
Discovery of a Non-Nucleoside SETD2 Methyltransferase Inhibitor against Acute Myeloid Leukemia

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
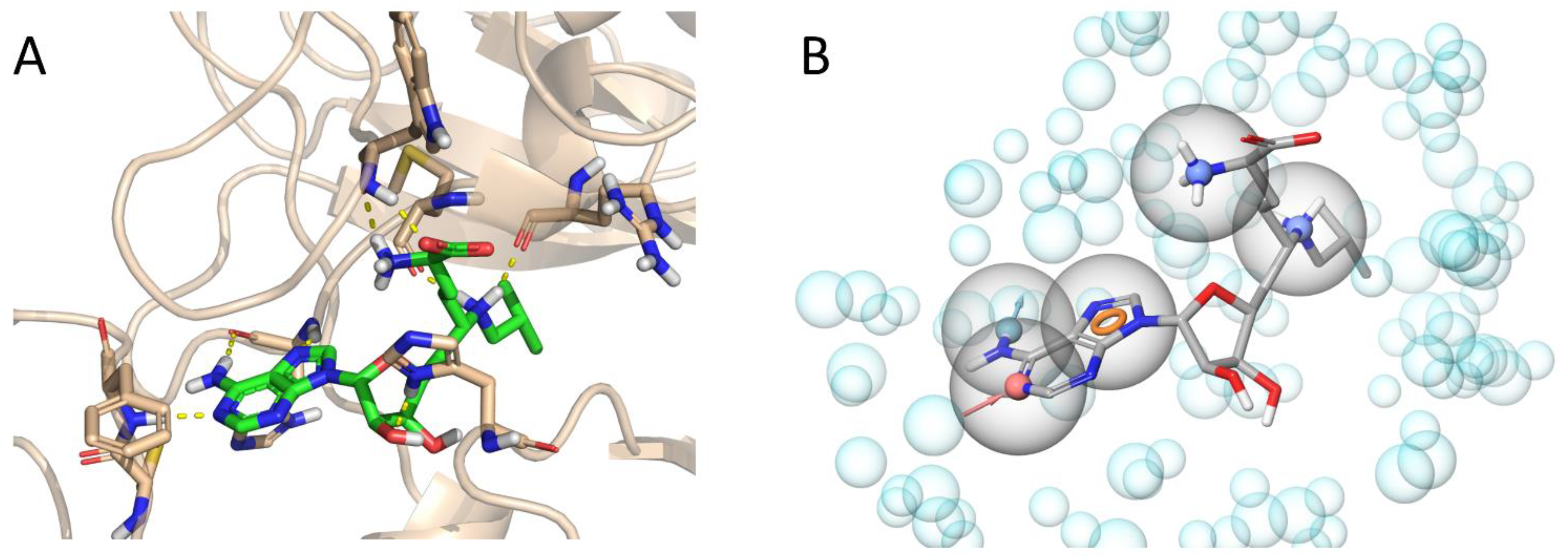
2.1. Virtual Screening Workflow and Retrospective Validation
2.2. Prospective Screening and Hit Selection
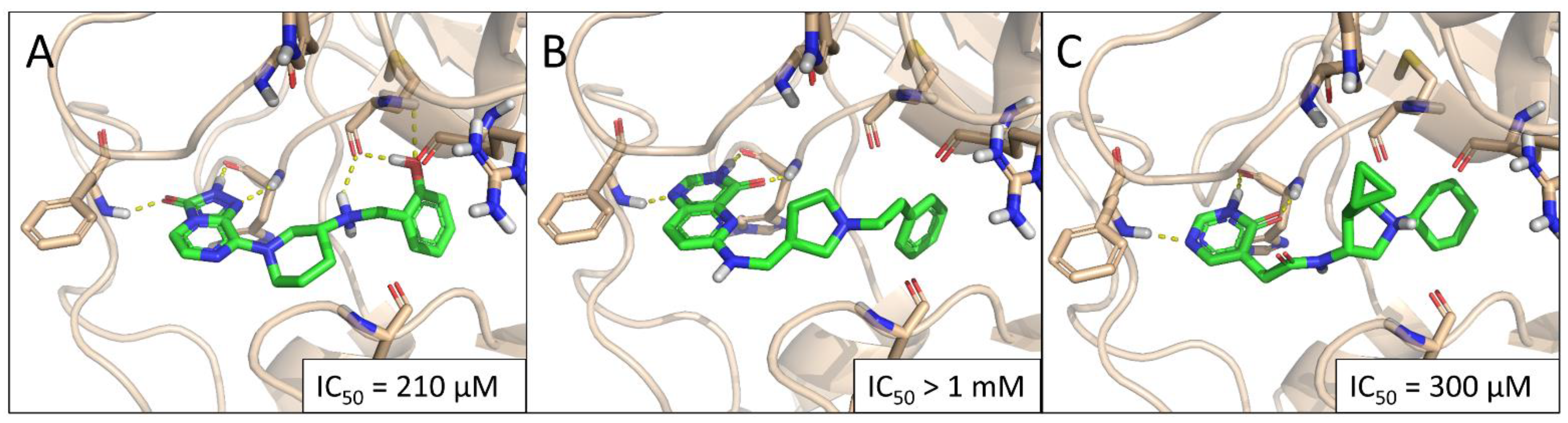
2.3. Experimental Hit Confirmation and Characterization
3. Discussion
4. Materials and Methods
4.1. Datasets and Ligand Preparation
4.2. Pharmacophore Screening
4.3. Ligand Docking
4.4. Post-Processing and Hit Selection
4.5. Measurement of SETD2 Enzyme Activity
4.6. Evaluation of Antiproliferative Effects of C13 in Leukemia Cell Lines
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Copeland, R.A.; Solomon, M.E.; Richon, V.M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 2009, 8, 724–732. [Google Scholar] [CrossRef]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008, 27, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Ibáñez, G.; Wu, H.; Blum, G.; Zeng, H.; Dong, A.; Li, F.; Hajian, T.; Allali-Hassani, A.; Amaya, M.F.; et al. Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J. Am. Chem. Soc. 2012, 134, 18004–18014. [Google Scholar] [CrossRef] [Green Version]
- Skucha, A.; Ebner, J.; Grebien, F. Roles of SETD2 in Leukemia—Transcription, DNA-Damage, and Beyond. Int. J. Mol. Sci. 2019, 20, 1029. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Luo, H.; Lu, J. Genetically Altered Cancer Epigenome; Elsevier Inc.: Amsterdam, The Netherlands, 2016; ISBN 9780127999586. [Google Scholar]
- Al Sarakbi, W.; Sasi, W.; Jiang, W.G.; Roberts, T.; Newbold, R.F.; Mokbel, K. The mRNA expression of SETD2 in human breast cancer: Correlation with clinico-pathological parameters. BMC Cancer 2009, 9, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duns, G.; van den Berg, E.; van Duivenbode, I.; Osinga, J.; Hollema, H.; Hofstra, R.M.W.; Kok, K. Histone Methyltransferase Gene SETD2 Is a Novel Tumor Suppressor Gene in Clear Cell Renal Cell Carcinoma. Cancer Res. 2010, 70, 4287–4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, G.; Mancini, M.; De Benedittis, C.; Rondoni, M.; Papayannidis, C.; Manfrini, M.; Meggendorfer, M.; Calogero, R.; Guadagnuolo, V.; Fontana, M.C.; et al. SETD2 and histone H3 lysine 36 methylation deficiency in advanced systemic mastocytosis. Leukemia 2018, 32, 139–148. [Google Scholar] [CrossRef]
- Bagger, F.O.; Sasivarevic, D.; Sohi, S.H.; Laursen, L.G.; Pundhir, S.; Sønderby, C.K.; Winther, O.; Rapin, N.; Porse, B.T. BloodSpot: A database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 2016, 44, D917–D924. [Google Scholar] [CrossRef] [PubMed]
- Mar, B.G.; Bullinger, L.B.; McLean, K.M.; Grauman, P.V.; Harris, M.H.; Stevenson, K.; Neuberg, D.S.; Sinha, A.U.; Sallan, S.E.; Silverman, L.B.; et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat. Commun. 2014, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Mar, B.G.; Chu, S.H.; Kahn, J.D.; Krivtsov, A.V.; Koche, R.; Castellano, C.A.; Kotlier, J.L.; Zon, R.L.; McConkey, M.E.; Chabon, J.; et al. SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood 2017, 130, 2631–2641. [Google Scholar] [CrossRef]
- Skucha, A.; Ebner, J.; Schmöllerl, J.; Roth, M.; Eder, T.; César-Razquin, A.; Stukalov, A.; Vittori, S.; Muhar, M.; Lu, B.; et al. MLL-fusion-driven leukemia requires SETD2 to safeguard genomic integrity. Nat. Commun. 2018, 9, 1983. [Google Scholar] [CrossRef]
- Yebra, M.J.; Sanchez, J.; Martin, C.G.; Hardisson, C.; Barbes, C. The effect of sinefungin and synthetic analogues on RNA and DNA methyltransferases from streptomyces. J. Antibiot. 1991, 44, 1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richon, V.M.; Johnston, D.; Sneeringer, C.J.; Jin, L.; Majer, C.R.; Elliston, K.; Jerva, L.F.; Scott, M.P.; Copeland, R.A. Chemogenetic Analysis of Human Protein Methyltransferases. Chem. Biol. Drug Des. 2011, 78, 199–210. [Google Scholar] [CrossRef]
- Tisi, D.; Chiarparin, E.; Tamanini, E.; Pathuri, P.; Coyle, J.E.; Hold, A.; Holding, F.P.; Amin, N.; Martin, A.C.L.; Rich, S.J.; et al. Structure of the epigenetic oncogene MMSET and inhibition by N-alkyl sinefungin derivatives. ACS Chem. Biol. 2016, 11, 3093–3105. [Google Scholar] [CrossRef]
- Kiss, R.; Sándor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J. Cheminform. 2012, 4 (Suppl. 1), 17. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, A. Confidence limits, error bars and method comparison in molecular modeling. Part 1: The calculation of confidence intervals. J. Comput. Aided. Mol. Des. 2014, 28, 887–918. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zheng, X.; Lu, C.; Li, G.M.; Allis, C.D.; Li, H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 2016, 30, 1611–1616. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Rácz, A.; Bajusz, D.; Héberger, K. Life beyond the Tanimoto coefficient: Similarity measures for interaction fingerprints. J. Cheminform. 2018, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Thomenius, M.J.; Totman, J.; Cosmopoulos, K.; Brach, D.; Ci, L.; Farrow, N.; Smith, J.J.; Chesworth, R.; Duncan, K.W.; Tang, C.; et al. Identification of a First-in-Class SETD2 Inhibitor That Shows Potent and Selective Anti-Proliferative Activity in t(4;14) Multiple Myeloma: T(4;14) Multiple Myeloma Cells Are Dependent on Both H3K36 Di and Tri-Methylation. Blood 2018, 132, 3207. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—the Konstanz Information Miner: Version 2.0 and Beyond. SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Instant JChem 19.4.0; ChemAxon LLC: Budapest, Hungary, 2019; Available online: http://www.chemaxon.com (accessed on 1 August 2021).
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-4: Schrödinger Suite 2020-4 Protein Preparation Wizard, LigPrep, Epik, ConfGen, Phase, Glide, Canvas; Schrödinger, LLC: New York, NY, USA, 2020.
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A Conformational Search Method for Efficient Generation of Bioactive Conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef]
- Salam, N.K.; Nuti, R.; Sherman, W. Novel Method for Generating Structure-Based Pharmacophores Using Energetic Analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef]
- Loving, K.; Salam, N.K.; Sherman, W. Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Aided. Mol. Des. 2009, 23, 541–554. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajusz, D.; Bognár, Z.; Ebner, J.; Grebien, F.; Keserű, G.M. Discovery of a Non-Nucleoside SETD2 Methyltransferase Inhibitor against Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 10055. https://doi.org/10.3390/ijms221810055
Bajusz D, Bognár Z, Ebner J, Grebien F, Keserű GM. Discovery of a Non-Nucleoside SETD2 Methyltransferase Inhibitor against Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2021; 22(18):10055. https://doi.org/10.3390/ijms221810055
Chicago/Turabian StyleBajusz, Dávid, Zsolt Bognár, Jessica Ebner, Florian Grebien, and György M. Keserű. 2021. "Discovery of a Non-Nucleoside SETD2 Methyltransferase Inhibitor against Acute Myeloid Leukemia" International Journal of Molecular Sciences 22, no. 18: 10055. https://doi.org/10.3390/ijms221810055