
Identification and Characterization of Novel circRNAs Involved in Muscle Growth of Blunt Snout Bream (Megalobrama amblycephala)


Abstract
:1. Introduction
2. Results
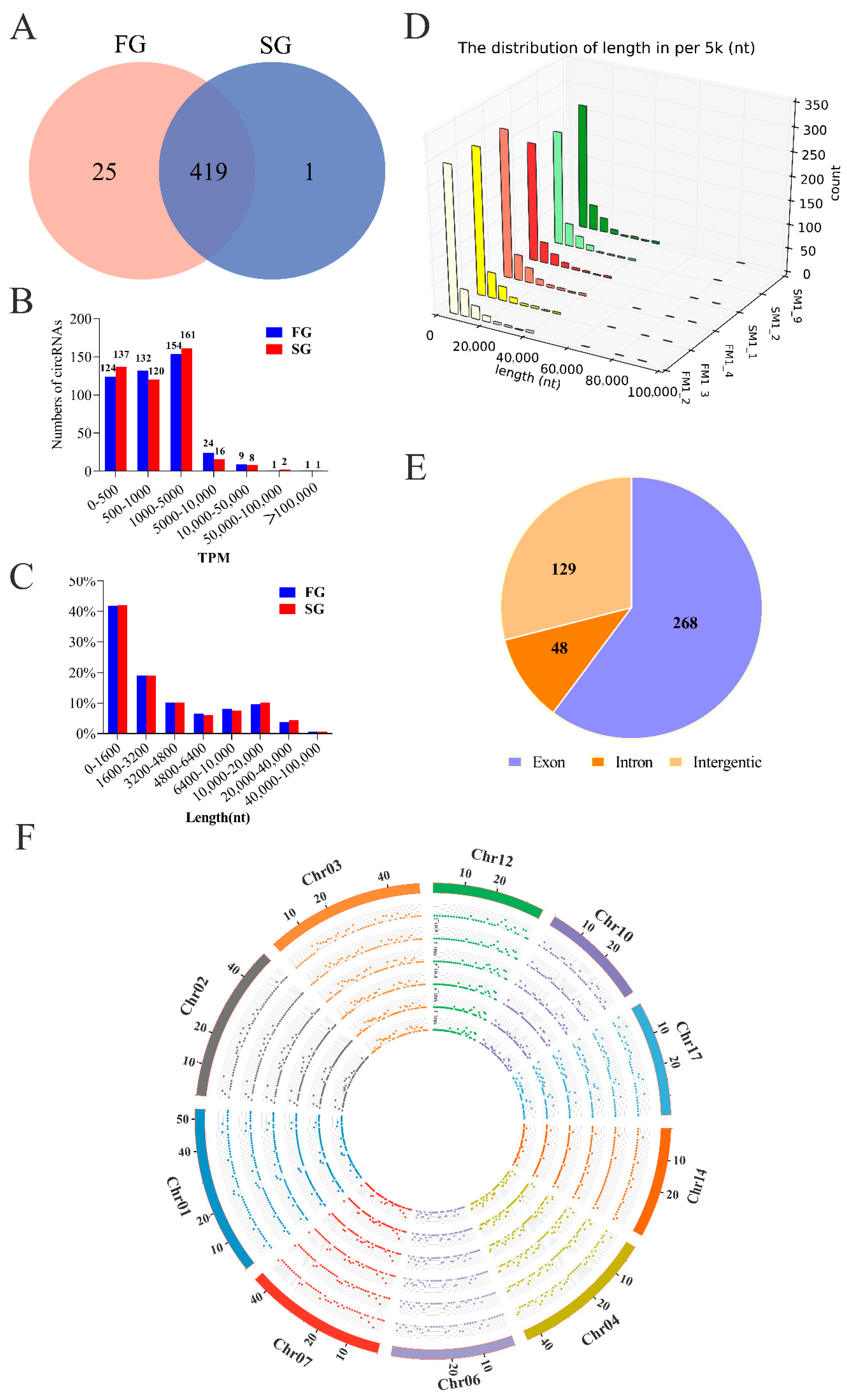
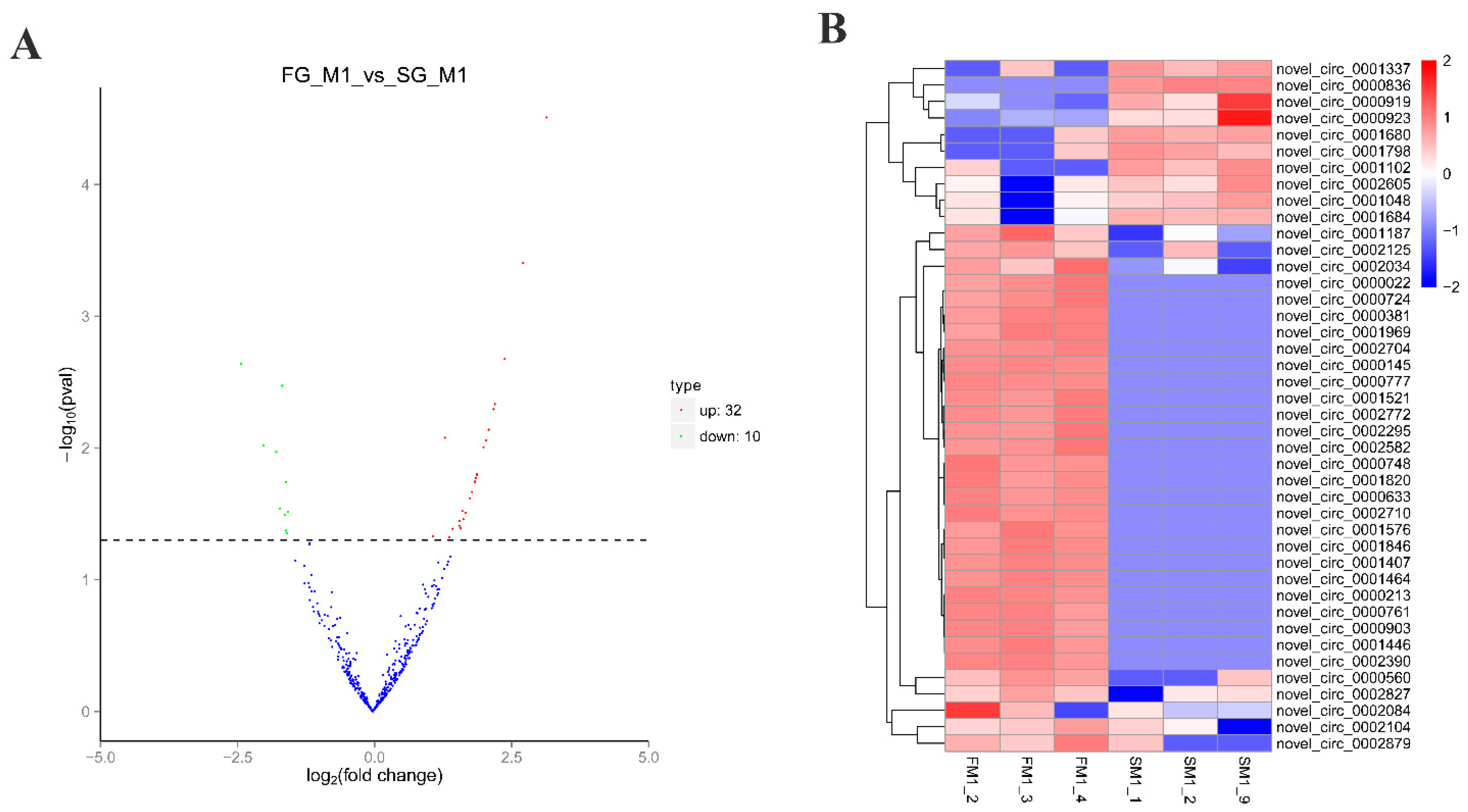
2.1. Identification of circRNAs and Differential Expression Analysis
2.2. Correlation Analysis between DE circRNAs and Their Source Genes
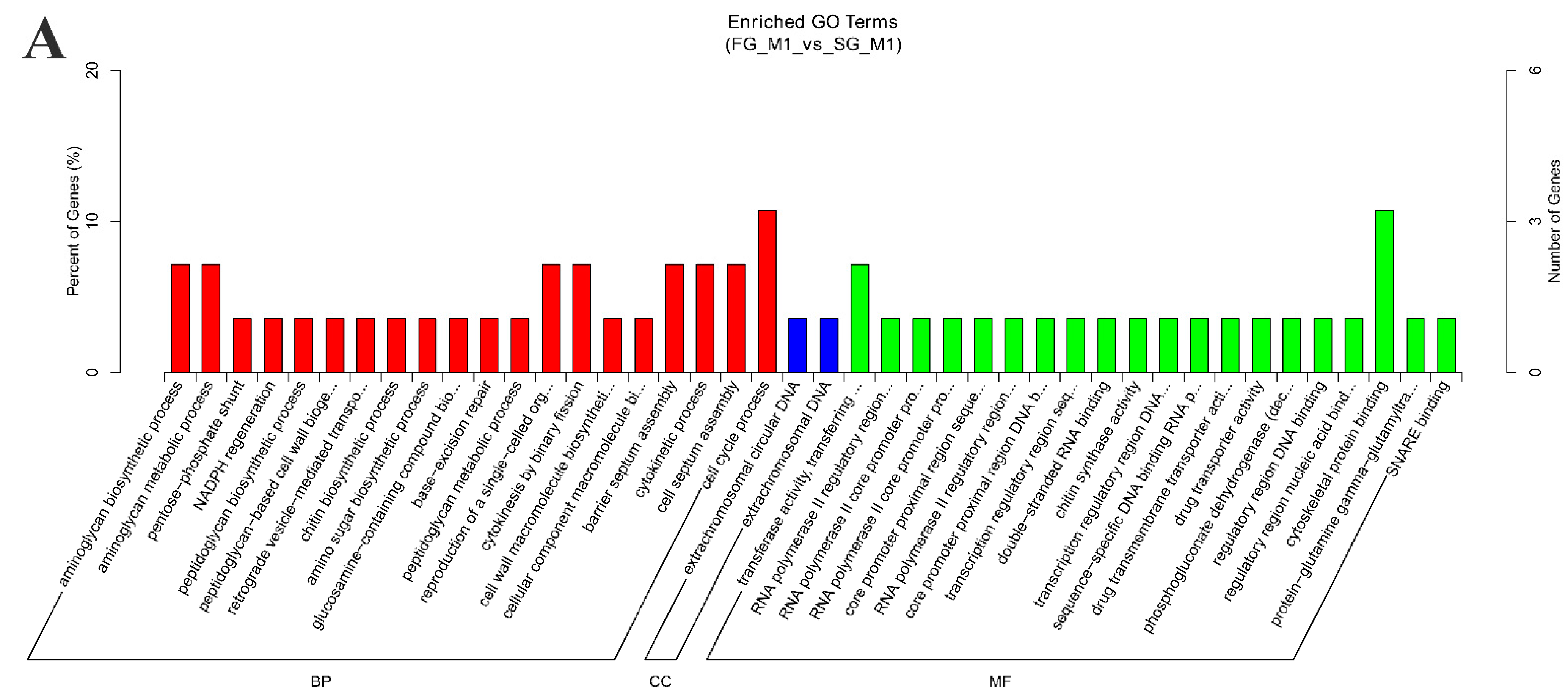
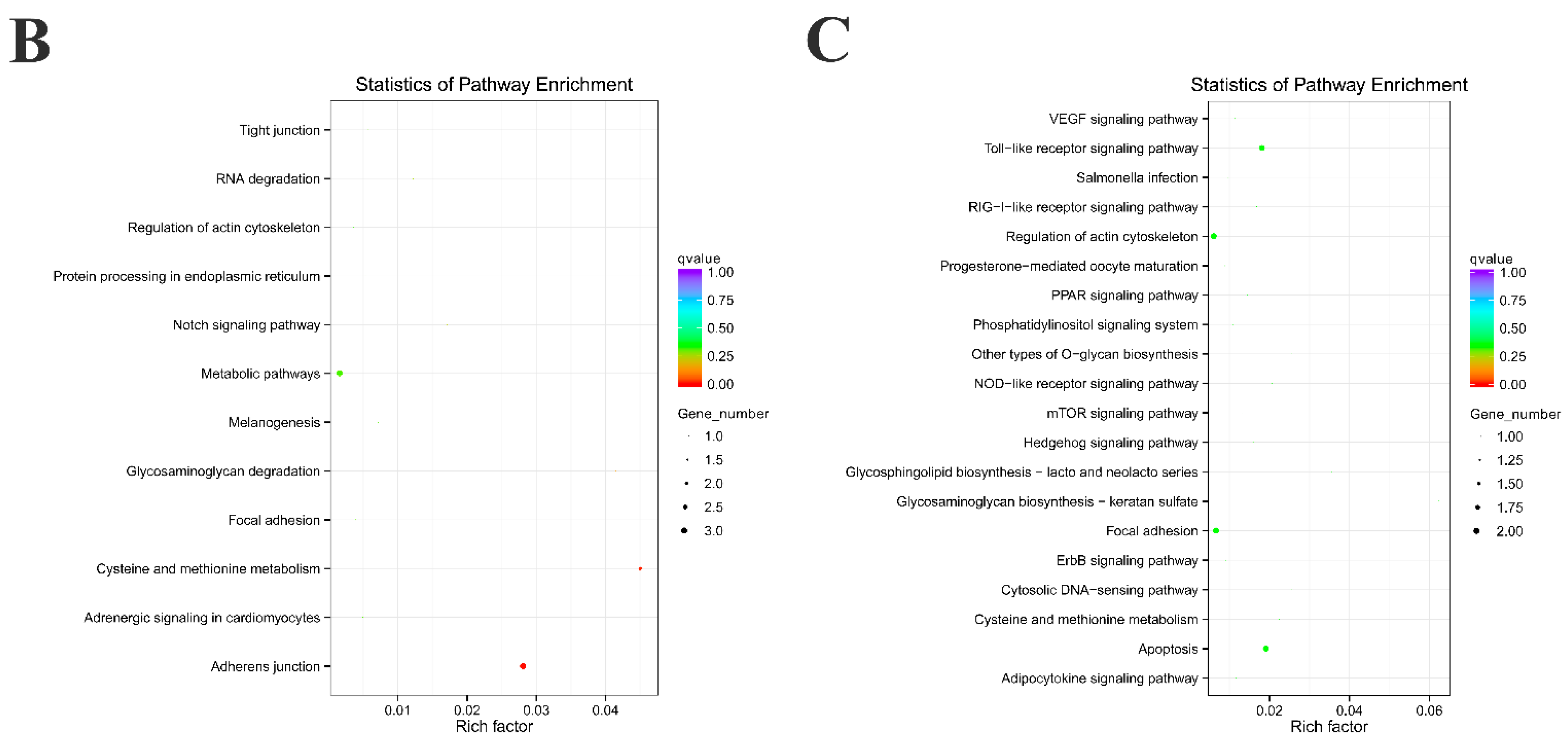
2.3. Functional Enrichment Analysis of Source Genes and Target miRNAs Prediction
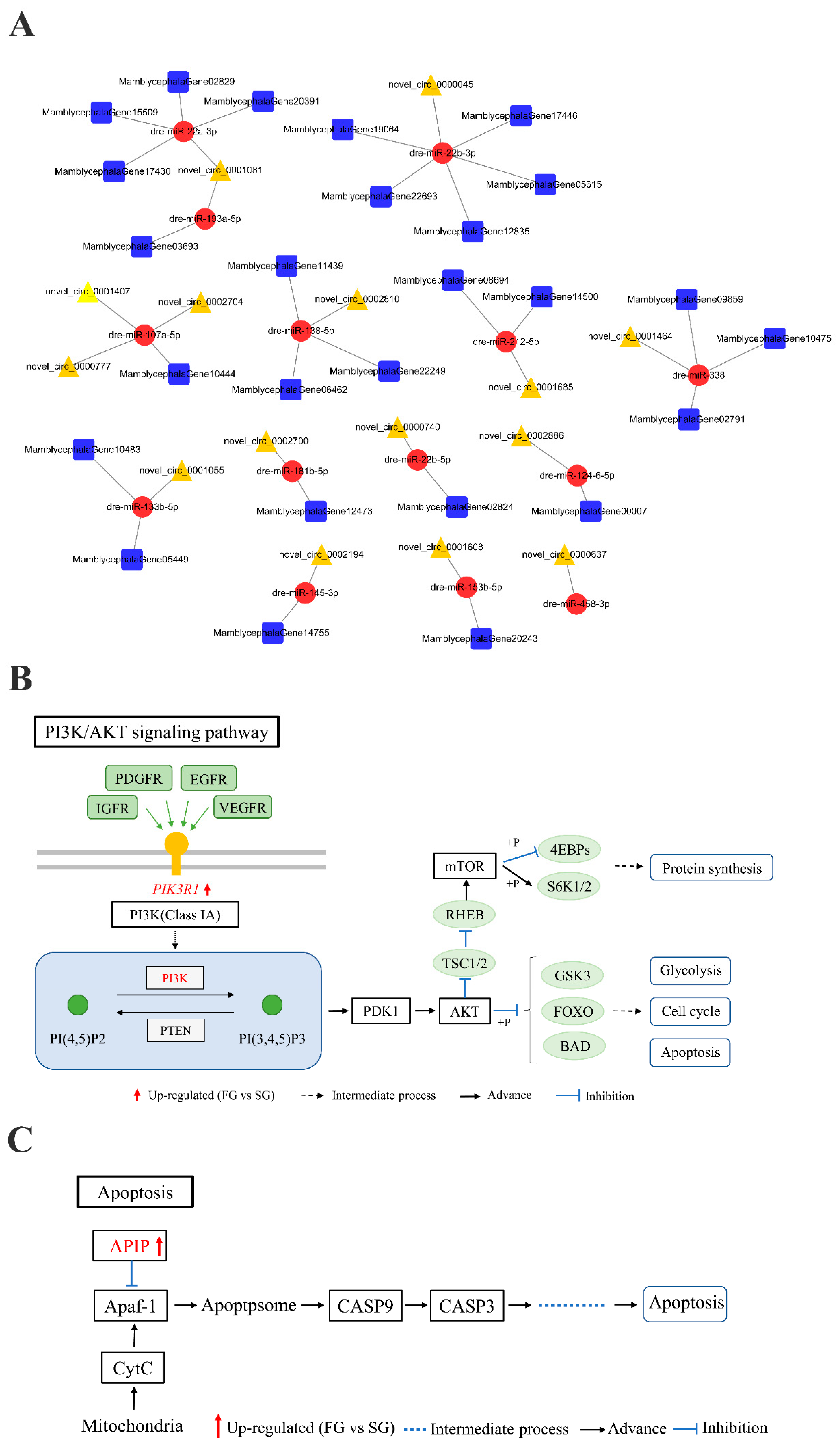
2.4. circRNA–miRNA–mRNA Regulation Networks Construction
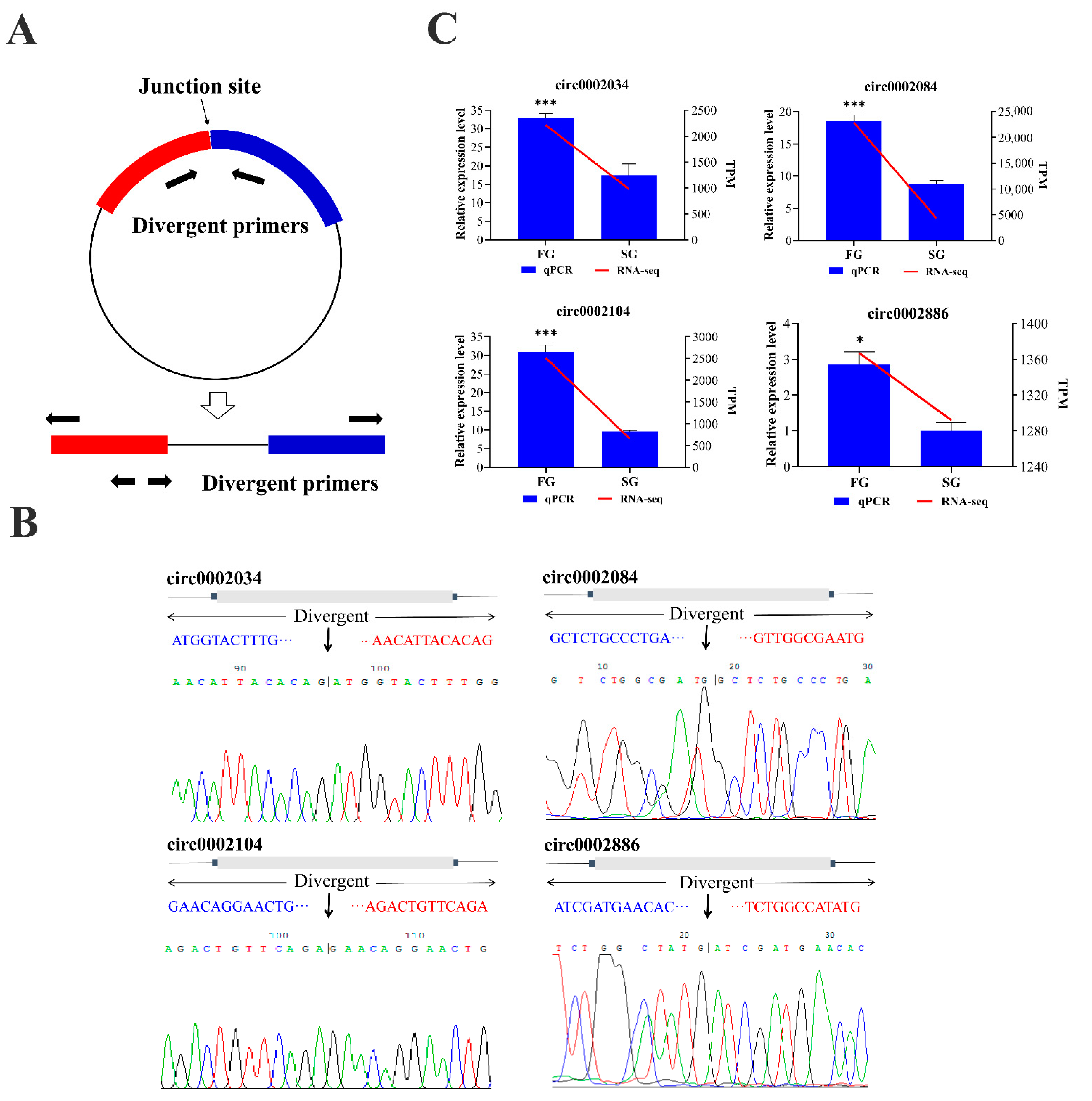
2.5. Validation of circRNAs by Quantitative Real Time PCR (qPCR)
3. Discussion
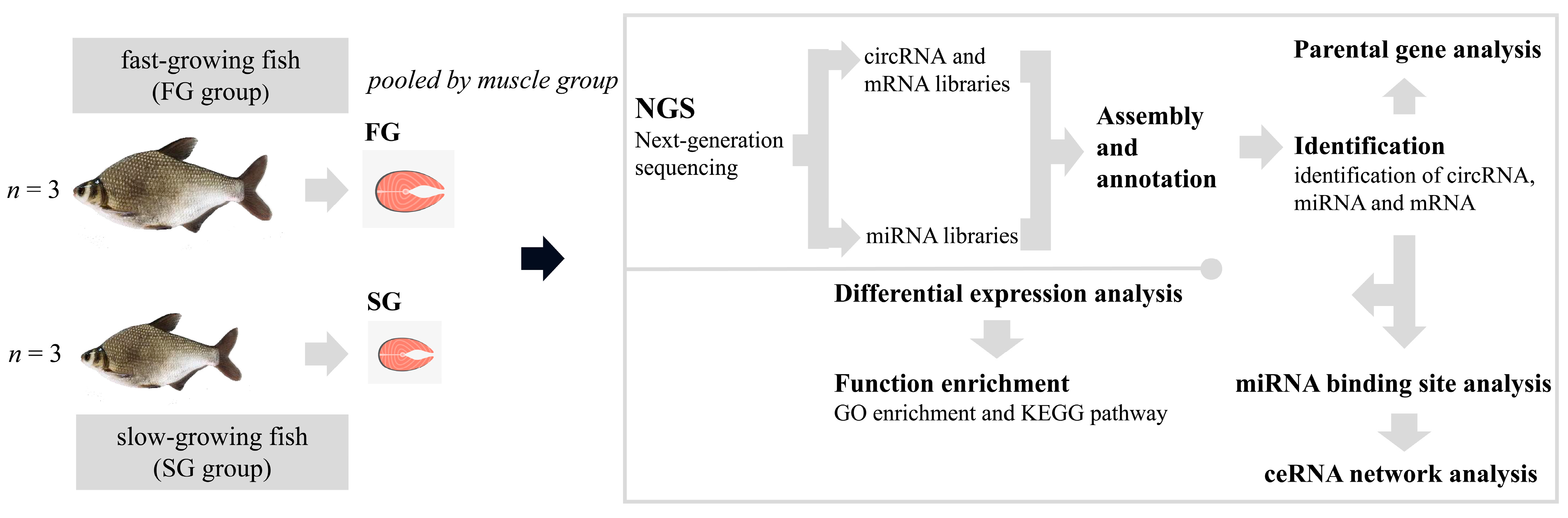
4. Materials and Methods
4.1. Sample Preparation and RNA Extraction
4.2. Library Construction and Whole Transcriptomic Sequencing
4.3. Quality Control
4.4. circRNA Identification
4.5. Differential Expression Analysis
4.6. GO and KEGG Enrichment Analysis
4.7. circRNA–miRNA–mRNA Network Analysis
4.8. Validation of circRNA with qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. MiRNA-dependent gene silencing involving ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011, 30, 4414–4422. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Memczak, S. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Chen, T.; Xiang, J.; Yin, Q.; Xing, Y.; Zhu, S.; Yang, L.; Chen, L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Kotb, A.; Panda, A.C.; Supriyo, D.; Ioannis, G.; Jiyoung, K.; Jun, D.; Noh, J.H.; Kim, K.M.; Mattison, J.A.; Cabo, R.D.; et al. Circular RNAs in monkey muscle: Age-dependent changes. Aging 2015, 7, 903–910. [Google Scholar]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Legnini, I.; Timoteo, G.D.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol. Cell 2017, 66, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, H.; Chen, X.; Li, W.; Li, Z.; Nie, Q.; Zhang, X. Circular RNA circSVIL promotes myoblast proliferation and differentiation by sponging miR-203 in chicken. Front. Genet. 2018, 9, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Li, H.; Yang, J.; Hao, D.; Dong, D.; Huang, Y.; Lan, X.; Plath, M.; Lei, C.; Lin, F.; et al. Circular RNA profiling reveals an abundant circLMO7 that regulates myoblasts differentiation and survival by sponging miR-378a-3p. Cell Death Dis. 2017, 8, e3153. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, A.; Doose, G.; Tafer, H.; Robinson, M.; Saha, N.R.; Gerdol, M.; Canapa, A.; Hoffmann, S.; Amemiya, C.T.; Stadler, P.F. Atypical RNAs in the coelacanth transcriptome. J. Exp. Zool. B Mol. Dev. Evol. 2014, 322, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, Y.; Yin, J.; Yan, X.Y.; Chen, W.J.; Jiang, C.Y.; Hu, X.S.; Wang, X.Y.; Zhu, J.G.; Yu, Z.B.; et al. Profiles analysis reveals circular RNAs involving zebrafish physiological development. J. Cell. Physiol. 2019, 234, 15922–15933. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dai, Y.; Zhang, X.; Dai, K.; Liu, B.; Yuan, R.; Feng, Y.; Liang, Z.; Zhu, M.; Zhang, M.; et al. Identification and characterization of novel type of RNAs, circRNAs in crucian carp Carassius auratus gibelio. Fish Shellfish Immunol. 2019, 94, 50–57. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, A.; Xiong, L.; Li, Y.; Huang, R.; Liao, L.; Zhu, Z.; Wang, Y. Deep circular RNA sequencing provides insights into the mechanism underlying grass carp reovirus infection. Int. J. Mol. Sci. 2017, 18, 1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Yuan, R.; Liang, Z.; Zhang, T. Comprehensive analysis of circRNA expression pattern and circRNA-mRNA-miRNA network in Ctenopharyngodon idellus kidney (CIK) cells after grass carp reovirus (GCRV) infection. Aquaculture 2019, 512, 734349. [Google Scholar] [CrossRef]
- Xiu, Y.; Jiang, G.; Zhou, S.; Diao, J.; Liu, H.; Su, B.; Li, C. Identification of potential immune-related circRNA-miRNA-mRNA regulatory network in intestine of Paralichthys olivaceus during Edwardsiella tarda infection. Front. Genet. 2019, 10, 731. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.; Chen, F.; Li, Y.; Wang, Z.; Wang, Z.; Lu, Y.; Wu, Z.; Jian, J.; Wang, B. A comprehensive profile of the tilapia (Oreochromis niloticus) circular RNA and circRNA-miRNA network in the pathogenesis of meningoencephalitis of teleosts. Mol. Omics 2019, 15, 233–246. [Google Scholar] [CrossRef]
- Chen, J.; Cao, M.; Zhang, A.; Shi, M.; Tao, B.; Li, Y.; Wang, Y.; Zhu, Z.; Trudeau, V.; Hu, W. Growth hormone overexpression disrupts reproductive status through actions on Leptin. Front. Endocrinol. 2018, 9, 131. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Luo, W.; Liu, H.; Zeng, C.; Liu, X.; Yi, S.; Wang, W. Transcriptome analysis and SSR/SNP markers information of the blunt snout bream (Megalobrama Amblycephala). PLoS ONE 2012, 7, e42637. [Google Scholar]
- Wang, W. The aquaculture status of blunt snout bream (Megalobrama Amblycephala). Sci. Fish Farming 2009, 4, 44–45. [Google Scholar]
- Yi, S.; Gao, Z.; Zhao, H.; Zeng, C.; Luo, W.; Chen, B.; Wang, W. Identification and characterization of microRNAs involved in growth of blunt snout bream (Megalobrama Amblycephala) by Solexa sequencing. BMC Genom. 2013, 14, 754. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Wang, H.; Wang, H.; Gul, Y.; Yang, M.; Zeng, C.; Wang, W. Characterization of muscle morphology and satellite cells, and expression of muscle-related genes in skeletal muscle of juvenile and adult Megalobrama Amblycephala. Micron 2014, 64, 66–75. [Google Scholar] [CrossRef]
- Shen, Y.; Guo, X.; Wang, W. Identification and characterization of circular RNAs in zebrafish. FEBS Lett. 2017, 591, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Hao, Z.; Zhu, Y.; Zhang, H.; Li, G. Genome-wide identification and functional analysis of circRNAs in Zea mays. PLoS ONE 2018, 13, e0202375. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Zhao, Y.; Wang, S.; Ma, J.; Wang, X.; Qian, L. Identification, characterization, and functional investigation of circular RNAs in subventricular zone of adult rat brain. J. Cell. Biochem. 2018, 120, 3428–3437. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ren, Y.; Lin, T.; Cui, D. Identification and characterization of circRNAs involved in the regulation of wheat root length. Biol. Res. 2019, 52, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Reckman, Y.J.; Aufiero, S.; van den Hoogenhof, M.M.; van der Made, I.; Beqqali, A.; Koolbergen, D.R.; Rasmussen, T.B.; Velden, J.V.D.; Creemers, E.E.; et al. Rbm20 regulates circular RNA production from the titin gene. Circ. Res. 2016, 119, 996–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zheng, Q.; Bao, C.; Li, S.; Guo, W.; Zhao, J.; Chen, D.; Gu, J.; He, X.; Huang, S. Circular RNA is enriched and stable in exosomes: A promising biomarker for cancer diagnosis. Cell Res. 2015, 25, 981–984. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, H.; Chen, X.; Wang, Z.; Yu, J.; Jia, X.; Li, Z.; Luo, W.; Abdalla, B.A.; Jebessa, E.; Nie, Q.; et al. Circular RNAs are abundant and dynamically expressed during embryonic muscle development in chickens. DNA Res. 2018, 25, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with alu repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, S.; Jost, I.; Rossbach, O.; Schneider, T.; Schreiner, S.; Hung, L.; Bindereif, A. Exon circularization requires canonical splice signals. Cell Rep. 2015, 10, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Ooshio, T.; Irie, K.; Morimoto, K.; Fukuhara, A.; Imai, T.; Takai, Y. Involvement of LMO7 in the association of two cell-cell adhesion molecules, nectin and E-cadherin, through afadin and alpha-actinin in epithelial cells. J. Biol. Chem. 2004, 279, 31365–31373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holaska, J.M.; Rais-Bahrami, S.; Wilson, K.L. Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum. Mol. Genet. 2006, 15, 3459–3472. [Google Scholar] [CrossRef]
- Dedeic, Z.; Cetera, M.; Cohen, T.V.; Holaska, J.M. Emerin inhibits Lmo7 binding to the Pax3 and MyoD promoters and expression of myoblast proliferation genes. J. Cell Sci. 2011, 124, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- Geiger, B.; Dutton, A.H.; Tokuyasu, K.T.; Singer, S.J. Immunoelectron microscope studies of membrane-microfilament interactions: Distributions of alpha-actinin, tropomyosin, and vinculin in intestinal epithelial brush border and chicken gizzard smooth muscle cells. J. Cell Biol. 1981, 91, 614–628. [Google Scholar] [CrossRef] [Green Version]
- Eddinger, T.J.; Schiebout, J.D.; Swartz, D.R. Smooth muscle adherens junctions associated proteins are stable at the cell periphery during relaxation and activation. Am. J. Physiol.-Cell Physiol. 2005, 289, C1379–C1387. [Google Scholar] [CrossRef] [Green Version]
- Naito, M.; Mori, M.; Inagawa, M.; Miyata, K.; Hashimoto, N.; Tanaka, S.; Asahara, H. Dnmt3a regulates proliferation of muscle satellite cells via p57Kip2. PLoS Genet. 2016, 12, e1006167. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.; Sharpless, N. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Li, H.; Wei, X.; Yang, J.; Dong, D.; Hao, D.; Huang, Y.; Lan, X.; Plath, M.; Lei, C.; Ma, Y.; et al. CircFGFR4 promotes differentiation of myoblasts via binding miR-107 to relieve its inhibition of Wnt3a. Mol. Ther.-Nucleic Acids 2018, 11, 272–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Yang, J.; Wei, X.; Song, C.; Dong, D.; Huang, Y.; Lan, X.; Plath, M.; Lei, C.; Ma, Y.; et al. CircFUT10 reduces proliferation and facilitates differentiation of myoblasts by sponging miR-133a. J. Cell. Physiol. 2017, 233, 4643–4651. [Google Scholar] [CrossRef]
- Liu, J.J.; Zhang, H.; Xing, F.; Tang, B.; Wu, S.L.; Xuan, L.; Kang, P.F.; Xu, Q.; Wang, H.J.; Zhang, N.R.; et al. microRNA-138 promotes proliferation and suppresses mitochondrial depolarization in human pulmonary artery smooth muscle cells through targeting TASK-1. Mol. Med. Rep. 2018, 17, 3021–3027. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, L.; Yun, H.F.; Han, Y.S. MiR-138 promotes smooth muscle cells proliferation and migration in db/db mice through down-regulation of SIRT1. Biochem. Biophys. Res. Commun. 2015, 463, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Liu, W.; Zhang, J.; Xiang, D. MiR-153 regulates apoptosis and autophagy of cardiomyocytes by targeting Mcl-1. Mol. Med. Rep. 2016, 14, 1033–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Zhao, Y.; Hou, W.; Guo, L. MiR-153 regulates cardiomyocyte apoptosis by targeting Nrf2/HO-1 signaling. Chromosome Res. 2019, 27, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Duan, P.; Guo, P.; Li, D.; Li, S.; Xu, Y.; Zhou, Q. Downregulation of miR-223 and miR-153 mediates mechanical stretch-stimulated proliferation of venous smooth muscle cells via activation of the insulin-like growth factor-1 receptor. Arch. Biochem. Biophys. 2012, 528, 204–211. [Google Scholar] [CrossRef]
- Luo, J.; Sobkiw, C.L.; Hirshman, M.F.; Logsdon, M.N.; Li, T.Q.; Goodyear, L.J.; Cantley, L.C. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006, 3, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Vogt, P.K.; Hart, J.R. Domain analysis reveals striking functional differences between the regulatory subunits of phosphatidylinositol 3-kinase (PI3K), p85α and p85β. Oncotarget 2017, 8, 55863–55876. [Google Scholar] [CrossRef] [Green Version]
- Mellor, P.; Furber, L.A.; Nyarko, J.N.K.; Anderson, D.H. Multiple roles for the p85α isoform in the regulation and function of PI3K signalling and receptor trafficking. Biochem. J. 2012, 441, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cui, L.; Yuan, J.; Zhang, Y.; Sang, H. Circular RNA WDR77 target FGF-2 to regulate vascular smooth muscle cells proliferation and migration by sponging miR-124. Biochem. Biophys. Res. Commun. 2017, 494, 126–132. [Google Scholar] [CrossRef]
- Choe, N.; Kwon, D.H.; Shin, S.; Kim, Y.S.; Kim, Y.K.; Kim, J.; Ahn, Y.; Eom, G.H.; Kook, H. The microRNA miR-124 inhibits vascular smooth muscle cell proliferation by targeting S100 calcium-binding protein A4 (S100A4). FEBS Lett. 2017, 591, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Yu, S.; Liu, Y.; Zhang, J.; Han, L.; Xu, Z. microRNA-124 control human vascular smooth muscle cell phenotypic switch via Sp1. Am. J. Physiol.-Heart Circ. Physiol. 2017, 313, H641–H649. [Google Scholar] [CrossRef] [PubMed]
- Qadir, A.S.; Woo, K.M.; Ryoo, H.M.; Yi, T.G.; Song, S.U.; Baek, J.H. MiR-124 inhibits myogenic differentiation of mesenchymal stem cells via targeting Dlx5. J. Cell. Biochem. 2014, 115, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Hong, Y.M.; Lee, H.J.; Woo, H.N.; Pyo, J.O.; Mak, T.W.; Jung, Y.K. Induced inhibition of ischemic/hypoxic injury by APIP, a novel apaf-1-interacting protein. J. Biol. Chem. 2004, 279, 39942–39950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2017, 19, 803–810. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef] [PubMed]
- Young, M.; Wakefield, M.; Smyth, G.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Pasquinelli, A. microRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Z.; Shen, X.; Cui, X.; Guo, Y. Identification of novel biomarkers and small molecule drugs in human colorectal cancer by microarray and bioinformatics analysis. Mol. Genet. Genom. Med. 2019, 7, e00713. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | FM1_2 | FM1_3 | FM1_4 | SM1_1 | SM1_2 | SM1_9 |
|---|---|---|---|---|---|---|
| Raw reads | 115,662,842 | 91,517,836 | 91,790,546 | 84,720,390 | 94,082,116 | 93,420,104 |
| Clean reads | 110,866,216 | 88,187,208 | 89,386,380 | 81,888,776 | 89,640,072 | 89,874,446 |
| Mapped reads | 99,858,773 | 78,623,397 | 82,180,304 | 72,820,911 | 77,340,146 | 82,612,180 |
| Mapping ratio | 90.07% | 89.16% | 91.94% | 88.93% | 86.28% | 91.92% |
| Uniquely mapped reads | 91,308,444 | 72,141,305 | 75,688,357 | 67,246,303 | 70,603,603 | 74,848,301 |
| Unique mapping ratio | 82.36% | 81.80% | 84.68% | 82.12% | 78.76% | 83.28% |
| circRNA ID | Source Gene | FG(TPM) | SG(TPM) | log2(FG/SG) | p-Value |
|---|---|---|---|---|---|
| novel_circ_0002772 | - | 1619.74 | 0 | 3.17 | 3.09 × 10−5 |
| novel_circ_0001521 | dnmt3a | 1048.5 | 0 | 2.75 | 3.93 × 10−4 |
| novel_circ_0001576 | txlnb | 795.39 | 0 | 2.41 | 2.11 × 10−3 |
| novel_circ_0002295 | vcl | 653.66 | 0 | 2.23 | 4.64 × 10−3 |
| novel_circ_0001446 | myom2 | 623.26 | 0 | 2.21 | 5.07 × 10−3 |
| novel_circ_0000633 | fam189a2 | 556.98 | 0 | 2.12 | 7.27 × 10−3 |
| novel_circ_0001187 | - | 5522.04 | 2070.39 | 1.32 | 8.36 × 10−3 |
| novel_circ_0000145 | antxr1 | 524.4 | 0 | 2.07 | 8.78 × 10−3 |
| novel_circ_0002879 | pla2g4c | 8396.64 | 605.37 | 2.02 | 9.87 × 10−3 |
| novel_circ_0002390 | wdr90 | 453.84 | 0 | 1.91 | 1.58 × 10−2 |
| novel_circ_0001407 | cep170 | 449.26 | 0 | 1.9 | 1.59 × 10−2 |
| novel_circ_0001464 | nbas | 449.26 | 0 | 1.9 | 1.59 × 10−2 |
| novel_circ_0000777 | slc25a21 | 441.98 | 0 | 1.9 | 1.60 × 10−2 |
| novel_circ_0000022 | - | 468.1 | 0 | 1.9 | 1.61 × 10−2 |
| novel_circ_0000724 | - | 468.1 | 0 | 1.9 | 1.61 × 10−2 |
| novel_circ_0002704 | wdr62 | 437.4 | 0 | 1.89 | 1.69 × 10−2 |
| novel_circ_0001969 | sik2 | 444.68 | 0 | 1.87 | 1.80 × 10−2 |
| novel_circ_0001820 | chchd3 | 434.7 | 0 | 1.87 | 1.81 × 10−2 |
| novel_circ_0000213 | - | 406.7 | 0 | 1.81 | 2.16 × 10−2 |
| novel_circ_0000748 | ktn1 | 399.41 | 0 | 1.78 | 2.41 × 10−2 |
| novel_circ_0002125 | - | 2643.91 | 459.68 | 1.64 | 3.02 × 10−2 |
| novel_circ_0002710 | kif1b | 359.56 | 0 | 1.7 | 3.10 × 10−2 |
| novel_circ_0000381 | rbpj | 362.26 | 0 | 1.7 | 3.11 × 10−2 |
| novel_circ_0002582 | - | 350.4 | 0 | 1.66 | 3.48 × 10−2 |
| novel_circ_0002084 | tgm1 | 22909.41 | 4312.85 | 1.59 | 3.58 × 10−2 |
| novel_circ_0000560 | efnb1 | 743.67 | 106.89 | 1.58 | 3.89 × 10−2 |
| novel_circ_0000761 | dnmt3a | 331.56 | 0 | 1.61 | 4.03 × 10−2 |
| novel_circ_0000903 | gtdc1 | 331.56 | 0 | 1.61 | 4.03 × 10−2 |
| novel_circ_0001846 | creb3l2 | 326.98 | 0 | 1.6 | 4.08 × 10−2 |
| novel_circ_0002104 | - | 2509.35 | 651.04 | 1.46 | 4.10 × 10−2 |
| novel_circ_0002034 | yes | 2212.3 | 976.14 | 1.1 | 4.67 × 10−2 |
| novel_circ_0002827 | cisd1 | 1134.23 | 291.67 | 1.4 | 4.77 × 10−2 |
| novel_circ_0000836 | - | 0 | 627.8 | −2.4 | 2.29 × 10−3 |
| novel_circ_0000923 | lmo7 | 1533.34 | 6240.78 | −1.65 | 3.37 × 10−3 |
| novel_circ_0001102 | - | 79.72 | 915.02 | −1.99 | 9.54 × 10−3 |
| novel_circ_0001684 | grip2 | 229.99 | 1329.04 | −1.76 | 1.07 × 10−2 |
| novel_circ_0000919 | lmo7 | 324.28 | 1442.7 | −1.58 | 1.82 × 10−2 |
| novel_circ_0001337 | hpse2 | 94.28 | 701.25 | −1.69 | 2.88 × 10−2 |
| novel_circ_0001048 | limch1 | 265.27 | 1303.58 | −1.54 | 3.04 × 10−2 |
| novel_circ_0002605 | sec24b | 185.56 | 1106.57 | −1.6 | 3.22 × 10−2 |
| novel_circ_0001798 | ddx6 | 70.56 | 606.58 | −1.58 | 4.24 × 10−2 |
| novel_circ_0001680 | - | 70.56 | 572.13 | −1.56 | 4.46 × 10−2 |
| Primer Name | Sequences (5′→3′) | Application |
|---|---|---|
| circ0002034-F | TACGGGACTGGGATGAGATGA | qPCR |
| circ0002034-R | GCCCCTTTAGTGGTCTCACTTT | |
| circ0002084-F | TGATGAAGGTTCTGCTGC | |
| circ0002084-R | CAACTTCCTCCAGGTCTG | |
| circ0002104-F | AAAGCGGATGGATTGGCGTA | |
| circ0002104-R | GGGATGAACCTGTCTCCGTG | |
| circ0002886-F | ACACCAAGGAAGTATGCAACAGT | |
| circ0002886-R | ACAGGGGCCTCCGATATTGT | |
| β-actin-F | ACCCACACCGTGCCCATCTA | |
| β-actin-R | CGGACAATTTCTCTTTCGGCTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Chen, Y.; Diao, J.; Luo, L.; Gao, Z. Identification and Characterization of Novel circRNAs Involved in Muscle Growth of Blunt Snout Bream (Megalobrama amblycephala). Int. J. Mol. Sci. 2021, 22, 10056. https://doi.org/10.3390/ijms221810056
Liu L, Chen Y, Diao J, Luo L, Gao Z. Identification and Characterization of Novel circRNAs Involved in Muscle Growth of Blunt Snout Bream (Megalobrama amblycephala). International Journal of Molecular Sciences. 2021; 22(18):10056. https://doi.org/10.3390/ijms221810056
Chicago/Turabian StyleLiu, Lifang, Yulong Chen, Jinghan Diao, Lifei Luo, and Zexia Gao. 2021. "Identification and Characterization of Novel circRNAs Involved in Muscle Growth of Blunt Snout Bream (Megalobrama amblycephala)" International Journal of Molecular Sciences 22, no. 18: 10056. https://doi.org/10.3390/ijms221810056