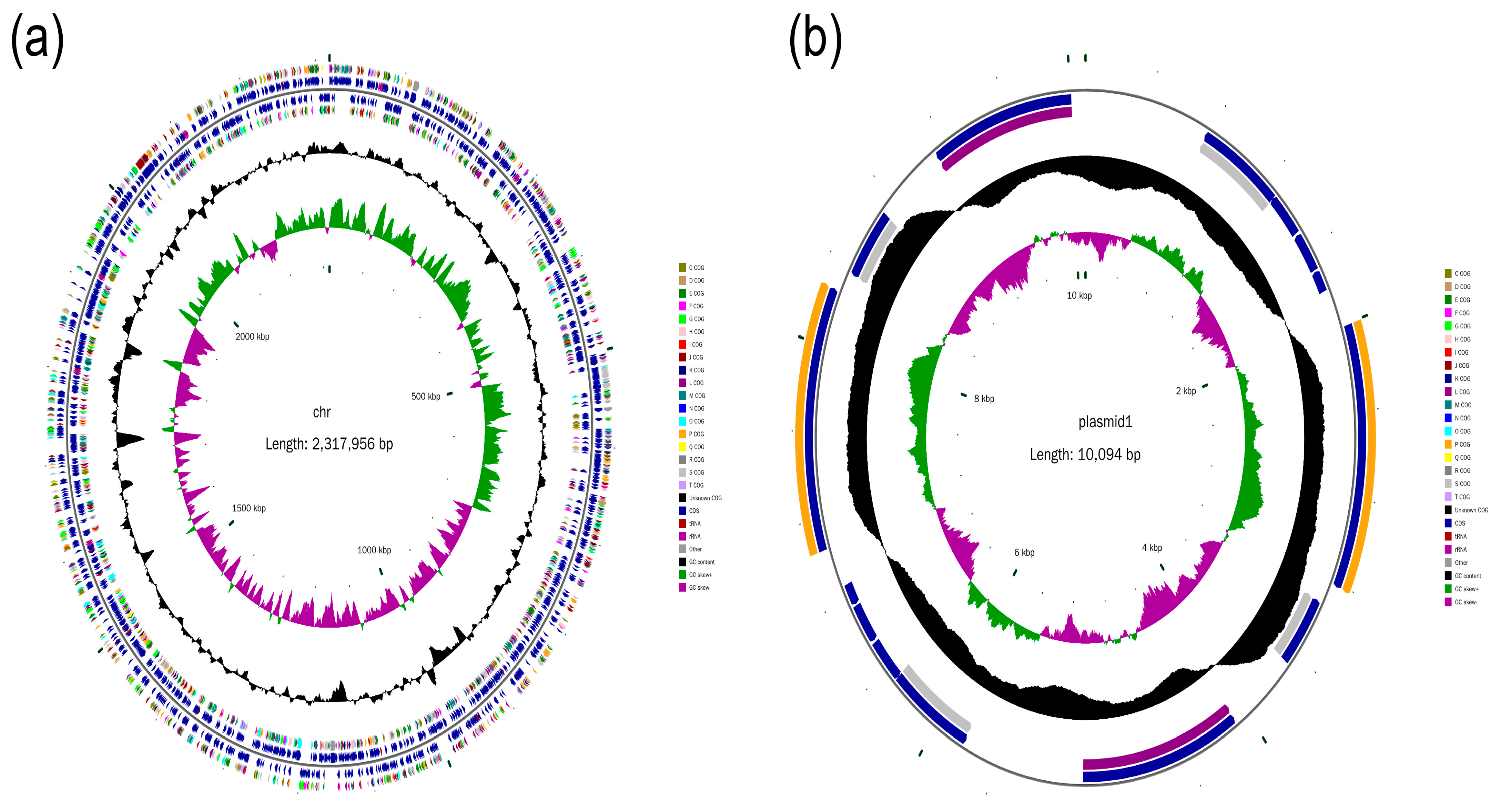
2.1. Genome Assembly
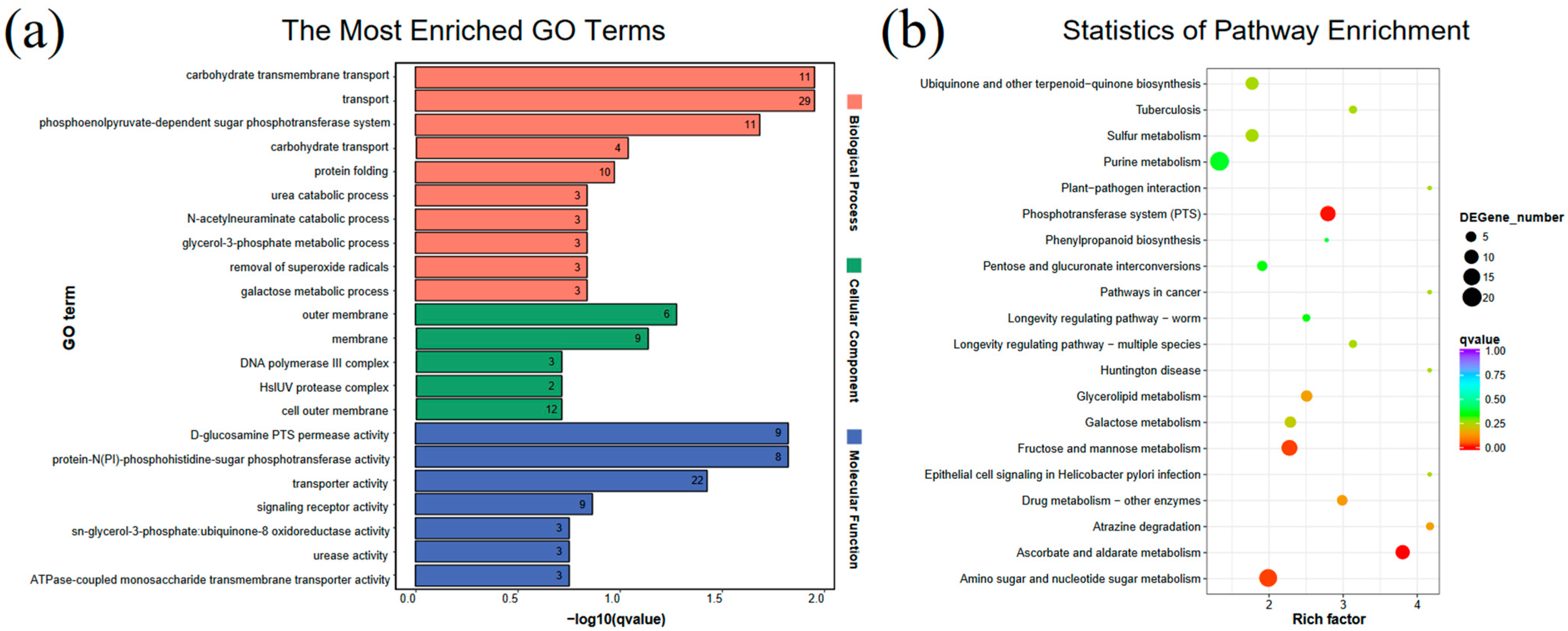
2.2. Transcriptome Results
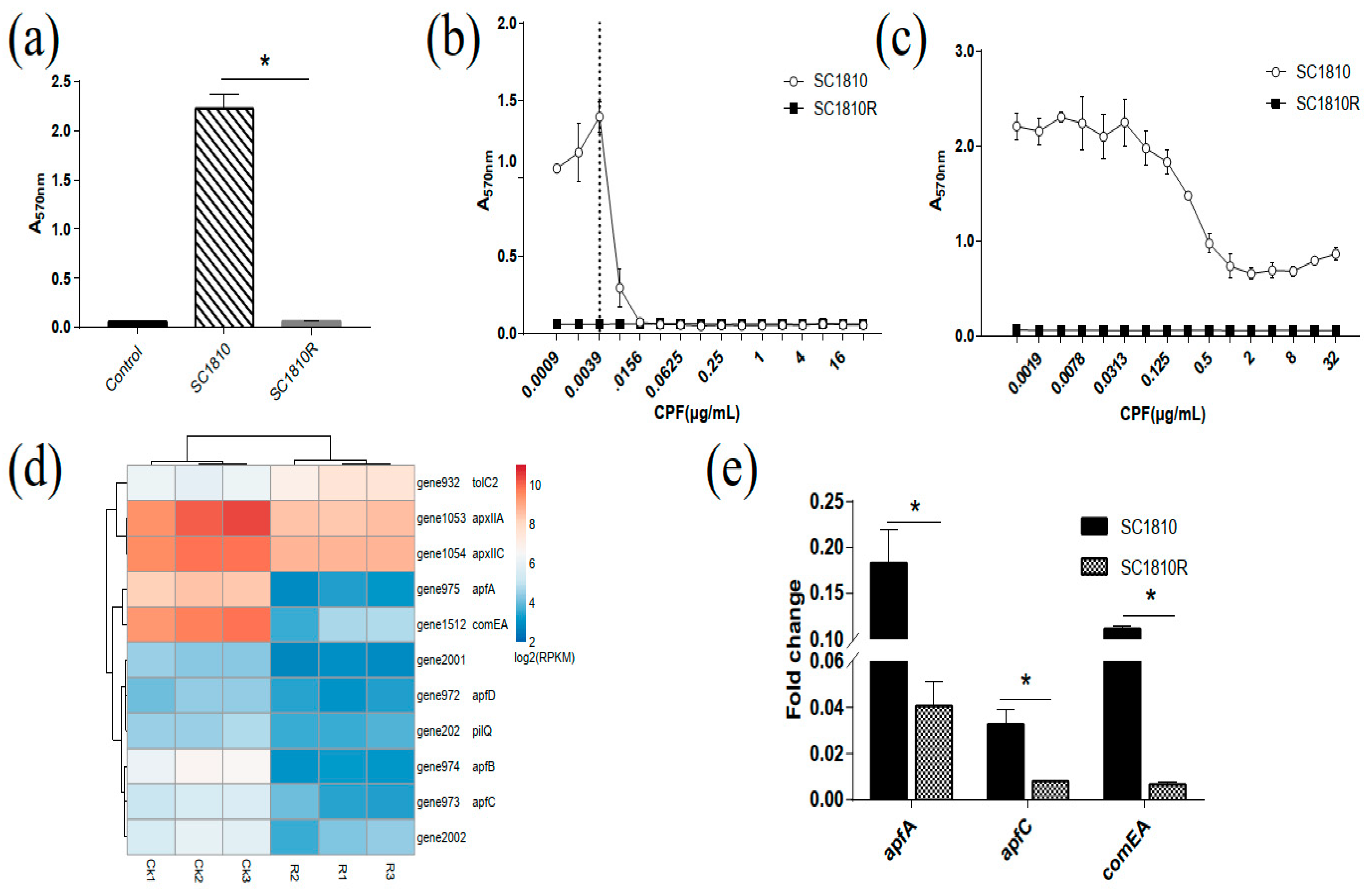
2.3. Analysis of Bofilm
2.3.1. Biofilm-Forming Capacity of SC1810 and SC1810R
2.3.2. DEGs in Biofilm
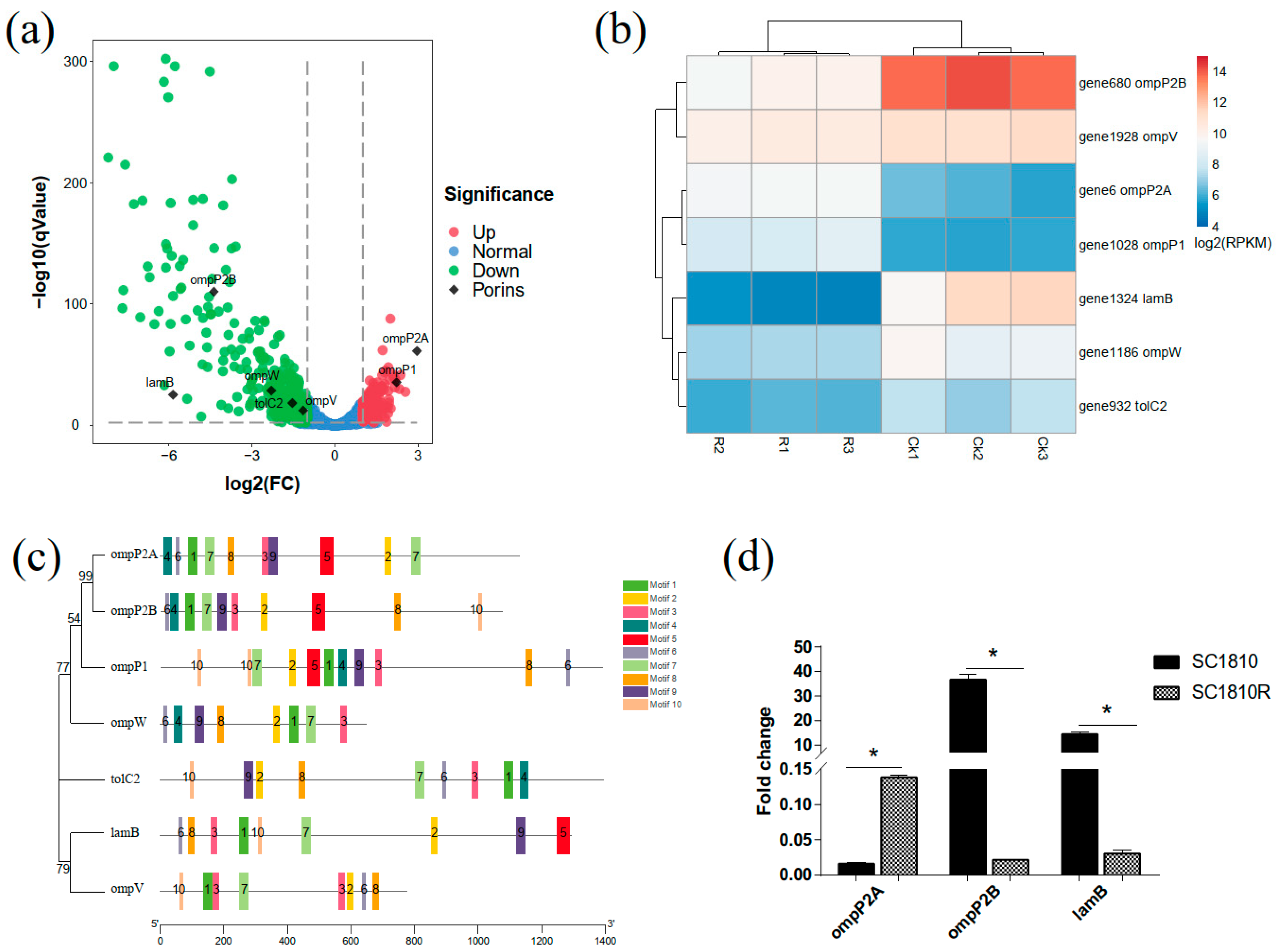
2.4. Analysis of Porins
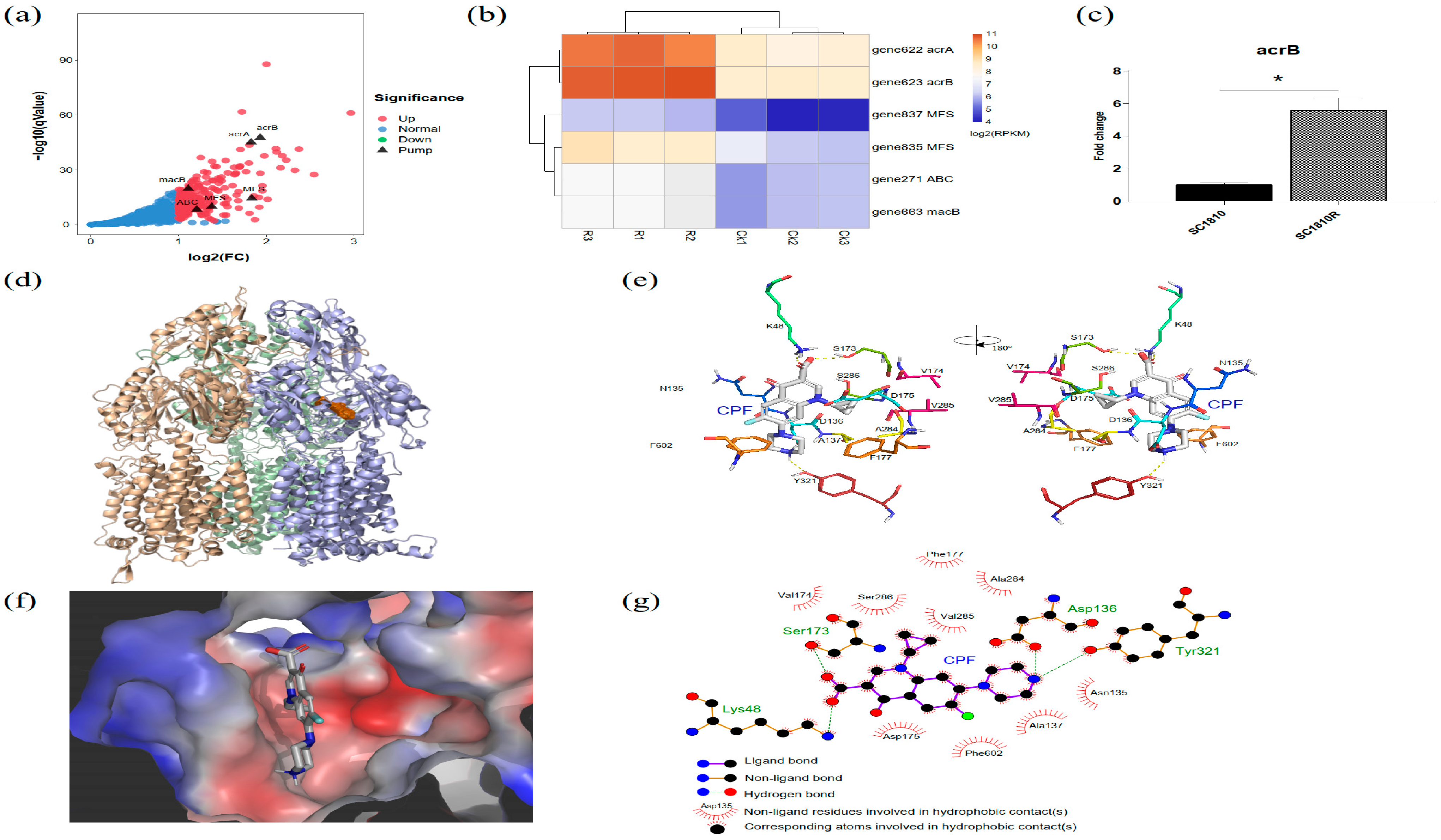
2.5. Analysis of Efflux Pumps
2.6. Analysis of QRDRs
2.6.1. Mutations in QRDRs of SC1810R
2.6.2. Structural Comparison of GyrA and ParC
2.7. Plasmid
3. Discussion
3.1. CPF Inhibits Biofilm Formation of APP
3.2. OmpP2B and LamB Mediate the Influx of CPF
3.3. AcrB Is the Main Efflux Pump Involved in CPF Efflux
3.4. QRDR and PMQR
4. Materials and Methods
4.1. Isolates and Growth Conditions
4.2. Antibiotic Susceptibility Assays
4.3. Genome Sequencing of Strain SC1810
4.4. RNA-Seq Analysis
4.4.1. RNA Isolation, RNA-Seq Library Construction and Sequencing
4.4.2. Analysis of RNA-Seq Data
4.4.3. And KEGG Pathway Enrichment Analyses of DEGs
4.5. RNA Extraction and qRT-PCR
4.6. Biofilm Test
4.6.1. Biofilm-Forming Capacity of Isolates
4.6.2. Biofilm Inhibition and Eradication Assay
4.7. Analysis of Porins and Efflux Pumps in Transcriptome Data
4.8. Quinolone Resistance Mutation Identification
4.9. PCR Detection of Plasmid-Mediated Resistance Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sassu, E.L.; Bosse, J.T.; Tobias, T.J.; Gottschalk, M.; Langford, P.R.; Hennig-Pauka, I. Update on Actinobacillus pleuropneumoniae-knowledge, gaps and challenges. Transbound. Emerg. Dis. 2018, 65 (Suppl. 1), 72–90. [Google Scholar] [CrossRef] [Green Version]
- Stringer, O.W.; Bosse, J.T.; Lacouture, S.; Gottschalk, M.; Fodor, L.; Angen, O.; Velazquez, E.; Penny, P.; Lei, L.; Langford, P.R.; et al. Proposal of Actinobacillus pleuropneumoniae serovar 19, and reformulation of previous multiplex PCRs for capsule-specific typing of all known serovars. Vet. Microbiol. 2021, 255, 109021. [Google Scholar] [CrossRef]
- Michael, G.B.; Bosse, J.T.; Schwarz, S. Antimicrobial Resistance in Pasteurellaceae of Veterinary Origin. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Nguyen, T.D.; HB, L.; Dong, T.O.; Pham, T.D. Determination of Fluoroquinolones in Pharmaceutical Formulations by Extractive Spectrophotometric Methods Using Ion-Pair Complex Formation with Bromothymol Blue. J. Anal. Methods Chem. 2018, 2018, 8436948. [Google Scholar] [CrossRef] [PubMed]
- Archambault, M.; Harel, J.; Goure, J.; Tremblay, Y.D.; Jacques, M. Antimicrobial susceptibilities and resistance genes of Canadian isolates of Actinobacillus pleuropneumoniae. Microb. Drug Resist. 2012, 18, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Vanni, M.; Merenda, M.; Barigazzi, G.; Garbarino, C.; Luppi, A.; Tognetti, R.; Intorre, L. Antimicrobial resistance of Actinobacillus pleuropneumoniae isolated from swine. Vet. Microbiol. 2012, 156, 172–177. [Google Scholar] [CrossRef]
- Drlica, K.; Hiasa, H.; Kerns, R.; Malik, M.; Mustaev, A.; Zhao, X. Quinolones: Action and resistance updated. Curr. Top. Med. Chem. 2009, 9, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Price, L.B.; Vogler, A.; Pearson, T.; Busch, J.D.; Schupp, J.M.; Keim, P. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob. Agents Chemother. 2003, 47, 2362–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, P.; Kaur, G.; Rampal, S. Impact of marbofloxacin administration on the emergence of marbofloxacin-resistant E. coli in faecal flora of goats and elucidation of molecular basis of resistance. J. Glob. Antimicrob. Resist. 2020, 21, 116–123. [Google Scholar] [CrossRef]
- Morgan-Linnell, S.K.; Becnel Boyd, L.; Steffen, D.; Zechiedrich, L. Mechanisms accounting for fluoroquinolone resistance in Escherichia coli clinical isolates. Antimicrob. Agents Chemother. 2009, 53, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwama, M.; Yamaguchi, A.; Nishino, K. Phylogenetic and functional characterisation of the Haemophilus influenzae multidrug efflux pump AcrB. Commun. Biol. 2019, 2, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, S.; Poeta, P.; Hebraud, M.; Capelo, J.L.; Igrejas, G. Mechanisms of quinolone action and resistance: Where do we stand? J. Med. Microbiol. 2017, 66, 551–559. [Google Scholar] [CrossRef]
- Fernandez, L.; Hancock, R.E. Adaptive and mutational resistance: Role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 2012, 25, 661–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, U.; Lee, C.R. Distinct Roles of Outer Membrane Porins in Antibiotic Resistance and Membrane Integrity in Escherichia coli. Front. Microbiol. 2019, 10, 953. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Plesiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zgurskaya, H.I.; Krishnamoorthy, G.; Ntreh, A.; Lu, S. Mechanism and Function of the Outer Membrane Channel TolC in Multidrug Resistance and Physiology of Enterobacteria. Front. Microbiol. 2011, 2, 189. [Google Scholar] [CrossRef] [Green Version]
- Mah, T.F. Biofilm-specific antibiotic resistance. Future Microbiol. 2012, 7, 1061–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.C.; Chan, J.P.; Yeh, K.S.; Chang, C.C.; Hsuan, S.L.; Hsieh, Y.M.; Chang, Y.C.; Lai, T.C.; Lin, W.H.; Chen, T.H. Molecular characterization of enrofloxacin resistant Actinobacillus pleuropneumoniae isolates. Vet. Microbiol. 2010, 142, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, S.; Zhang, L.; Lau, G.W.; Wen, Y.; Wu, R.; Zhao, Q.; Huang, X.; Yan, Q.; Huang, Y.; et al. A TolC-Like Protein of Actinobacillus pleuropneumoniae Is Involved in Antibiotic Resistance and Biofilm Formation. Front. Microbiol. 2016, 7, 1618. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Yang, T.Y.; Lu, P.L.; Tseng, S.P. Update on fosfomycin-modified genes in Enterobacteriaceae. J. Microbiol. Immunol. Infect. 2019, 52, 9–21. [Google Scholar] [CrossRef]
- Kaplan, J.B.; Velliyagounder, K.; Ragunath, C.; Rohde, H.; Mack, D.; Knobloch, J.K.; Ramasubbu, N. Genes involved in the synthesis and degradation of matrix polysaccharide in Actinobacillus actinomycetemcomitans and Actinobacillus pleuropneumoniae biofilms. J. Bacteriol. 2004, 186, 8213–8220. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Xu, Z.; Zhou, Y.; Li, T.; Sun, L.; Chen, H.; Zhou, R. Analysis on Actinobacillus pleuropneumoniae LuxS regulated genes reveals pleiotropic roles of LuxS/AI-2 on biofilm formation, adhesion ability and iron metabolism. Microb. Pathog. 2011, 50, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Li, G.; Zhang, W.; Zhang, Y.; Zhou, L.; Liu, S.; Liu, S.; Wang, C. Outer membrane lipoprotein VacJ is required for the membrane integrity, serum resistance and biofilm formation of Actinobacillus pleuropneumoniae. Vet. Microbiol. 2016, 183, 1–8. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, Y.; Li, G.; Zhou, L.; Liu, S.; Wang, C. The ClpP protease is required for the stress tolerance and biofilm formation in Actinobacillus pleuropneumoniae. PLoS ONE 2013, 8, e53600. [Google Scholar] [CrossRef]
- Li, L.; Xu, Z.; Zhou, Y.; Sun, L.; Liu, Z.; Chen, H.; Zhou, R. Global effects of catecholamines on Actinobacillus pleuropneumoniae gene expression. PLoS ONE 2012, 7, e31121. [Google Scholar] [CrossRef] [PubMed]
- Mullen, L.M.; Bosse, J.T.; Nair, S.P.; Ward, J.M.; Rycroft, A.N.; Robertson, G.; Langford, P.R.; Henderson, B. Pasteurellaceae ComE1 proteins combine the properties of fibronectin adhesins and DNA binding competence proteins. PLoS ONE 2008, 3, e3991. [Google Scholar] [CrossRef] [Green Version]
- Caiazza, N.C.; O’Toole, G.A. Alpha-toxin is required for biofilm formation by Staphylococcus aureus. J. Bacteriol. 2003, 185, 3214–3217. [Google Scholar] [CrossRef] [Green Version]
- Shak, J.R.; Ludewick, H.P.; Howery, K.E.; Sakai, F.; Yi, H.; Harvey, R.M.; Paton, J.C.; Klugman, K.P.; Vidal, J.E. Novel role for the Streptococcus pneumoniae toxin pneumolysin in the assembly of biofilms. mBio 2013, 4, e00655-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Cao, S.; Zhang, L.; Yuan, J.; Yang, Y.; Zhu, Z.; Wen, Y.; Wu, R.; Zhao, Q.; Huang, X.; et al. TolC2 is required for the resistance, colonization and virulence of Actinobacillus pleuropneumoniae. J. Med. Microbiol. 2017, 66, 1170–1176. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Hayashi, K.; Nagata, C.; Hoshino, K.; Onodera, Y.; Nishino, K.; Yamaguchi, A. Structural basis for the inhibition of bacterial multidrug exporters. Nature 2013, 500, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Vuotto, C.; Donelli, G. Novel Treatment Strategies for Biofilm-Based Infections. Drugs 2019, 79, 1635–1655. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, J.; Hao, Z.; Wang, S.; Liang, C. Biodegradable ciprofloxacin-incorporated waterborne polyurethane polymers prevent bacterial biofilm formation in vitro. Exp. Ther. Med. 2019, 17, 1831–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Wang, Y.; Liu, Y.N. Activity of ciprofloxacin and azithromycin on biofilms produced in vitro by Haemophilus influenzae. Chin. Med. J. 2009, 122, 1305–1310. [Google Scholar]
- Garcia-Sureda, L.; Juan, C.; Domenech-Sanchez, A.; Alberti, S. Role of Klebsiella pneumoniae LamB Porin in antimicrobial resistance. Antimicrob. Agents Chemother. 2011, 55, 1803–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.M.; Yang, M.J.; Li, H.; Wang, C.; Peng, X.X. Decreased expression of LamB and Odp1 complex is crucial for antibiotic resistance in Escherichia coli. J. Proteom. 2014, 98, 244–253. [Google Scholar] [CrossRef]
- Li, W.; Wang, G.; Zhang, S.; Fu, Y.; Jiang, Y.; Yang, X.; Lin, X. An integrated quantitative proteomic and metabolomics approach to reveal the negative regulation mechanism of LamB in antibiotics resistance. J. Proteom. 2019, 194, 148–159. [Google Scholar] [CrossRef]
- Likhatskaya, G.N.; Solov’Eva, T.F.; Novikova, O.D.; Issaeva, M.P.; Gusev, K.V.; Kryzhko, I.B.; Trifonov, E.V.; Nurminski, E.A. Homology models of the Yersinia pseudotuberculosis and Yersinia pestis general porins and comparative analysis of their functional and antigenic regions. J. Biomol. Struct. Dyn. 2005, 23, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Eicher, T.; Cha, H.J.; Seeger, M.A.; Brandstatter, L.; El-Delik, J.; Bohnert, J.A.; Kern, W.V.; Verrey, F.; Grutter, M.G.; Diederichs, K.; et al. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci. USA 2012, 109, 5687–5692. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Nishino, K.; Yamaguchi, A. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 2011, 480, 565–569. [Google Scholar] [CrossRef]
- Oswald, C.; Tam, H.K.; Pos, K.M. Transport of lipophilic carboxylates is mediated by transmembrane helix 2 in multidrug transporter AcrB. Nat. Commun. 2016, 7, 13819. [Google Scholar] [CrossRef]
- Bosse, J.T.; Li, Y.; Fernandez, C.R.; Lacouture, S.; Gottschalk, M.; Sarkozi, R.; Fodor, L.; Casas, A.M.; Angen, O.; Nedbalcova, K.; et al. Comparative sequence analysis of the capsular polysaccharide loci of Actinobacillus pleuropneumoniae serovars 1–18, and development of two multiplex PCRs for comprehensive capsule typing. Vet. Microbiol. 2018, 220, 83–89. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hathroubi, S.; Hancock, M.A.; Bossé, J.T.; Langford, P.R.; Tremblay, Y.D.N.; Labrie, J.; Jacques, M. Surface Polysaccharide Mutants Reveal that Absence of O Antigen Reduces Biofilm Formation of Actinobacillus pleuropneumoniae. Infect. Immun. 2016, 84, 127–137. [Google Scholar] [CrossRef] [Green Version]
- She, P.; Luo, Z.; Chen, L.; Wu, Y. Efficacy of levofloxacin against biofilms of Pseudomonas aeruginosa isolated from patients with respiratory tract infections in vitro. Microbiologyopen 2019, 8, e00720. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Whitington, T.; Machanick, P. The value of position-specific priors in motif discovery using MEME. BMC Bioinform. 2010, 11, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehnal, D.; Svobodova, V.R.; Berka, K.; Pravda, L.; Navratilova, V.; Banas, P.; Ionescu, C.M.; Otyepka, M.; Koca, J. MOLE 2.0: Advanced approach for analysis of biomacromolecular channels. J. Cheminform. 2013, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavaco, L.M.; Hasman, H.; Xia, S.; Aarestrup, F.M. qnrD, a novel gene conferring transferable quinolone resistance in Salmonella enterica serovar Kentucky and Bovismorbificans strains of human origin. Antimicrob. Agents Chemother. 2009, 53, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.L.; Wu, S.L.; Fu, J.L.; Jiang, H.X.; Ding, H.Z. Research Note: Epidemiological cutoff values and acquired resistance mechanisms of three veterinary antibiotics against Escherichia coli from chicken respiratory tract infections. Poult. Sci. 2021, 100, 1093–1097. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Agent | MIC (μg/mL) | |
|---|---|---|
| SC1810 | SC1810R | |
| Enrofloxacin | 0.125 | 32 |
| Norfloxacin | 0.3125 | 128 |
| Levofloxacin | 0.0039 | 16 |
| Ciprofloxacin | 0.0039 | 32 |
| Doxycycline | 16 | 16 |
| Gentamicin | 0.5 | 2 |
| Erythromycin | 2 | 2 |
| Florfenicol | 64 | 128 |
| Sulfamethoxazole | >128 | >128 |
| Gene ID | Gene | Description | Log2FC |
|---|---|---|---|
| gene975 | apfA | Possible prepilin peptidase dependent protein D | −5.49 |
| gene974 | apfB | Protein transport protein HofB-like protein, Pili/fimbriae biogenesis protein | −3.61 |
| gene973 | apfC | Protein transport protein HofC-like protein | −2.05 |
| gene972 | apfD | Type IV prepillin dependent peptidase | −1.36 |
| gene202 | pilQ | Type IV pilus secretin PilQ | −1.26 |
| gene2002 | - | prepilin peptidase dependent protein A | −1.98 |
| gene2001 | - | prepilin peptidase dependent protein B | −1.82 |
| gene1512 | comEA | DNA uptake protein and related DNA-binding protein | −5.39 |
| gene1053 | apxIIA | RTX-II toxin determinant A | −1.84 |
| gene1054 | apxIIC | RTX-II toxin-activating lysine-acyltransferase ApxIIC | −1.33 |
| gene932 | tolC2 | TolC family protein | −1.54 |
| Gene ID | Name | Rad (Å) | Hdry | Hdp | Pol | Residues | log2FC |
|---|---|---|---|---|---|---|---|
| gene6 | OmpP2A | 1.68 | −2.93 | −0.54 | 34.96 | R40/K42/N149 | 2.96 |
| gene1324 | LamB | 2.25 | −2.03 | 0.39 | 17.64 | Y142/D140/Y29 | −5.85 |
| gene680 | OmpP2B | 3.54 | −1.5 | 0.32 | 34.71 | R70/R37/I144 | −4.38 |
| gene1028 | OmpP1 | 0.98 | −1.03 | 0.18 | 2.13 | Y40/N43/N55/V31 | 2.23 |
| gene1186 | OmpW | 1.17 | 2.37 | 1.33 | 0.79 | T178/Le77/V206 | −2.3 |
| gene1928 | OmpV | 1.46 | −1.73 | −0.15 | 2.79 | Y255/G224/N212 | −1.15 |
| gene932 | TolC2 | 6.77 | −0.14 | 0.3 | 1.76 | Q318c/L115c/N114c/Y129c/L339b | −1.54 |
| Gene ID | Pump Family | Description | Log2FC |
|---|---|---|---|
| gene835 | MFS | hypothetical protein | 1.83 |
| gene837 | MFS transporter YcaD | 1.38 | |
| gene663 | ABC | MacB family efflux pump subunit | 1.11 |
| gene271 | ABC transporter permease | 1.21 | |
| gene622 | RND | acrA-like transporter periplasmic adaptor | 1.83 |
| gene623 | acrB-like transporter | 1.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Zheng, B.; Wang, J.; Li, G.; Cao, S.; Wen, Y.; Huang, X.; Zuo, Z.; Zhong, Z.; Gu, Y. Quinolone Resistance of Actinobacillus pleuropneumoniae Revealed through Genome and Transcriptome Analyses. Int. J. Mol. Sci. 2021, 22, 10036. https://doi.org/10.3390/ijms221810036
Ma X, Zheng B, Wang J, Li G, Cao S, Wen Y, Huang X, Zuo Z, Zhong Z, Gu Y. Quinolone Resistance of Actinobacillus pleuropneumoniae Revealed through Genome and Transcriptome Analyses. International Journal of Molecular Sciences. 2021; 22(18):10036. https://doi.org/10.3390/ijms221810036
Chicago/Turabian StyleMa, Xiaoping, Bowen Zheng, Jiafan Wang, Gen Li, Sanjie Cao, Yiping Wen, Xiaobo Huang, Zhicai Zuo, Zhijun Zhong, and Yu Gu. 2021. "Quinolone Resistance of Actinobacillus pleuropneumoniae Revealed through Genome and Transcriptome Analyses" International Journal of Molecular Sciences 22, no. 18: 10036. https://doi.org/10.3390/ijms221810036