
Profiling of Human Neural Crest Chemoattractant Activity as a Replacement of Fetal Bovine Serum for In Vitro Chemotaxis Assays


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
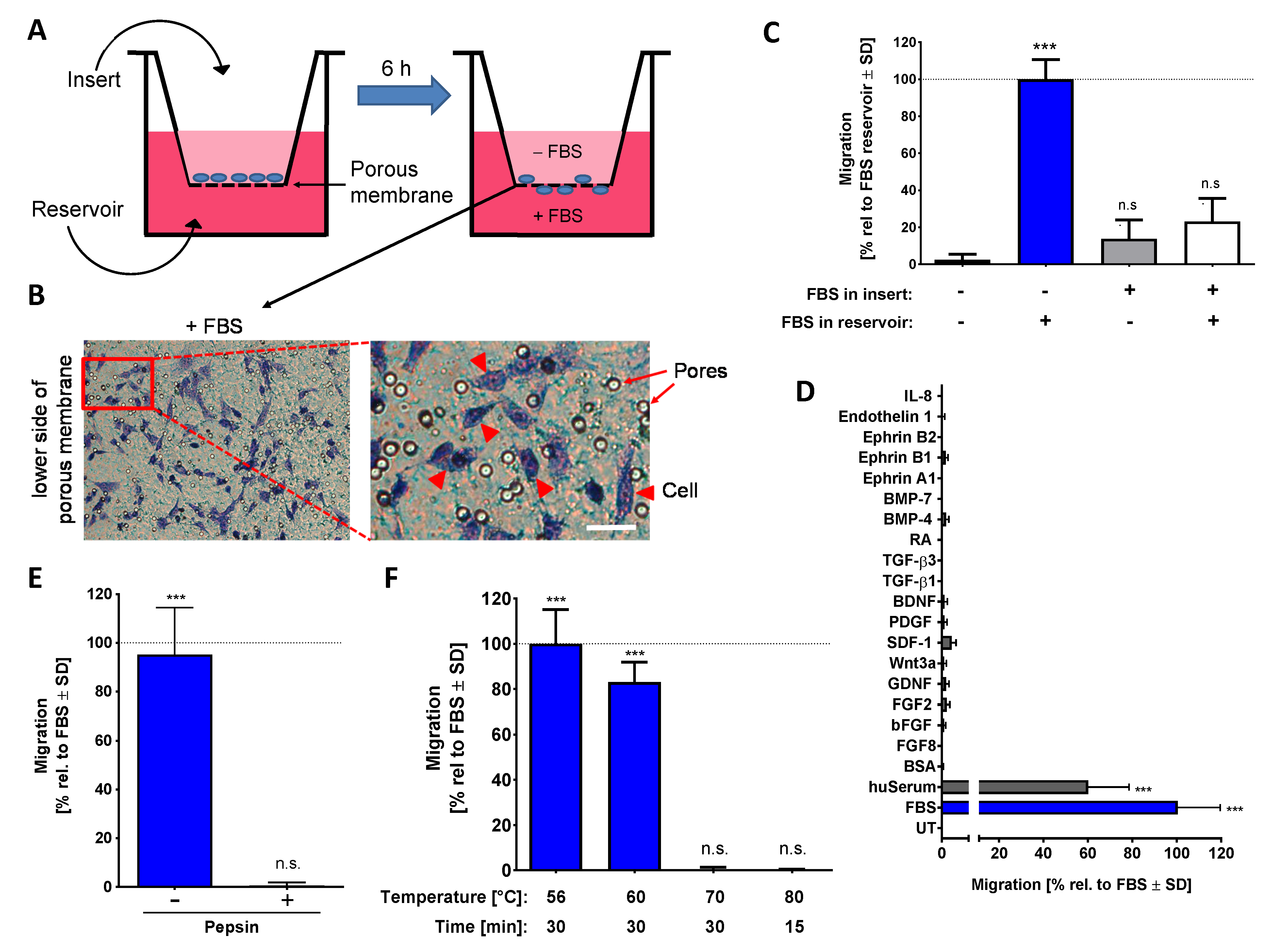
2.1. Establishment of a Chemotaxis Assay Based on Human NCCs
2.2. Characterization of the Chemotaxis-Triggering Factor in FBS
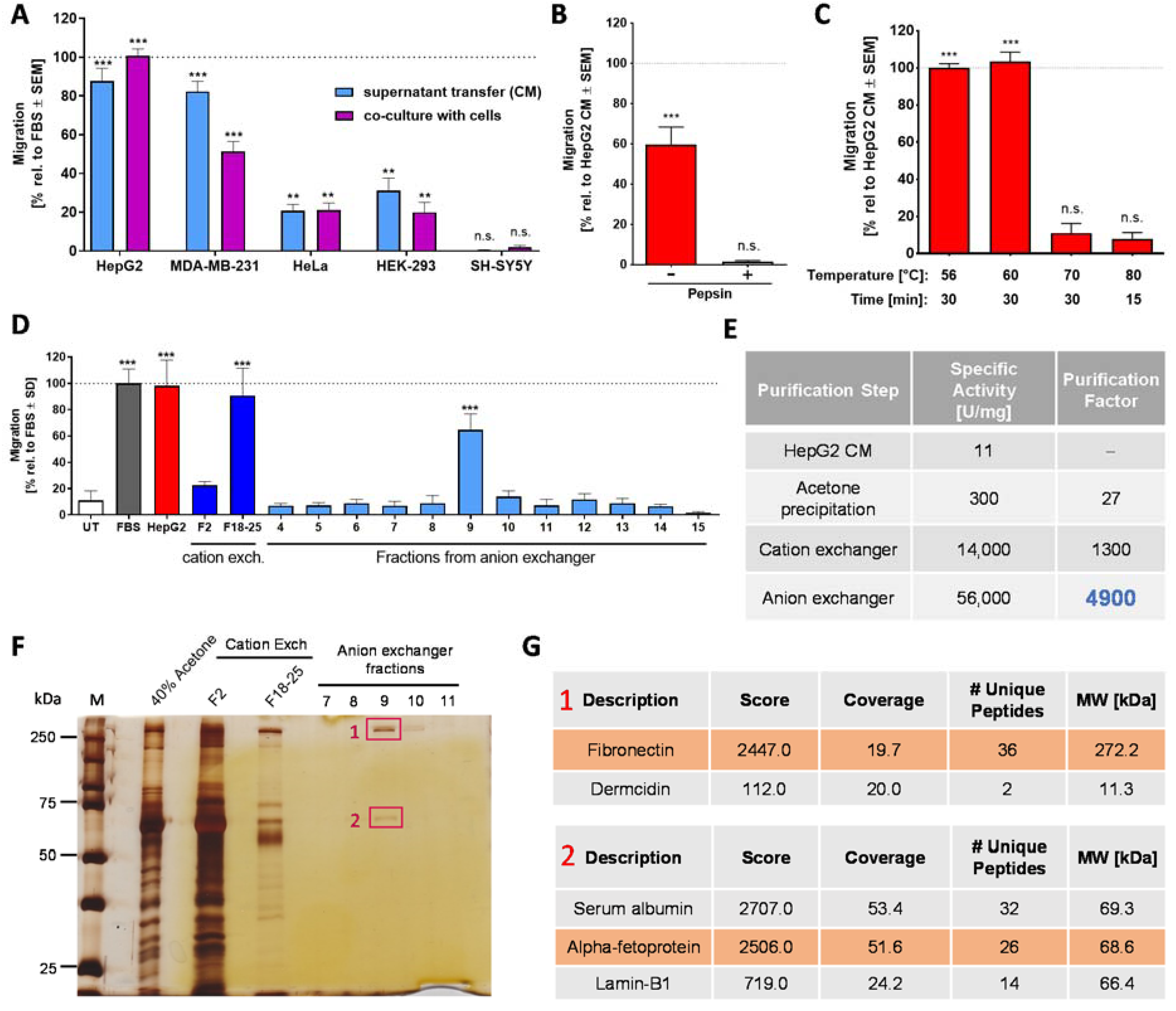
2.3. Chemotaxis-Triggering Activity Present in Conditioned Medium of HepG2 Cells
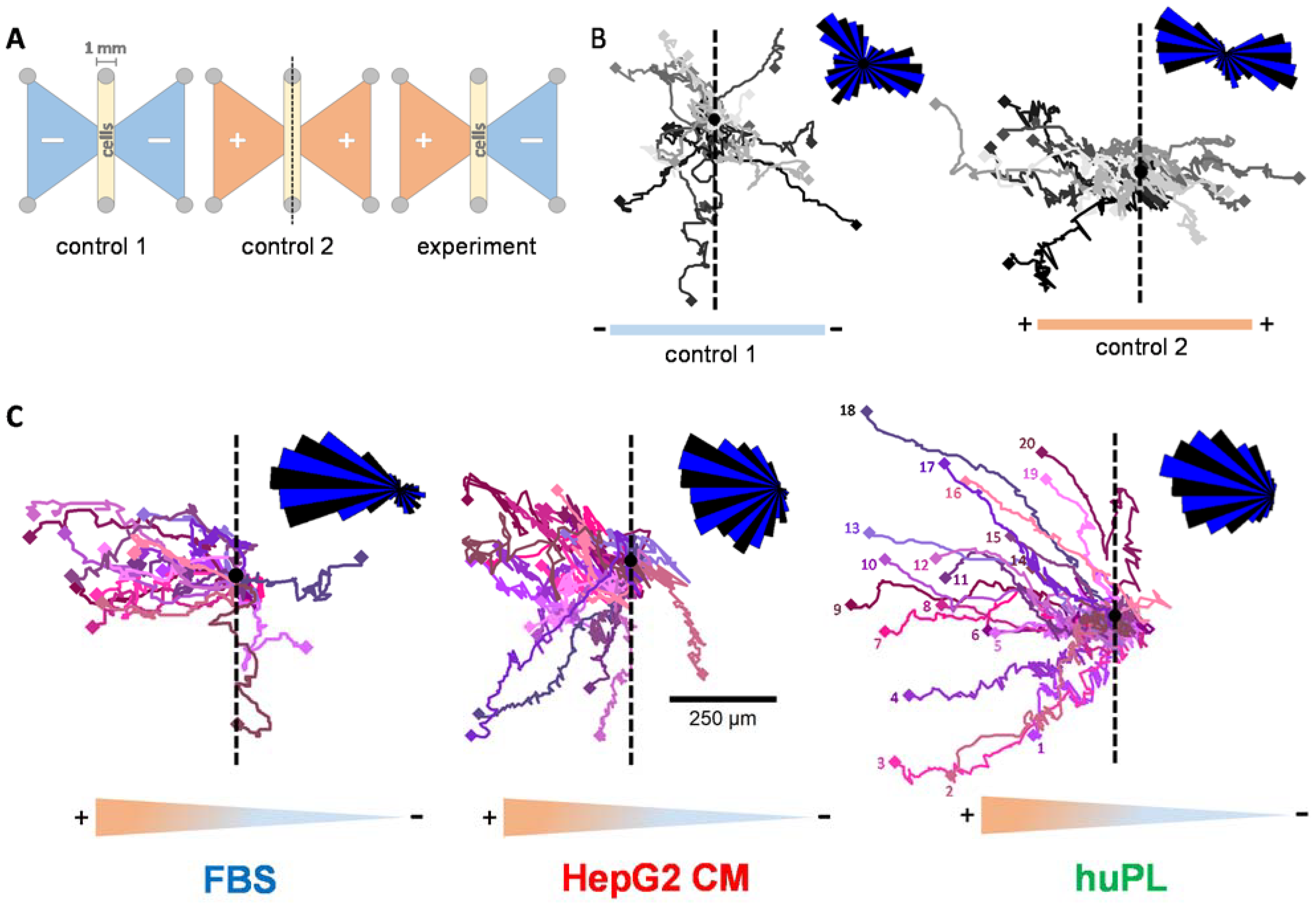
2.4. Human Platelet Lysate as Animal-Free CTA Alternative
2.5. Time-Lapse Analysis of the Chemotactic Activity of FBS, HepG2 CM and huPL
3. Materials and Methods
3.1. Neural Crest Cell Differentiation
3.2. Migration Assay (cMINC)
3.3. Neural Crest Membrane Translocation (NC-MT) Assay
3.4. Determination of Chemotactic Behaviour by Cell Tracking
3.5. Co-Culture and Conditioned Medium Preparation
3.6. Human Serum and Platelet Lysate Preparation
3.7. Acetone Precipitation
3.8. Protein Purification
3.9. Protein Separation and Detection
3.10. Mass Spectrometry (MS) for Protein Identification
3.11. Heat Inactivation and Pepsin Digestion
3.12. Stability Control Tests
3.13. Fractionation of Proteins According to Their Molecular Weight
3.14. Data Handling and Statistics
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cMINC | circular MINC |
| DMSO | dimethyl sulfoxide |
| DNT | developmental neurotoxicity |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| FBS | fetal bovine serum |
| FGF | fibroblast growth factor |
| FPLC | fast protein liquid chromatography |
| HTS | high-throughput screening |
| huPL | human platelet lysate |
| iPSC | induced pluripotent stem cell |
| MINC | migration inhibition of neural crest cell |
| MW | molecular weight |
| NAM | new approach method |
| NCC | neural crest cell |
| NC-MT | neural crest membrane translocation |
| NGRA | next generation risk assessment |
| PBS | phosphate buffered saline |
| PCB | polychlorinated biphenyl |
| PDGF | platelet derived growth factor |
| RA | retinoic acid |
| SDF-1 | stromal cell derived factor 1 |
| VEGF | vascular endothelium growth factor |
| VPA | valproic acid |
References
- Vega-Lopez, G.A.; Cerrizuela, S.; Tribulo, C.; Aybar, M.J. Neurocristopathies: New insights 150 years after the neural crest discovery. Dev. Biol. 2018, 444, S110–S143. [Google Scholar] [CrossRef] [PubMed]
- Serrano, F.; Bernard, W.G.; Granata, A.; Iyer, D.; Steventon, B.; Kim, M.; Vallier, L.; Gambardella, L.; Sinha, S. A novel human pluripotent stem cell-derived neural crest model of treacher collins syndrome shows defects in cell death and migration. Stem Cells Dev. 2019, 28, 81–100. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, G.; Lin, Z.; Wu, Y.; Zhang, J.; Liu, M.; Lee, K.K.H.; Chuai, M.; Yang, X. Alcohol exposure induces chick craniofacial bone defects by negatively affecting cranial neural crest development. Toxicol. Lett. 2017, 281, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menegola, E.; Broccia, M.L.; Di Renzo, F.; Massa, V.; Giavini, E. Craniofacial and axial skeletal defects induced by the fungicide triadimefon in the mouse. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2005, 74, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.L.; Bohnsack, B.L. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis 2019, 57, e23308. [Google Scholar] [CrossRef] [PubMed]
- Le Douarin, N.M. The avian embryo as a model to study the development of the neural crest: A long and still ongoing story. Mech. Dev. 2004, 121, 1089–1102. [Google Scholar] [CrossRef]
- Prasad, M.S.; Charney, R.M.; Garcia-Castro, M.I. Specification and formation of the neural crest: Perspectives on lineage segregation. Genesis 2019, 57, e23276. [Google Scholar] [CrossRef] [Green Version]
- Sieber-Blum, M. Cardiac neural crest stem cells. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2004, 276, 34–42. [Google Scholar] [CrossRef]
- Giovannone, D.; Ortega, B.; Reyes, M.; El-Ghali, N.; Rabadi, M.; Sao, S.; de Bellard, M.E. Chicken trunk neural crest migration visualized with hnk1. Acta Histochem. 2015, 117, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Miller, M.L.; McHenry, L.K.; Zheng, T.; Zhen, Q.; Ilkhanizadeh, S.; Conklin, B.R.; Bronner, M.E.; Weiss, W.A. Generating trunk neural crest from human pluripotent stem cells. Sci. Rep. 2016, 6, 19727. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.W.; Jacquemet, G. Cell matrix adhesion in cell migration. Essays Biochem. 2019, 63, 535–551. [Google Scholar]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Shellard, A.; Mayor, R. Chemotaxis during neural crest migration. Semin. Cell Dev. Biol. 2016, 55, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Kay, R.R.; Langridge, P.; Traynor, D.; Hoeller, O. Changing directions in the study of chemotaxis. Nat. Rev. Mol. Cell Biol. 2008, 9, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Wu, D. Signaling mechanisms for regulation of chemotaxis. Cell Res. 2005, 15, 52–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Capuana, L.; Boström, A.; Etienne-Manneville, S. Multicellular scale front-to-rear polarity in collective migration. Curr. Opin. Cell Biol. 2020, 62, 114–122. [Google Scholar] [CrossRef]
- Merchant, B.; Feng, J.J. A rho-gtpase based model explains group advantage in collective chemotaxis of neural crest cells. Phys. Biol. 2020, 17, 036002. [Google Scholar] [CrossRef]
- Mayor, R.; Etienne-Manneville, S. The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 2016, 17, 97–109. [Google Scholar] [CrossRef] [Green Version]
- McLennan, R.; Schumacher, L.J.; Morrison, J.A.; Teddy, J.M.; Ridenour, D.A.; Box, A.C.; Semerad, C.L.; Li, H.; McDowell, W.; Kay, D.; et al. Vegf signals induce trailblazer cell identity that drives neural crest migration. Dev. Biol. 2015, 407, 12–25. [Google Scholar] [CrossRef] [PubMed]
- McLennan, R.; Teddy, J.M.; Kasemeier-Kulesa, J.C.; Romine, M.H.; Kulesa, P.M. Vascular endothelial growth factor (vegf) regulates cranial neural crest migration in vivo. Dev. Biol. 2010, 339, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Eberhart, J.K.; He, X.; Swartz, M.E.; Yan, Y.L.; Song, H.; Boling, T.C.; Kunerth, A.K.; Walker, M.B.; Kimmel, C.B.; Postlethwait, J.H. Microrna mirn140 modulates pdgf signaling during palatogenesis. Nat. Genet. 2008, 40, 290–298. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y.; Ito, K. Chemotactic migration of mesencephalic neural crest cells in the mouse. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2000, 217, 170–179. [Google Scholar] [CrossRef]
- Kasemeier-Kulesa, J.C.; McLennan, R.; Romine, M.H.; Kulesa, P.M.; Lefcort, F. Cxcr4 controls ventral migration of sympathetic precursor cells. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 13078–13088. [Google Scholar] [CrossRef] [PubMed]
- Bahm, I.; Barriga, E.H.; Frolov, A.; Theveneau, E.; Frankel, P.; Mayor, R. Pdgf controls contact inhibition of locomotion by regulating n-cadherin during neural crest migration. Development 2017, 144, 2456–2468. [Google Scholar] [PubMed] [Green Version]
- Theveneau, E.; Marchant, L.; Kuriyama, S.; Gull, M.; Moepps, B.; Parsons, M.; Mayor, R. Collective chemotaxis requires contact-dependent cell polarity. Dev. Cell 2010, 19, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackland, J.O.S.; Frith, T.J.R.; Thompson, O.; Marin Navarro, A.; Garcia-Castro, M.I.; Unger, C.; Andrews, P.W. Top-down inhibition of bmp signaling enables robust induction of hpscs into neural crest in fully defined, xeno-free conditions. Stem Cell Rep. 2017, 9, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Hackland, J.O.S.; Frith, T.J.R.; Andrews, P.W. Fully defined and xeno-free induction of hpscs into neural crest using top-down inhibition of bmp signaling. Methods Mol. Biol. 2019, 1976, 49–54. [Google Scholar]
- Tchieu, J.; Zimmer, B.; Fattahi, F.; Amin, S.; Zeltner, N.; Chen, S.; Studer, L. A modular platform for differentiation of human pscs into all major ectodermal lineages. Cell Stem Cell 2017, 21, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, B.; Lee, G.; Balmer, N.V.; Meganathan, K.; Sachinidis, A.; Studer, L.; Leist, M. Evaluation of developmental toxicants and signaling pathways in a functional test based on the migration of human neural crest cells. Environ. Health Perspect. 2012, 120, 1116–1122. [Google Scholar] [CrossRef]
- Chambers, S.M.; Mica, Y.; Lee, G.; Studer, L.; Tomishima, M.J. Dual-smad inhibition/wnt activation-based methods to induce neural crest and derivatives from human pluripotent stem cells. Methods Mol. Biol. 2016, 1307, 329–343. [Google Scholar]
- Srinivasan, A.; Toh, Y.C. Human pluripotent stem cell-derived neural crest cells for tissue regeneration and disease modeling. Front. Mol. Neurosci. 2019, 12, 39. [Google Scholar] [CrossRef] [Green Version]
- Workman, M.J.; Mahe, M.M.; Trisno, S.; Poling, H.M.; Watson, C.L.; Sundaram, N.; Chang, C.F.; Schiesser, J.; Aubert, P.; Stanley, E.G.; et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat. Med. 2017, 23, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Papapetrou, E.P.; Kim, H.; Chambers, S.M.; Tomishima, M.J.; Fasano, C.A.; Ganat, Y.M.; Menon, J.; Shimizu, F.; Viale, A.; et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific ipscs. Nature 2009, 461, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Zeltner, N.; Fattahi, F.; Dubois, N.C.; Saurat, N.; Lafaille, F.; Shang, L.; Zimmer, B.; Tchieu, J.; Soliman, M.A.; Lee, G.; et al. Capturing the biology of disease severity in a psc-based model of familial dysautonomia. Nat. Med. 2016, 22, 1421–1427. [Google Scholar] [CrossRef] [Green Version]
- Nyffeler, J.; Dolde, X.; Krebs, A.; Pinto-Gil, K.; Pastor, M.; Behl, M.; Waldmann, T.; Leist, M. Combination of multiple neural crest migration assays to identify environmental toxicants from a proof-of-concept chemical library. Arch. Toxicol. 2017, 91, 3613–3632. [Google Scholar] [CrossRef] [Green Version]
- Hartung, T. Toxicology for the twenty-first century. Nature 2009, 460, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Hartung, T.; Leist, M. Food for thought... On the evolution of toxicology and the phasing out of animal testing. Altex 2008, 25, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, F.S.; Gray, G.M.; Bucher, J.R. Toxicology. Transforming environmental health protection. Science 2008, 319, 906–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crofton, K.M.; Mundy, W.R.; Shafer, T.J. Developmental neurotoxicity testing: A path forward. Congenit. Anom. 2012, 52, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Ceccatelli, S.; Daneshian, M.; Fritsche, E.; Hasiwa, N.; Hartung, T.; Hogberg, H.T.; Leist, M.; Li, A.; Mundi, W.R.; et al. Reference compounds for alternative test methods to indicate developmental neurotoxicity (dnt) potential of chemicals: Example lists and criteria for their selection and use. Altex 2017, 34, 49–74. [Google Scholar] [CrossRef] [Green Version]
- Bal-Price, A.; Hogberg, H.T.; Crofton, K.M.; Daneshian, M.; FitzGerald, R.E.; Fritsche, E.; Heinonen, T.; Hougaard Bennekou, S.; Klima, S.; Piersma, A.H.; et al. Recommendation on test readiness criteria for new approach methods in toxicology: Exemplified for developmental neurotoxicity. Altex 2018, 35, 306–352. [Google Scholar] [CrossRef]
- Krebs, A.; van Vugt-Lussenburg, B.M.A.; Waldmann, T.; Albrecht, W.; Boei, J.; Ter Braak, B.; Brajnik, M.; Braunbeck, T.; Brecklinghaus, T.; Busquet, F.; et al. The eu-toxrisk method documentation, data processing and chemical testing pipeline for the regulatory use of new approach methods. Arch. Toxicol. 2020, 94, 2435–2461. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, L.; Hogberg, H.T.; Leist, M.; Hartung, T. Developmental neurotoxicity—challenges in the 21st century and in vitro opportunities. Altex 2014, 31, 129–156. [Google Scholar]
- Zimmer, B.; Pallocca, G.; Dreser, N.; Foerster, S.; Waldmann, T.; Westerhout, J.; Julien, S.; Krause, K.H.; van Thriel, C.; Hengstler, J.G.; et al. Profiling of drugs and environmental chemicals for functional impairment of neural crest migration in a novel stem cell-based test battery. Arch. Toxicol. 2014, 88, 1109–1126. [Google Scholar] [CrossRef] [Green Version]
- Baltazar, M.T.; Cable, S.; Carmichael, P.L.; Cubberley, R.; Cull, T.; Delagrange, M.; Dent, M.P.; Hatherell, S.; Houghton, J.; Kukic, P.; et al. A next-generation risk assessment case study for coumarin in cosmetic products. Toxicol. Sci. Off. J. Soc. Toxicol. 2020, 176, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Benfenati, E.; Busquet, F.; Castell, J.; Clevert, D.A.; de Kok, T.; Dirven, H.; Fritsche, E.; Geris, L.; Gozalbes, R.; et al. Safer chemicals using less animals: Kick-off of the european ontox project. Toxicology 2021, 458, 152846. [Google Scholar] [CrossRef] [PubMed]
- Moné, M.J.; Pallocca, G.; Escher, S.E.; Exner, T.; Herzler, M.; Bennekou, S.H.; Kamp, H.; Kroese, E.D.; Leist, M.; Steger-Hartmann, T.; et al. Setting the stage for next-generation risk assessment with non-animal approaches: The eu-toxrisk project experience. Arch. Toxicol. 2020, 94, 3581–3592. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Chambers, S.M.; Tomishima, M.J.; Studer, L. Derivation of neural crest cells from human pluripotent stem cells. Nat. Protoc. 2010, 5, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Nyffeler, J.; Karreman, C.; Leisner, H.; Kim, Y.J.; Lee, G.; Waldmann, T.; Leist, M. Design of a high-throughput human neural crest cell migration assay to indicate potential developmental toxicants. Altex 2017, 34, 75–94. [Google Scholar] [CrossRef] [Green Version]
- Dreser, N.; Zimmer, B.; Dietz, C.; Sugis, E.; Pallocca, G.; Nyffeler, J.; Meisig, J.; Bluthgen, N.; Berthold, M.R.; Waldmann, T.; et al. Grouping of histone deacetylase inhibitors and other toxicants disturbing neural crest migration by transcriptional profiling. Neurotoxicology 2015, 50, 56–70. [Google Scholar] [CrossRef] [Green Version]
- Nyffeler, J.; Chovancova, P.; Dolde, X.; Holzer, A.K.; Purvanov, V.; Kindinger, I.; Kerins, A.; Higton, D.; Silvester, S.; van Vugt-Lussenburg, B.M.A.; et al. A structure-activity relationship linking non-planar pcbs to functional deficits of neural crest cells: New roles for connexins. Arch. Toxicol. 2018, 92, 1225–1247. [Google Scholar] [CrossRef] [Green Version]
- Boyden, S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 1962, 115, 453–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastyugin, V.; McWhinnie, E.; Labow, M.; Buxton, F. A quantitative high-throughput endothelial cell migration assay. J. Biomol. Screen. 2004, 9, 712–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallocca, G.; Nyffeler, J.; Dolde, X.; Grinberg, M.; Gstraunthaler, G.; Waldmann, T.; Rahnenfuhrer, J.; Sachinidis, A.; Leist, M. Impairment of human neural crest cell migration by prolonged exposure to interferon-beta. Arch. Toxicol. 2017, 91, 3385–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, J.; Pahl, M.; Bartmann, K.; Bendt, F.; Blum, J.; Dolde, X.; Förster, N.; Holzer, A.K.; Hübenthal, U.; Keßel, H.E.; et al. Neurodevelopmental toxicity assessment of flame retardants using a human dnt in vitro testing battery. Cell Biol. Toxicol. 2021, 1–27. [Google Scholar]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. MCP 2002, 1, 845–867. [Google Scholar] [CrossRef] [Green Version]
- Morinaga, T.; Sakai, M.; Wegmann, T.G.; Tamaoki, T. Primary structures of human alpha-fetoprotein and its mrna. Proc. Natl. Acad. Sci. USA 1983, 80, 4604–4608. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.A.; Zhu, S.; Pagano-Mirarchi, A.; Kellom, T.A.; Jacobson, H.I. Alpha-fetoprotein derived from a human hepatoma prevents growth of estrogen-dependent human breast cancer xenografts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1998, 4, 2877–2884. [Google Scholar]
- Chou, M.L.; Burnouf, T. Current methods to manufacture human platelet lysates for cell therapy and tissue engineering: Possible trends in product safety and standardization. ISBT Sci. Ser. 2017, 12, 168–175. [Google Scholar] [CrossRef]
- Horn, P.; Bokermann, G.; Cholewa, D.; Bork, S.; Walenda, T.; Koch, C.; Drescher, W.; Hutschenreuther, G.; Zenke, M.; Ho, A.D.; et al. Impact of individual platelet lysates on isolation and growth of human mesenchymal stromal cells. Cytotherapy 2010, 12, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Baud, A.; Wessely, F.; Mazzacuva, F.; McCormick, J.; Camuzeaux, S.; Heywood, W.E.; Little, D.; Vowles, J.; Tuefferd, M.; Mosaku, O.; et al. Multiplex high-throughput targeted proteomic assay to identify induced pluripotent stem cells. Anal. Chem. 2017, 89, 2440–2448. [Google Scholar] [CrossRef] [Green Version]
- Mica, Y.; Lee, G.; Chambers, S.M.; Tomishima, M.J.; Studer, L. Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hescs and patient-specific ipscs. Cell Rep. 2013, 3, 1140–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiegler, N.V.; Krug, A.K.; Matt, F.; Leist, M. Assessment of chemical-induced impairment of human neurite outgrowth by multiparametric live cell imaging in high-density cultures. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 121, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.K.; Balmer, N.V.; Matt, F.; Schonenberger, F.; Merhof, D.; Leist, M. Evaluation of a human neurite growth assay as specific screen for developmental neurotoxicants. Arch. Toxicol. 2013, 87, 2215–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Franko, A.; Hartwig, S.; Kotzka, J.; Ruoss, M.; Nussler, A.K.; Konigsrainer, A.; Haring, H.U.; Lehr, S.; Peter, A. Identification of the secreted proteins originated from primary human hepatocytes and hepg2 cells. Nutrients 2019, 11, 1795. [Google Scholar] [CrossRef] [Green Version]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. CB 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Mahoney, J.; Le Trang, N.; Pekala, P.; Cerami, A. Purification of cachectin, a lipoprotein lipase-suppressing hormone secreted by endotoxin-induced raw 264.7 cells. J. Exp. Med. 1985, 161, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Lewis, K.A.; Perrin, M.H.; Vale, W.W. Expression cloning of a human corticotropin-releasing-factor receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 8967–8971. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D.; Lodish, H.F.; Wong, G.G. Expression cloning of the murine erythropoietin receptor. Cell 1989, 57, 277–285. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolde, X.; Karreman, C.; Wiechers, M.; Schildknecht, S.; Leist, M. Profiling of Human Neural Crest Chemoattractant Activity as a Replacement of Fetal Bovine Serum for In Vitro Chemotaxis Assays. Int. J. Mol. Sci. 2021, 22, 10079. https://doi.org/10.3390/ijms221810079
Dolde X, Karreman C, Wiechers M, Schildknecht S, Leist M. Profiling of Human Neural Crest Chemoattractant Activity as a Replacement of Fetal Bovine Serum for In Vitro Chemotaxis Assays. International Journal of Molecular Sciences. 2021; 22(18):10079. https://doi.org/10.3390/ijms221810079
Chicago/Turabian StyleDolde, Xenia, Christiaan Karreman, Marianne Wiechers, Stefan Schildknecht, and Marcel Leist. 2021. "Profiling of Human Neural Crest Chemoattractant Activity as a Replacement of Fetal Bovine Serum for In Vitro Chemotaxis Assays" International Journal of Molecular Sciences 22, no. 18: 10079. https://doi.org/10.3390/ijms221810079