
Specific Engineered G Protein Coupling to Histamine Receptors Revealed from Cellular Assay Experiments and Accelerated Molecular Dynamics Simulations

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
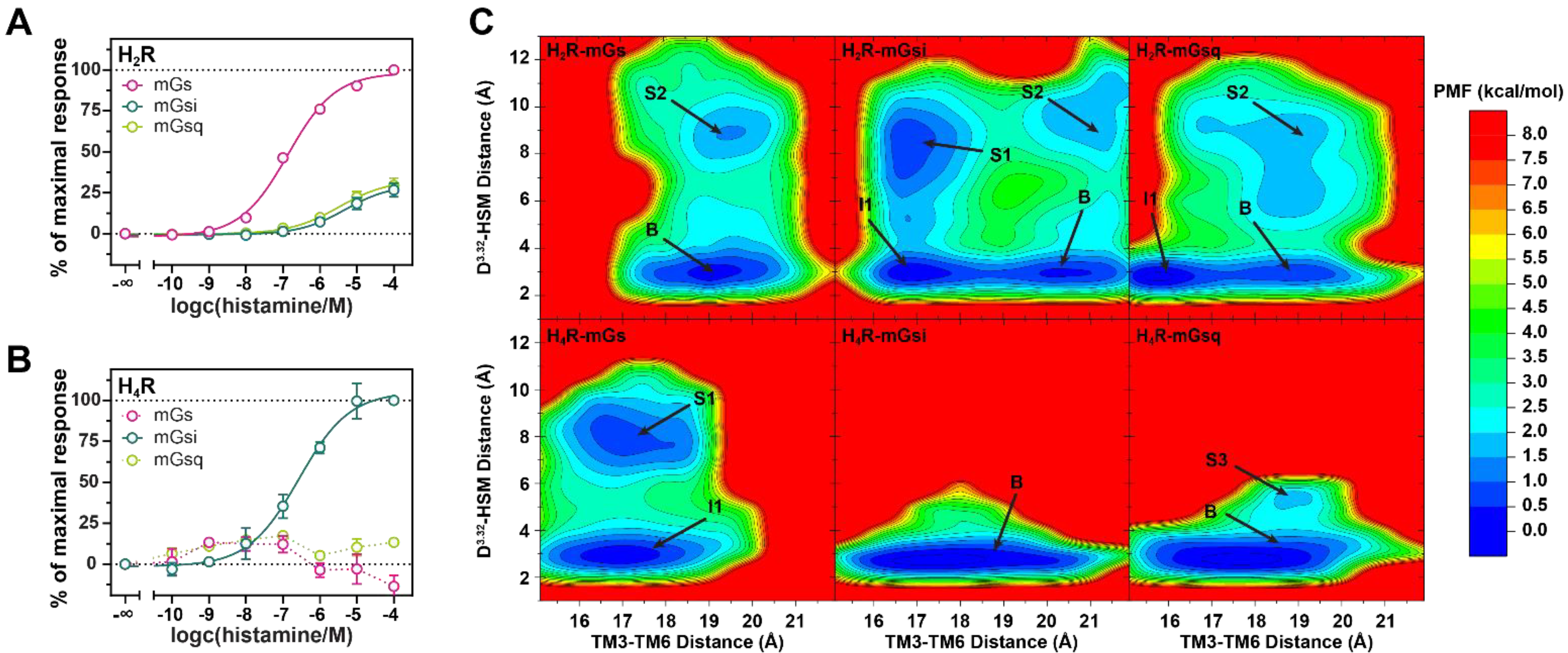
2.1. Functional Characterization of Histamine at H2R– and H4R–Mini-G Protein Complexes
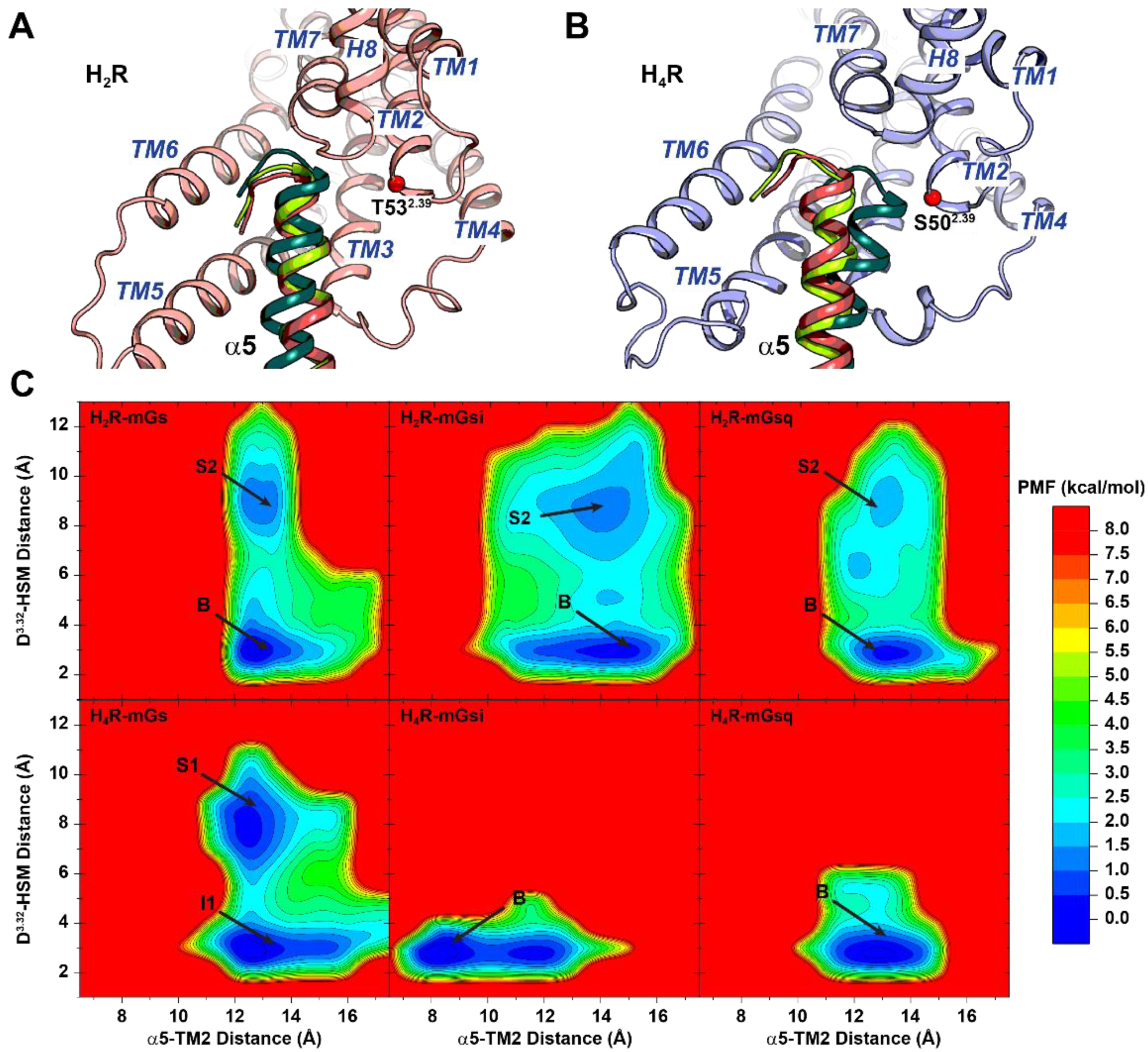
2.2. Free Energy Profiles of HR–mG Protein Complexes Were Calculated from GaMD Simulations
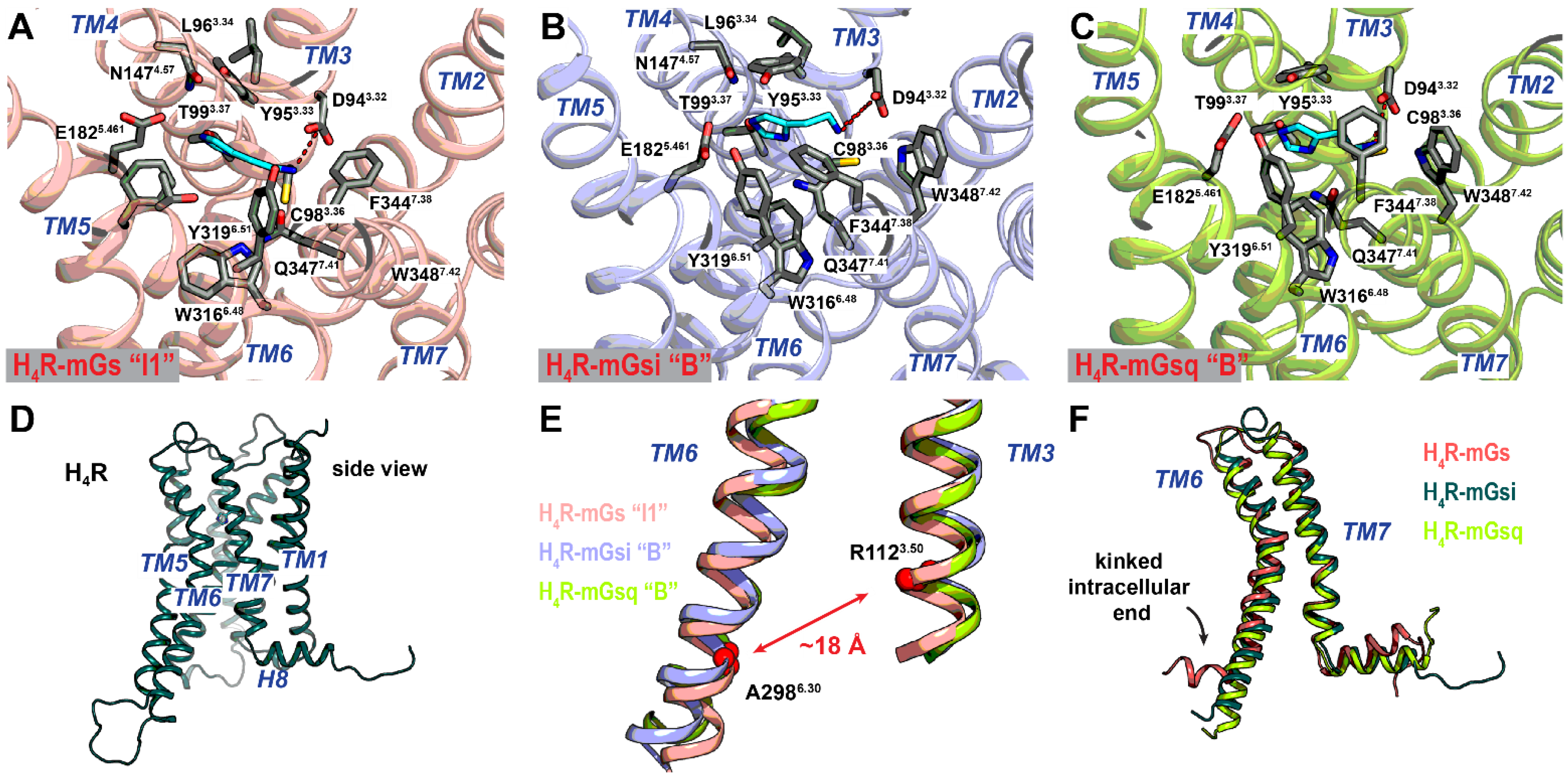
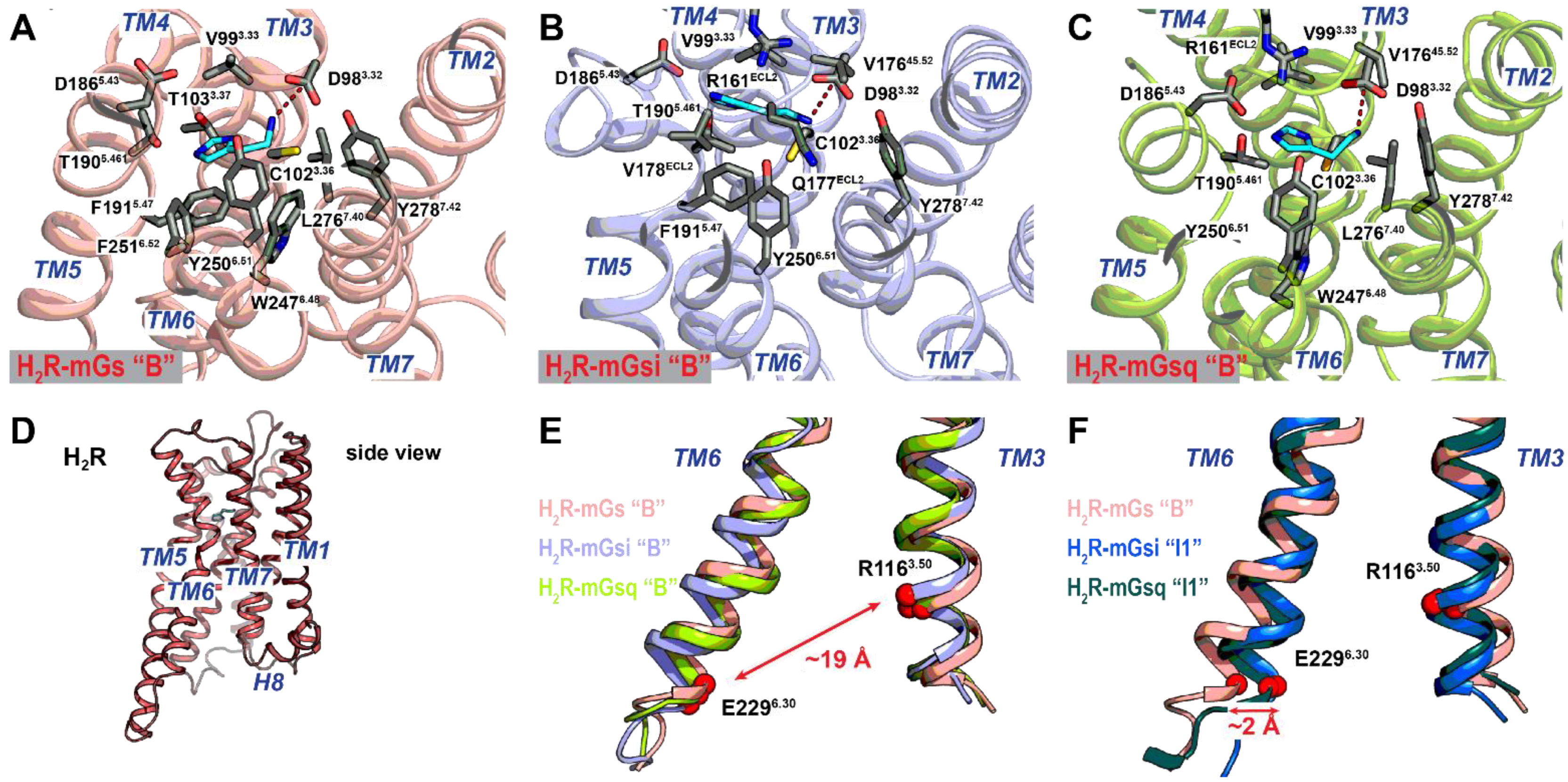
2.3. Different Affinities and Binding Conformations of Histamine in H2R and H4R Complexes
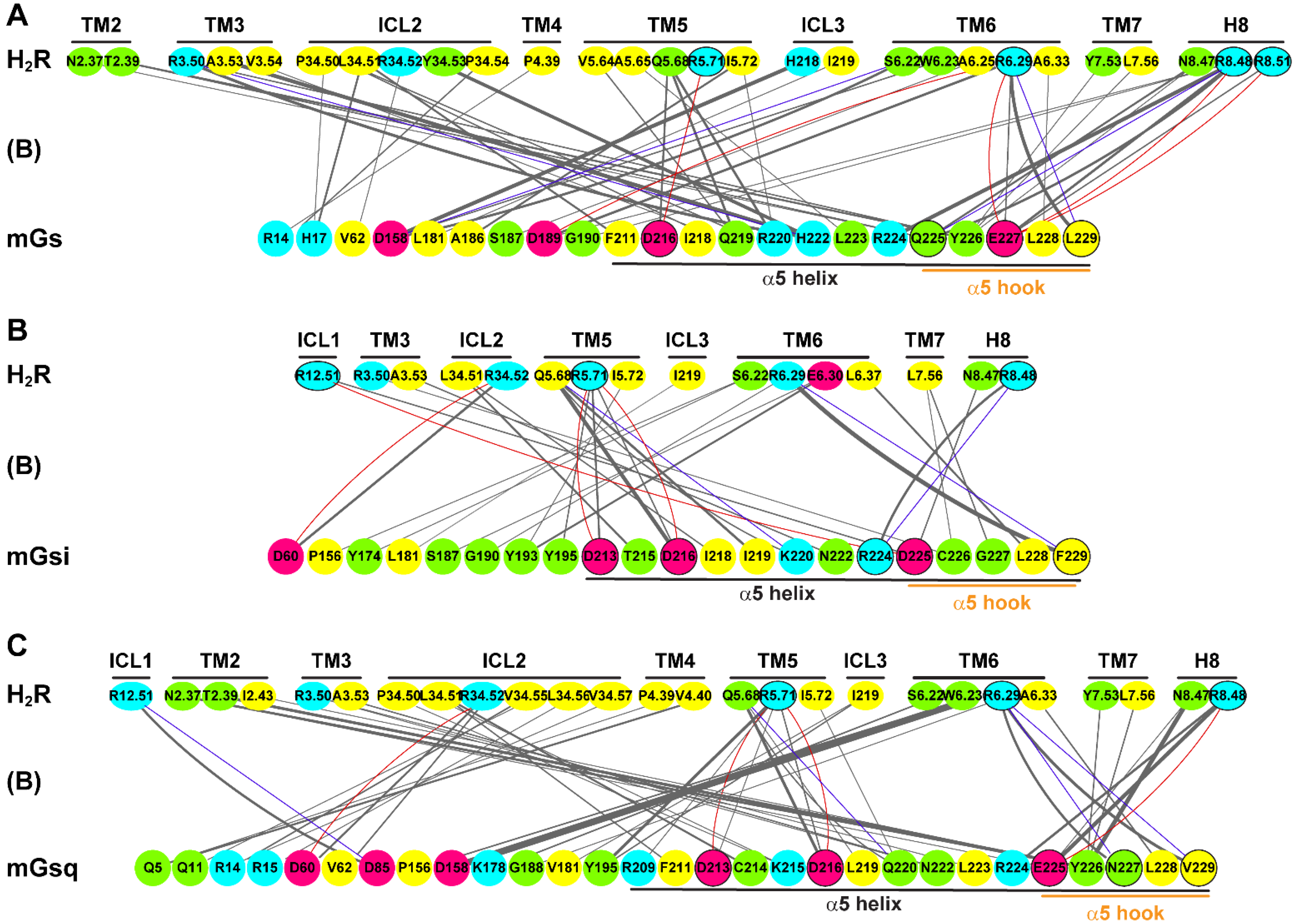
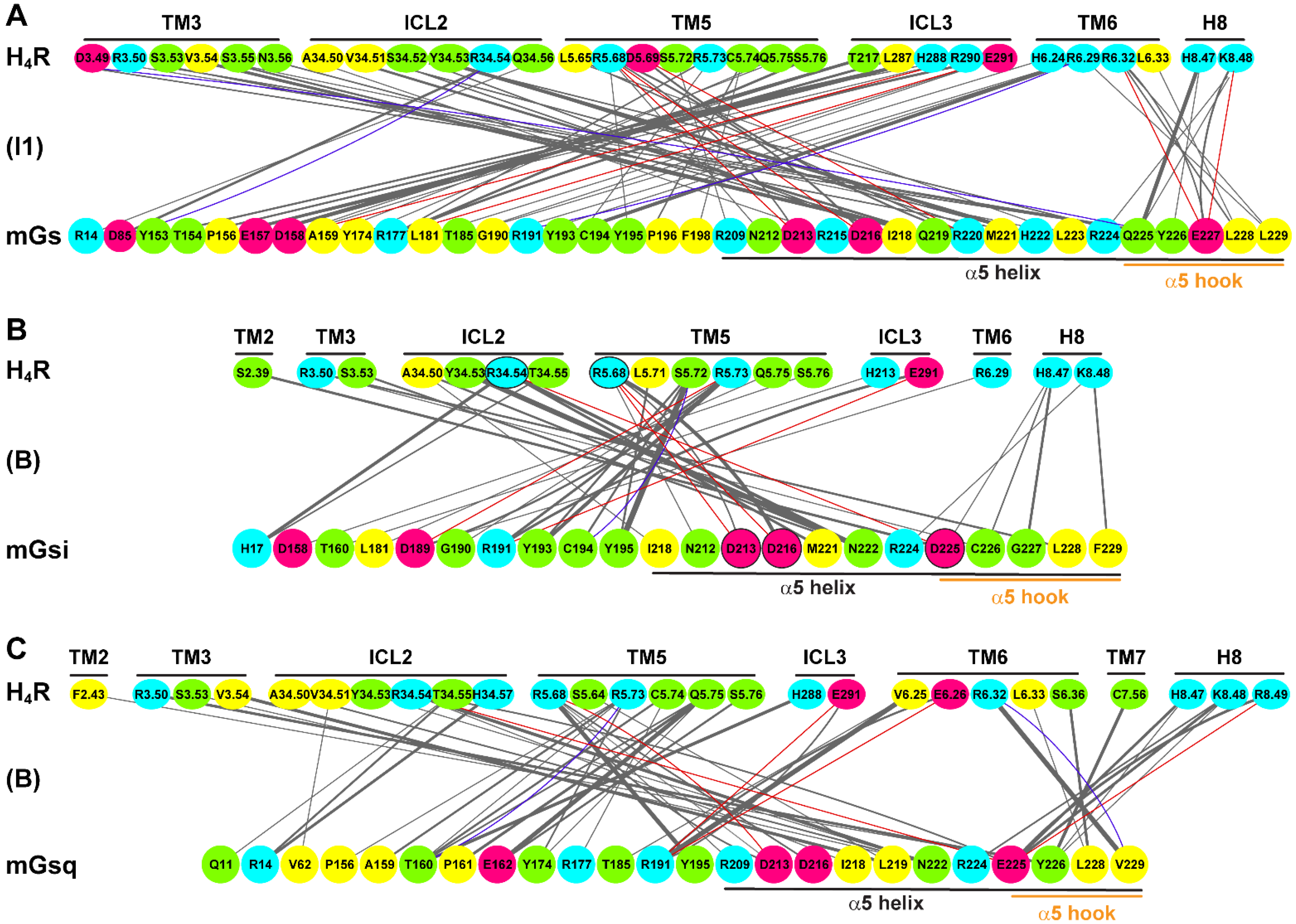
2.4. Residue Contacts at the Protein Binding Interface in the H2R and H4R Systems
2.5. α5 Hook Orientation within the Binding Interface of H2R and H4R Complexes
3. Materials and Methods
3.1. Generation of Transfectants
3.2. Mini-G Protein Recruitment Assay
3.3. Preparation of the H2R and H4R Complexes
3.4. Gaussian Accelerated Molecular Dynamics (GaMD) Simulations
3.5. Energetic Reweighting of GaMD Simulations
3.6. Simulation Protocol
3.7. Structural Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.S.; Stark, H.; Thurmond, R.L.; Haas, H.L. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, E.; Bouhelal, R.; Gerspacher, M.; Seuwen, K. The 7 TM G-Protein-Coupled Receptor Target Family. ChemMedChem 2006, 1, 760–782. [Google Scholar] [CrossRef]
- Lagerström, M.C.; Schiöth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Van Der Goot, H. Selective ligands as tools to study histamine receptors. Eur. J. Med. Chem. 2000, 35, 5–20. [Google Scholar] [CrossRef]
- Pockes, S.; Wifling, D.; Keller, M.; Buschauer, A.; Elz, S. Highly Potent, Stable, and Selective Dimeric Hetarylpropylguanidine-Type Histamine H2 Receptor Agonists. ACS Omega 2018, 3, 2865–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tropmann, K.; Bresinsky, M.; Forster, L.; Mönnich, D.; Buschauer, A.; Wittmann, H.-J.; Hübner, H.; Gmeiner, P.; Pockes, S.; Strasser, A. Abolishing Dopamine D2long/D3 Receptor Affinity of Subtype-Selective Carbamoylguanidine-Type Histamine H2 Receptor Agonists. J. Med. Chem. 2021, 64, 8684–8709. [Google Scholar] [CrossRef]
- Hirasawa, N.; Ohsawa, Y.; Katoh, G.; Shibata, K.; Ishihara, K.; Seyama, T.; Tamura, S.; Hong, J.; Ohuchi, K. Modification of the Picryl Chloride-Induced Allergic Dermatitis Model in Mouse Ear Lobes by 12-O-Tetradecanoylphorbol 13-Acetate, and Analysis of the Role of Histamine in the Modified Model. Int. Arch. Allergy Immunol. 2009, 148, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Zampeli, E.; Tiligada, E. The role of histamine H4 receptor in immune and inflammatory disorders. Br. J. Pharmacol. 2009, 157, 24–33. [Google Scholar] [CrossRef]
- Cowden, J.M.; Riley, J.P.; Ma, J.Y.; Thurmond, R.L.; Dunford, P.J. Histamine H4 receptor antagonism diminishes existing airway inflammation and dysfunction via modulation of Th2 cytokines. Respir. Res. 2010, 11, 86. [Google Scholar] [CrossRef] [Green Version]
- Hofstra, C.L.; Desai, P.J.; Thurmond, R.L.; Fung-Leung, W.-P. Histamine H4Receptor Mediates Chemotaxis and Calcium Mobilization of Mast Cells. J. Pharmacol. Exp. Ther. 2003, 305, 1212–1221. [Google Scholar] [CrossRef] [Green Version]
- Ling, P.; Ngo, K.; Nguyen, S.; Thurmond, R.; Edwards, J.P.; Karlsson, L.; Fung-Leung, W.-P. Histamine H4 receptor mediates eosinophil chemotaxis with cell shape change and adhesion molecule upregulation. Br. J. Pharmacol. 2004, 142, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutzmer, R.; Mommert, S.; Gschwandtner, M.; Zwingmann, K.; Stark, H.; Werfel, T. The histamine H4 receptor is functionally expressed on TH2 cells. J. Allergy Clin. Immunol. 2009, 123, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Braathen, L.R.; Simon, H.-U. Eosinophils and atopic dermatitis. Allergy 2004, 59, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-T.; Goodarzi, H.; Chen, H.-Y. IgE, Mast Cells, and Eosinophils in Atopic Dermatitis. Clin. Rev. Allergy Immunol. 2011, 41, 298–310. [Google Scholar] [CrossRef]
- Werfel, T.; Layton, G.; Yeadon, M.; Whitlock, L.; Osterloh, I.; Jimenez, P.; Liu, W.; Lynch, V.; Asher, A.; Tsianakas, A.; et al. Efficacy and safety of the histamine H4 receptor antagonist ZPL-3893787 in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2018, 143, 1830–1837.e4. [Google Scholar] [CrossRef]
- Milligan, G.; Kostenis, E. Heterotrimeric G-proteins: A short history. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S46–S55. [Google Scholar] [CrossRef] [Green Version]
- Wettschureck, N.; Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefkowitz, R.J. Seven transmembrane receptors—A brief personal retrospective. Biochim. Biophys. Acta (BBA)—Biomembr. 2007, 1768, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Milligan, G. Is promiscuity of G protein interaction an issue in the classification of receptors? Ann. N. Y. Acad. Sci. 1997, 812, 126–132. [Google Scholar] [CrossRef]
- Albert, P.R.; Robillard, L. G protein specificity: Traffic direction required. Cell. Signal. 2002, 14, 407–418. [Google Scholar] [CrossRef]
- Harding, S.D.; Sharman, J.; Faccenda, E.; Southan, C.; Pawson, A.J.; Ireland, S.; Gray, A.; Bruce, L.; Alexander, S.; Anderton, S.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018, 46, D1091–D1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176, S21–S141. [Google Scholar] [CrossRef] [Green Version]
- Woehler, A.; Ponimaskin, E. G Protein—Mediated Signaling: Same Receptor, Multiple Effectors. Curr. Mol. Pharmacol. 2009, 2, 237–248. [Google Scholar] [CrossRef]
- Gantz, I.; Munzert, G.; Tashiro, T.; Schäffer, M.; Wang, L.; DelValle, J.; Yamada, T. Molecular cloning of the human histamine H2 receptor. Biochem. Biophys. Res. Commun. 1991, 178, 1386–1392. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, B.; Schmid, A.; Harteneck, C.; Gudermann, T.; Schultz, G. G proteins of the Gq family couple the H2 histamine receptor to phospholipase C. Mol. Endocrinol. 1996, 10, 1697–1707. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Itadani, H.; Hidaka, Y.; Ohta, M.; Tanaka, K. Molecular Cloning and Characterization of a New Human Histamine Receptor, HH4R. Biochem. Biophys. Res. Commun. 2000, 279, 615–620. [Google Scholar] [CrossRef]
- Liu, C.; Ma, X.-J.; Jiang, X.; Wilson, S.J.; Hofstra, C.L.; Blevitt, J.; Pyati, J.; Li, X.; Chai, W.; Carruthers, N.; et al. Cloning and Pharmacological Characterization of a Fourth Histamine Receptor (H4) Expressed in Bone Marrow. Mol. Pharmacol. 2001, 59, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Shapiro, D.A.; George, S.R.; Setola, V.; Lee, D.K.; Cheng, R.; Rauser, L.; Lee, S.P.; Lynch, K.R.; Roth, B.L.; et al. Discovery of a Novel Member of the Histamine Receptor Family. Mol. Pharmacol. 2001, 59, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, T.; Morikawa, N.; Saito, Y.; Masuho, Y.; Matsumoto, S.-I. Molecular Cloning and Characterization of a Novel Type of Histamine Receptor Preferentially Expressed in Leukocytes. J. Biol. Chem. 2000, 275, 36781–36786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, K.L.; Behan, J.; Laz, T.M.; West, R.E.; Greenfeder, S.A.; Anthes, J.C.; Umland, S.; Wan, Y.; Hipkin, R.W.; Gonsiorek, W.; et al. Cloning and characterization of a novel human histamine receptor. J. Pharmacol. Exp. Ther. 2001, 296, 1058–1066. [Google Scholar]
- Zhu, Y.; Michalovich, D.; Wu, H.-L.; Tan, K.B.; Dytko, G.M.; Mannan, I.J.; Boyce, R.; Alston, J.; Tierney, L.A.; Li, X.; et al. Cloning, Expression, and Pharmacological Characterization of a Novel Human Histamine Receptor. Mol. Pharmacol. 2001, 59, 434–441. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Alpert, R.; Jenkinson, S.; Gladue, R.P.; Foo, S.; Trim, S.; Peter, B.; Trevethick, M.; Fidock, M. Identification of a histamine h4receptor on human eosinophils—Role in eosinophil chemotaxis. J. Recept. Signal Transduct. Res. 2002, 22, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Buckland, K.F.; Williams, T.J.; Conroy, D.M. Histamine induces cytoskeletal changes in human eosinophils via the H4 receptor. Br. J. Pharmacol. 2003, 140, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkstra, D.; Leurs, R.; Chazot, P.; Shenton, F.C.; Stark, H.; Werfel, T.; Gutzmer, R. Histamine downregulates monocyte CCL2 production through the histamine H4 receptor. J. Allergy Clin. Immunol. 2007, 120, 300–307. [Google Scholar] [CrossRef]
- Jemima, E.A.; Prema, A.; Thangam, E.B. Functional characterization of histamine H4 receptor on human mast cells. Mol. Immunol. 2014, 62, 19–28. [Google Scholar] [CrossRef]
- Schneider, E.H.; Schnell, D.; Papa, D.; Seifert, R. High Constitutive Activity and a G-Protein-Independent High-Affinity State of the Human Histamine H4-Receptor. Biochemistry 2009, 48, 1424–1438. [Google Scholar] [CrossRef]
- Olsen, R.H.J.; DiBerto, J.F.; English, J.G.; Glaudin, A.M.; Krumm, B.E.; Slocum, S.T.; Che, T.; Gavin, A.C.; McCorvy, J.D.; Roth, B.L.; et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 2020, 16, 841–849. [Google Scholar] [CrossRef]
- Okashah, N.; Wan, Q.; Ghosh, S.; Sandhu, M.; Inoue, A.; Vaidehi, N.; Lambert, N.A. Variable G protein determinants of GPCR coupling selectivity. Proc. Natl. Acad. Sci. USA 2019, 116, 12054–12059. [Google Scholar] [CrossRef] [Green Version]
- Avet, C.; Mancini, A.; Breton, B.; Le Gouill, C.; Hauser, A.S.; Normand, C.; Kobayashi, H.; Gross, F.; Hogue, M.; Lukasheva, V.; et al. Selectivity Landscape of 100 Therapeutically Relevant GPCR Profiled by an Effector Translocation-Based BRET Platform. bioRxiv 2020. [Google Scholar] [CrossRef]
- Conklin, B.R.; Farfel, Z.; Lustig, K.D.; Julius, D.; Bourne, H.R. Substitution of three amino acids switches receptor specificity of Gqα to that of Giα. Nature 1993, 363, 274–276. [Google Scholar] [CrossRef]
- Flock, T.; Hauser, A.; Lund, N.; Gloriam, D.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR–G-protein binding. Nature 2017, 545, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.; DeVree, B.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Raimondi, F.; Kadji, F.M.N.; Singh, G.; Kishi, T.; Uwamizu, A.; Ono, Y.; Shinjo, Y.; Ishida, S.; Arang, N.; et al. Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 2019, 177, 1933–1947.e25. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Tate, C.G. Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Eng. Des. Sel. 2016, 29, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehmé, R.; Carpenter, B.; Singhal, A.; Strege, A.; Edwards, P.C.; White, C.F.; Du, H.; Grisshammer, R.; Tate, C.G. Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS ONE 2017, 12, e0175642. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Okashah, N.; Inoue, A.; Nehmé, R.; Carpenter, B.; Tate, C.G.; Lambert, N.A. Mini G protein probes for active G protein–coupled receptors (GPCRs) in live cells. J. Biol. Chem. 2018, 293, 7466–7473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, E.; Marín, A.R.; Dalton, J.A.R.; Giraldo, J.; Stove, C. Distinct Dopamine D₂ Receptor Antagonists Differentially Impact D₂ Receptor Oligomerization. Int. J. Mol. Sci. 2019, 20, 1686. [Google Scholar] [CrossRef] [Green Version]
- Wouters, E.; Walraed, J.; Robertson, M.J.; Meyrath, M.; Szpakowska, M.; Chevigné, A.; Skiniotis, G.; Stove, C. Assessment of Biased Agonism among Distinct Synthetic Cannabinoid Receptor Agonist Scaffolds. ACS Pharmacol. Transl. Sci. 2020, 3, 285–295. [Google Scholar] [CrossRef]
- Pottie, E.; Tosh, D.K.; Gao, Z.-G.; Jacobson, K.A.; Stove, C.P. Assessment of biased agonism at the A3 adenosine receptor using β-arrestin and miniGαi recruitment assays. Biochem. Pharmacol. 2020, 177, 113934. [Google Scholar] [CrossRef]
- Pottie, E.; Dedecker, P.; Stove, C.P. Identification of psychedelic new psychoactive substances (NPS) showing biased agonism at the 5-HT2AR through simultaneous use of β-arrestin 2 and miniGαq bioassays. Biochem. Pharmacol. 2020, 182, 114251. [Google Scholar] [CrossRef] [PubMed]
- Höring, C.; Seibel, U.; Tropmann, K.; Grätz, L.; Mönnich, D.; Pitzl, S.; Bernhardt, G.; Pockes, S.; Strasser, A. A Dynamic, Split-Luciferase-Based Mini-G Protein Sensor to Functionally Characterize Ligands at All Four Histamine Receptor Subtypes. Int. J. Mol. Sci. 2020, 21, 8440. [Google Scholar] [CrossRef] [PubMed]
- Nafria, J.G.; Lee, Y.; Bai, X.; Carpenter, B.; Tate, C.G. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 2018, 7. [Google Scholar] [CrossRef]
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.-P.; et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942.e18. [Google Scholar] [CrossRef]
- Lin, X.; Li, M.; Wang, N.; Wu, Y.; Luo, Z.; Guo, S.; Han, G.-W.; Li, S.; Yue, Y.; Wei, X.; et al. Structural basis of ligand recognition and self-activation of orphan GPR52. Nature 2020, 579, 152–157. [Google Scholar] [CrossRef]
- Nafria, J.G.; Nehmé, R.; Edwards, P.C.; Tate, C.G. Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. Nature 2018, 558, 620–623. [Google Scholar] [CrossRef]
- Kim, K.; Che, T.; Panova, O.; DiBerto, J.F.; Lyu, J.; Krumm, B.E.; Wacker, D.; Robertson, M.J.; Seven, A.B.; Nichols, D.E.; et al. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. Cell 2020, 182, 1574–1588.e19. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2020, 49, D335–D343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Johnson, R.M.; Drulyte, I.; Yu, L.; Kotecha, A.; Danev, R.; Wootten, D.; Sexton, P.M.; Belousoff, M.J. Evolving cryo-EM structural approaches for GPCR drug discovery. Structure 2021, 29, 963–974.e6. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Wang, N.; Xu, Z.; Lu, Y.; Song, J.; Zhang, A.; Guo, C.; He, Y. Cryo-EM structure of the human histamine H1 receptor/Gq complex. Nat. Commun. 2021, 12, 2086. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Rutkowska, E.; Bartuzi, D.; Targowska-Duda, K.M.; Matosiuk, D.; Selent, J. Computational methods for studying G protein-coupled receptors (GPCRs). Methods Cell Biol. 2016, 132, 359–399. [Google Scholar] [CrossRef]
- Wang, J.; Miao, Y. Recent advances in computational studies of GPCR-G protein interactions. Adv. Protein Chem. Struct. Biol. 2019, 116, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Fontanals, M.; Stepniewski, T.M.; Aranda-García, D.; Morales-Pastor, A.; Medel-Lacruz, B.; Selent, J. How do Molecular Dynamics Data Complement Static Structural Data of GPCRs. Int. J. Mol. Sci. 2020, 21, 5933. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.; Liu, C.W.; Fung, J.J.; Bokoch, M.; et al. The Dynamic Process of β2-Adrenergic Receptor Activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mafi, A.; Kim, S.-K.; Goddard, W.A., III. The atomistic level structure for the activated human κ-opioid receptor bound to the full Gi protein and the MP1104 agonist. Proc. Natl. Acad. Sci. USA 2020, 117, 5836–5843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.T.; Miao, Y.; Wang, Y.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics in NAMD. J. Chem. Theory Comput. 2016, 13, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Theory, Implementation, and Applications. Annu. Rep. Comput. Chem. 2017, 13, 231–278. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Arantes, P.R.; Bhattarai, A.; Hsu, R.V.; Pawnikar, S.; Huang, Y.M.; Palermo, G.; Miao, Y. Gaussian accelerated molecular dynamics: Principles and applications. WIREs Comput. Mol. Sci. 2021, 11, e1521. [Google Scholar] [CrossRef]
- Miao, Y.; Feixas, F.; Eun, C.; McCammon, J.A. Accelerated molecular dynamics simulations of protein folding. J. Comput. Chem. 2015, 36, 1536–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; McCammon, J.A. Graded activation and free energy landscapes of a muscarinic G-protein–coupled receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 12162–12167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; McCammon, J.A. Mechanism of the G-protein mimetic nanobody binding to a muscarinic G-protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 3036–3041. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Miao, Y. Mechanistic Insights into Specific G Protein Interactions with Adenosine Receptors. J. Phys. Chem. B 2019, 123, 6462–6473. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, W.; Frazer, J.; Unett, D. Functional assays for screening GPCR targets. Curr. Opin. Biotechnol. 2005, 16, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Söldner, C.A.; Miao, Y.; Sticht, H. Agonist Binding and G Protein Coupling in Histamine H2 Receptor: A Molecular Dynamics Study. Int. J. Mol. Sci. 2020, 21, 6693. [Google Scholar] [CrossRef] [PubMed]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.; Mordalski, S.; Harpsøe, K.; Hauser, A.; Bojarski, A.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2017, 46, D440–D446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, P.; Miszta, P.; Filipek, S. Molecular Modeling of Histamine Receptors—Recent Advances in Drug Discovery. Molecules 2021, 26, 1778. [Google Scholar] [CrossRef] [PubMed]
- Fleetwood, O.; Matricon, P.; Carlsson, J.; Delemotte, L. Energy landscapes reveal agonist’s control of GPCR activation via microswitches. bioRxiv 2019, 627026. [Google Scholar] [CrossRef]
- Dai, H.; Fu, Q.; Shen, Y.; Hu, W.; Zhang, Z.; Timmerman, H.; Leurs, R.; Chen, Z. The histamine H3 receptor antagonist clobenpropit enhances GABA release to protect against NMDA-induced excitotoxicity through the cAMP/protein kinase A pathway in cultured cortical neurons. Eur. J. Pharmacol. 2007, 563, 117–123. [Google Scholar] [CrossRef]
- Glukhova, A.; Draper-Joyce, C.J.; Sunahara, R.K.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol. Transl. Sci. 2018, 1, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. The ligand paradox between affinity and efficacy: Can you be there and not make a difference? Trends Pharmacol. Sci. 2002, 23, 275–280. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Tropmann, K.; Höring, C.; Plank, N.; Pockes, S. Discovery of a G Protein-Biased Radioligand for the Histamine H2 Receptor with Reversible Binding Properties. J. Med. Chem. 2020, 63, 13090–13102. [Google Scholar] [CrossRef] [PubMed]
- Weinhart, C.G.; Wifling, D.; Schmidt, M.F.; Neu, E.; Höring, C.; Clark, T.; Gmeiner, P.; Keller, M. Dibenzodiazepinone-type muscarinic receptor antagonists conjugated to basic peptides: Impact of the linker moiety and unnatural amino acids on M2R selectivity. Eur. J. Med. Chem. 2021, 213, 113159. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Söldner, C.; Horn, A.; Sticht, H. Binding of histamine to the H1 receptor—A molecular dynamics study. J. Mol. Model. 2018, 24, 346. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; et al. AMBER; University of California: San Francisco, CA, USA, 2017. [Google Scholar]
- Berendsen, H.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Siu, S.W.I.; Vácha, R.; Jungwirth, P.; Böckmann, R.A. Biomolecular simulations of membranes: Physical properties from different force fields. J. Chem. Phys. 2008, 128, 125103. [Google Scholar] [CrossRef]
- Toukan, K.; Rahman, A. Molecular-dynamics study of atomic motions in water. Phys. Rev. B 1985, 31, 2643–2648. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef]
- Miao, Y.; Sinko, W.; Pierce, L.; Bucher, D.; Walker, R.C.; McCammon, J.A. Improved Reweighting of Accelerated Molecular Dynamics Simulations for Free Energy Calculation. J. Chem. Theory Comput. 2014, 10, 2677–2689. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Goreishi, D.; Gilson, M.K.; et al. AMBER; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Baumeister, P.; Erdmann, D.; Biselli, S.; Kagermeier, N.; Elz, S.; Bernhardt, G.; Buschauer, A. [3H]UR-DE257: Development of a Tritium-Labeled Squaramide-Type Selective Histamine H2Receptor Antagonist. ChemMedChem 2014, 10, 83–93. [Google Scholar] [CrossRef]
- Lim, H.D.; van Rijn, R.M.; Ling, P.; Bakker, R.A.; Thurmond, R.L.; Leurs, R. Evaluation of Histamine H1-, H2-, and H3-Receptor Ligands at the Human Histamine H4 Receptor: Identification of 4-Methylhistamine as the First Potent and Selective H4 Receptor Agonist. J. Pharmacol. Exp. Ther. 2005, 314, 1310–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; I Shakhnovich, E.; et al. Common activation mechanism of class A GPCRs. eLife 2019, 8, e50279. [Google Scholar] [CrossRef] [PubMed]
- Seibel-Ehlert, U.; Plank, N.; Inoue, A.; Bernhardt, G.; Strasser, A. Label-Free Investigations on the G Protein Dependent Signaling Pathways of Histamine Receptors. Int. J. Mol. Sci. 2021, 22, 9739. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mini-G Protein Recruitment Assay | MM/GBSA | ||
|---|---|---|---|
| pEC50 ± SEM | Emax ± SEM (%) | Binding Energy ± SD (kcal/mol) | |
| H2R–mGs | 6.86 ± 0.04 | 100 | −17.23 ± 3.44 |
| H2R–mGsi | 5.30 ± 0.06 | 26.77 ± 3.47 | −16.57 ± 3.81 |
| H2R–mGsq | 5.48 ± 0.04 | 29.56 ± 3.66 | −16.25 ± 6.35 |
| H4R–mGs | n.d. | −13.51 ± 5.50 | −20.03 ± 5.44 |
| H4R–mGsi | 6.60 ± 0.10 | 100 | −27.91 ± 4.69 |
| H4R–mGsq | n.d. | 13.82 ± 0.28 | −20.49 ± 6.78 |
| System | Natoms | Simulation | Length (ns) | ∆Vpot (kcal/mol) | ∆Vdihedral (kcal/mol) |
|---|---|---|---|---|---|
| H2R–mGs | 128,856 | GaMD1 | 1000 | 7.53 ± 3.15 | 6.40 ± 2.65 |
| GaMD2 | 1000 | 7.50 ± 3.15 | 6.50 ± 2.68 | ||
| GaMD3 | 1000 | 7.53 ± 3.16 | 6.36 ± 2.64 | ||
| H2R–mGsi | 128,819 | GaMD1 | 1000 | 7.87 ± 3.23 | 5.93 ± 2.55 |
| GaMD2 | 1000 | 7.86 ± 3.23 | 5.88 ± 2.54 | ||
| GaMD3 | 1000 | 7.85 ± 3.22 | 5.98 ± 2.56 | ||
| H2R–mGsq | 128,864 | GaMD1 | 1000 | 7.39 ± 3.13 | 6.32 ± 2.63 |
| GaMD2 | 1000 | 7.36 ± 3.13 | 6.34 ± 2.64 | ||
| GaMD3 | 1000 | 7.37 ± 3.13 | 6.52 ± 2.68 | ||
| H4R–mGs | 129,747 | GaMD1 | 1000 | 7.87 ± 3.23 | 6.25 ± 2.62 |
| GaMD2 | 1000 | 7.86 ± 3.23 | 6.08 ± 2.58 | ||
| GaMD3 | 1000 | 7.84 ± 3.23 | 6.45 ± 2.66 | ||
| H4R–mGsi | 129,737 | GaMD1 | 1000 | 8.05 ± 3.26 | 6.03 ± 2.57 |
| GaMD2 | 1000 | 8.02 ± 3.25 | 6.42 ± 2.65 | ||
| GaMD3 | 1000 | 8.04 ± 3.26 | 6.19 ± 2.60 | ||
| H4R–mGsq | 129,788 | GaMD1 | 1000 | 8.09 ± 3.27 | 6.55 ± 2.68 |
| GaMD2 | 1000 | 8.21 ± 3.29 | 6.57 ± 2.68 | ||
| GaMD3 | 1000 | 8.22 ± 3.30 | 6.43 ± 2.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Höring, C.; Conrad, M.; Söldner, C.A.; Wang, J.; Sticht, H.; Strasser, A.; Miao, Y. Specific Engineered G Protein Coupling to Histamine Receptors Revealed from Cellular Assay Experiments and Accelerated Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 10047. https://doi.org/10.3390/ijms221810047
Höring C, Conrad M, Söldner CA, Wang J, Sticht H, Strasser A, Miao Y. Specific Engineered G Protein Coupling to Histamine Receptors Revealed from Cellular Assay Experiments and Accelerated Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2021; 22(18):10047. https://doi.org/10.3390/ijms221810047
Chicago/Turabian StyleHöring, Carina, Marcus Conrad, Christian A. Söldner, Jinan Wang, Heinrich Sticht, Andrea Strasser, and Yinglong Miao. 2021. "Specific Engineered G Protein Coupling to Histamine Receptors Revealed from Cellular Assay Experiments and Accelerated Molecular Dynamics Simulations" International Journal of Molecular Sciences 22, no. 18: 10047. https://doi.org/10.3390/ijms221810047
APA StyleHöring, C., Conrad, M., Söldner, C. A., Wang, J., Sticht, H., Strasser, A., & Miao, Y. (2021). Specific Engineered G Protein Coupling to Histamine Receptors Revealed from Cellular Assay Experiments and Accelerated Molecular Dynamics Simulations. International Journal of Molecular Sciences, 22(18), 10047. https://doi.org/10.3390/ijms221810047