
Exploiting B Cell Transfer for Cancer Therapy: Engineered B Cells to Eradicate Tumors



Abstract
1. Introduction
2. Adoptive Cell Therapies with B Cells Loaded with Tumor Antigens
2.1. B Cells Loaded with Tumor Antigen Peptides
2.2. B Cells Modification with RNA Encoding Tumor Antigen
2.3. B Cells Engineered with DNA Encoding Tumor Antigens
2.4. B Cells Transduced with Viral Vectors Encoding Tumor Antigens
3. Adoptive Cell Therapies of B Cells Harboring a BCR Specific for a Predetermined Tumor Antigen
3.1. B Cells with an Endogenous BCR Specific of Tumor Antigens
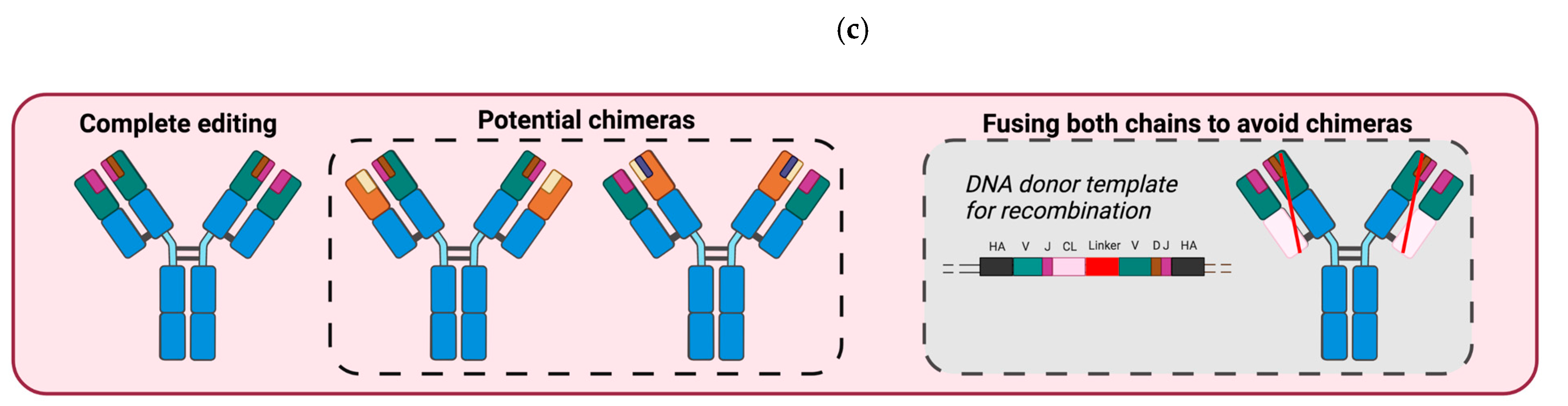
3.2. B Cells Engineered to Express a BCR Specific of Tumor Antigens
4. Adoptive Cell Therapies of B Cells with Enhanced IMMUNO-Regulatory Properties
4.1. Modulation of CoStimulatory Immune Cell Ligands
4.2. Modulation of Immuno-Regulatory Molecules Secretion or Recognition
5. Synthetic Circuit to Control Cellular Responses
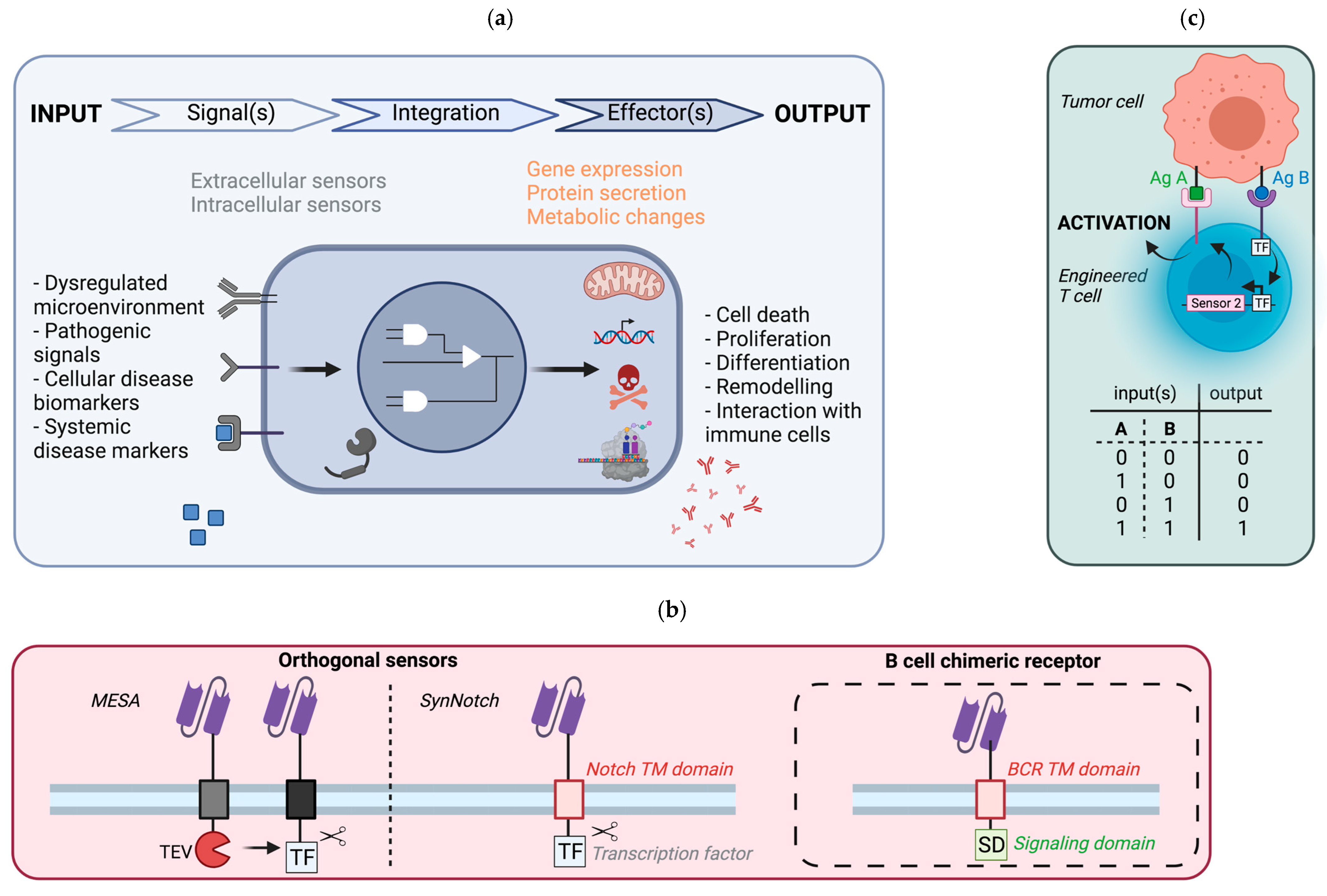
5.1. General Principle
5.2. Potential Implementation in B Cells
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Transgenic Mouse Strains with BCR Specific to a Given Antigen
Appendix B. Advantages of Immune Cells for Synthetic Biology
References
- Kondo, E.; Gryschok, L.; Klein-Gonzalez, N.; Rademacher, S.; Weihrauch, M.R.; Liebig, T.; Shimabukuro-Vornhagen, A.; Kochanek, M.; Draube, A.; Von Bergwelt-Baildon, M.S. CD40-activated B cells can be generated in high number and purity in cancer patients: Analysis of immunogenicity and homing potential. Clin. Exp. Immunol. 2009, 155, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Tittlbach, H.; Schneider, A.; Strobel, J.; Zimmermann, R.; Maas, S.; Gebhardt, B.; Rauser, G.; Mach, M.; Mackensen, A.; Winkler, T.H.; et al. GMP-production of purified human B lymphocytes for the adoptive transfer in patients after allogeneic hematopoietic stem cell transplantation. J. Transl. Med. 2017, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.M.; Buchholz, C.J. Surface-engineered lentiviral vectors for selective gene transfer into subtypes of lymphocytes. Mol. Ther. Methods Clin. Dev. 2018, 12, 19–31. [Google Scholar] [CrossRef]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Negre, D.; Lavillette, D.; Cosset, F.-L.; Verhoeyen, E. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Frecha, C.; Costa, C.; Lévy, C.; Negre, D.; Russell, S.J.; Maisner, A.; Salles, G.; Peng, K.-W.; Cosset, F.-L.; Verhoeyen, E. Efficient and stable transduction of resting B lymphocytes and primary chronic lymphocyte leukemia cells using measles virus gp displaying lentiviral vectors. Blood 2009, 114, 3173–3180. [Google Scholar] [CrossRef]
- Sandrin, V.; Boson, B.; Salmon, P.; Gay, W.; Negre, D.; Le Grand, R.; Trono, D.; Cosset, F.-L. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood 2002, 100, 823–832. [Google Scholar] [CrossRef]
- Laoharawee, K.; Johnson, M.J.; Lahr, W.S.; Peterson, J.J.; Webber, B.R.; Moriarity, B.S. Genome engineering of primary human B cells using CRISPR/Cas9. J. Vis. Exp. 2020, e61855. [Google Scholar] [CrossRef]
- Kubuschok, B.; Schmits, R.; Hartmann, F.; Cochlovius, C.; Breit, R.; König, J.; Pistorius, G.; Schilling, M.; Renner, C.; Pfreundschuh, M. Use of spontaneous epstein-barr virus-lymphoblastoid cell lines genetically modified to express tumor antigen as cancer vaccines: Mutated p21rasOncogene in pancreatic carcinoma as a model. Hum. Gene Ther. 2002, 13, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Trefzer, U.; Weingart, G.; Chen, Y.; Herberth, G.; Adrian, K.; Winter, H.; Audring, H.; Guo, Y.; Sterry, W.; Walden, P. Hybrid cell vaccination for cancer immune therapy: First clinical trial with metastatic melanoma. Int. J. Cancer 2000, 85, 618–626. [Google Scholar] [CrossRef]
- Kugler, A.; Seseke, F.; Thelen, P.; Kallerhoff, M.; Müller, G.A.; Stuhler, G.; Ringert, R.H. Autologous and allogenic hybrid cell vaccine in patients with metastatic renal cell carcinoma. BJU Int. 1998, 82, 487–493. [Google Scholar] [CrossRef]
- Winkler, J.; Tittlbach, H.; Roesler, W.; Strobel, J.; Zimmermann, R.; Maas, S.; Spriewald, B.; Wolff, D.; Repp, R.; Kordelas, L.; et al. Adoptive transfer of purified donor-b-lymphocytes after allogeneic stem cell transplantation: Results from a phase I/IIa clinical trial. Blood 2016, 128, 502. [Google Scholar] [CrossRef]
- Wennhold, K.; Shimabukuro-Vornhagen, A.; Von Bergwelt-Baildon, M. B cell-based cancer immunotherapy. Transfus. Med. Hemotherapy 2019, 46, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Tao, H.; Hu, Y.; Chen, Q.; Chen, X.; Xia, L.; Zhou, L.; Wang, Y.; Bao, Y.; Huang, S.; et al. IL-2 augments the therapeutic efficacy of adoptively transferred B cells which directly kill tumor cells via the CXCR4/CXCL12 and perforin pathways. Oncotarget 2016, 7, 60461–60474. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lao, X.-M.; Pan, Q.; Ning, N.; Yet, J.; Xu, Y.; Li, S.; Chang, A.E. Adoptive transfer of tumor reactive B cells confers host T-cell immunity and tumor regression. Clin. Cancer Res. 2011, 17, 4987–4995. [Google Scholar] [CrossRef]
- Li, Q.; Teitz-Tennenbaum, S.; Donald, E.J.; Li, M.; Chang, A.E. In vivo sensitized and in vitro activated B cells mediate tumor regression in cancer adoptive immunotherapy. J. Immunol. 2009, 183, 3195–3203. [Google Scholar] [CrossRef]
- Lee-Chang, C.; Miska, J.; Hou, D.; Rashidi, A.; Zhang, P.; Burga, R.A.; Jusué-Torres, I.; Xiao, T.; Arrieta, V.A.; Zhang, D.Y.; et al. Activation of 4-1BBL+ B cells with CD40 agonism and IFNγ elicits potent immunity against glioblastoma. J. Exp. Med. 2020, 218, e20200913. [Google Scholar] [CrossRef]
- Guo, S.; Xu, J.; Denning, W.; Hel, Z. Induction of protective cytotoxic T-cell responses by a B-cell-based cellular vaccine requires stable expression of antigen. Gene Ther. 2009, 16, 1300–1313. [Google Scholar] [CrossRef] [PubMed]
- Wennhold, K.; Thelen, M.; Schlößer, H.A.; Haustein, N.; Reuter, S.; Garcia-Marquez, M.; Lechner, A.; Kobold, S.; Rataj, F.; Utermöhlen, O.; et al. Using antigen-specific B cells to combine antibody and T cell-based cancer immunotherapy. Cancer Immunol. Res. 2017, 5, 730–743. [Google Scholar] [CrossRef]
- Moutai, T.; Yamana, H.; Nojima, T.; Kitamura, D. A novel and effective cancer immunotherapy mouse model using antigen-specific B cells selected in vitro. PLoS ONE 2014, 9, e92732. [Google Scholar] [CrossRef]
- Ren, H.; Zhao, S.; Li, W.; Dong, H.; Zhou, M.; Cao, M.; Hu, H.-M.; Wang, L.-X. Therapeutic antitumor efficacy of B cells loaded with tumor-derived autophagasomes vaccine (DRibbles). J. Immunother. 2014, 37, 383–393. [Google Scholar] [CrossRef]
- Wennhold, K.; Weber, T.M.; Klein-Gonzalez, N.; Thelen, M.; Garcia-Marquez, M.; Chakupurakal, G.; Fiedler, A.; Schlösser, H.A.; Fischer, R.; Theurich, S.; et al. CD40-activated B cells induce anti-tumor immunity in vivo. Oncotarget 2016, 8, 27740–27753. [Google Scholar] [CrossRef]
- Park, M.-Y.; Kim, H.-S.; Woo, S.-J.; Kim, C.-H.; Park, J.-S.; Sohn, H.-J.; Kim, H.-J.; Oh, S.-T.; Kim, T.-G. Efficient antitumor immunity in a murine colorectal cancer model induced by CEA RNA-electroporated B cells. Eur. J. Immunol. 2008, 38, 2106–2117. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Kim, Y.-J.; Lee, J.-M.; Han, S.-H.; Ko, H.-J.; Park, H.-J.; Pereboev, A.; Nguyen, H.H.; Kang, C.-Y. CD40-Targeted recombinant adenovirus significantly enhances the efficacy of antitumor vaccines based on dendritic cells and B cells. Hum. Gene Ther. 2010, 21, 1697–1706. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Han, S.-H.; Kang, H.-W.; Lee, J.-M.; Kim, Y.-S.; Seo, J.-H.; Seong, Y.-K.; Ko, H.-J.; Choi, T.H.; Moon, C.; et al. NKT ligand-loaded, antigen-expressing B cells function as long-lasting antigen presenting cells in vivo. Cell. Immunol. 2011, 270, 135–144. [Google Scholar] [CrossRef]
- Jeon, I.; Lee, J.-M.; Shin, K.-S.; Kang, T.; Park, M.H.; Seo, H.; Song, B.; Koh, C.-H.; Choi, J.; Shin, Y.K.; et al. Enhanced immunogenicity of engineered HER2 antigens potentiates antitumor immune responses. Vaccines 2020, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xiang, D.; Sun, J.; Ding, C.; Liu, M.; Hu, X.; Li, G.; Kloecker, G.; Zhang, H.-G.; Yan, J. Targeting of antigens to B lymphocytes via CD19 as a means for tumor vaccine development. J. Immunol. 2013, 190, 5588–5599. [Google Scholar] [CrossRef]
- Evans, D.E.; Munks, M.W.; Purkerson, J.M.; Parker, D.C. Resting B lymphocytes as APC for naive T lymphocytes: Dependence on CD40 ligand/CD40. J. Immunol. 2000, 164, 688–697. [Google Scholar] [CrossRef]
- Tao, H.; Lu, L.; Xia, Y.; Dai, F.; Wang, Y.; Bao, Y.; Lundy, S.; Ito, F.; Pan, Q.; Zhang, X.; et al. Antitumor effector B cells directly kill tumor cells via the Fas/FasL pathway and are regulated by IL-10. Eur. J. Immunol. 2014, 45, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Von Bergwelt-Baildon, M.S.; Vonderheide, R.H.; Maecker, B.; Hirano, N.; Anderson, K.S.; Butler, M.O.; Xia, Z.; Zeng, W.Y.; Wucherpfennig, K.W.; Nadler, L.M.; et al. Human primary and memory cytotoxic T lymphocyte responses are efficiently induced by means of CD40-activated B cells as antigen-presenting cells: Potential for clinical application. Blood 2002, 99, 3319–3325. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, R.; Bellemare-Pelletier, A.; Housseau, F.; Thibodeau, J.; Hwu, P. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003, 63, 2836–2843. [Google Scholar] [PubMed]
- Schultze, J.L.; Michalak, S.; Seamon, M.J.; Dranoff, G.; Jung, K.; Daley, J.; Delgado, J.C.; Gribben, J.G.; Nadler, L.M. CD40-activated human B cells: An alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J. Clin. Investig. 1997, 100, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, C.M.; Vonderheide, R.H. Targeting adult and pediatric cancers via cell-based vaccines and the prospect of activated B lymphocytes as a novel modality. Cancer Biol. Ther. 2003, 2, 466–470. [Google Scholar] [CrossRef][Green Version]
- Castiglioni, P.; Gerloni, M.; Zanetti, M. Genetically programmed B lymphocytes are highly efficient in inducing anti-virus protective immunity mediated by central memory CD8 T cells. Vaccine 2004, 23, 699–708. [Google Scholar] [CrossRef]
- Gerloni, M.; Rizzi, M.; Castiglioni, P.; Zanetti, M. T cell immunity using transgenic B lymphocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 3892–3897. [Google Scholar] [CrossRef] [PubMed]
- Colluru, V.T.; McNeel, D.G. B lymphocytes as direct antigen-presenting cells for anti-tumor DNA vaccines. Oncotarget 2016, 7, 67901–67918. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-K.; Seo, H.-S.; Chae, M.-J.; Jeon, I.-S.; Song, B.-Y.; Park, Y.-J.; Ahn, H.M.; Yun, C.-O.; Kang, C.-Y. Enhanced antitumor immunotherapeutic effect of B-cell-based vaccine transduced with modified adenoviral vector containing type 35 fiber structures. Gene Ther. 2013, 21, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Lizée, G.; Gonzales, M.I.; Topalian, S.L. Lentivirus vector-mediated expression of tumor-associated epitopes by human antigen presenting cells. Hum. Gene Ther. 2004, 15, 393–404. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Sicard, A.; Koenig, A.; Graff-Dubois, S.; Dussurgey, S.; Rouers, A.; Dubois, V.; Blanc, P.; Chartoire, D.; Errazuriz-Cerda, E.; Paidassi, H.; et al. B Cells loaded with synthetic particulate antigens: A versatile platform to generate antigen-specific helper T cells for cell therapy. Nano Lett. 2015, 16, 297–308. [Google Scholar] [CrossRef]
- Luo, X.; Maarschalk, E.; O’Connell, R.M.; Wang, P.; Yang, L.; Baltimore, D. Engineering human hematopoietic stem/progenitor cells to produce a broadly neutralizing anti-HIV antibody after in vitro maturation to human B lymphocytes. Blood 2009, 113, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, A.-S.; Haworth, K.G.; Barber-Axthelm, I.M.; Ironside, C.; Giese, M.A.; Peterson, C.W.; Kiem, H.-P. Long-term persistence of anti-HIV broadly neutralizing antibody-secreting hematopoietic cells in humanized mice. Mol. Ther. 2018, 27, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Fusil, F.; Calattini, S.; Amirache, F.; Mancip, J.; Costa, C.; Robbins, J.B.; Douam, F.; Lavillette, D.; Law, M.; Defrance, T.; et al. A lentiviral vector allowing physiologically regulated membrane-anchored and secreted antibody expression depending on B-cell maturation status. Mol. Ther. 2015, 23, 1734–1747. [Google Scholar] [CrossRef]
- Johnson, M.J.; Laoharawee, K.; Lahr, W.S.; Webber, B.R.; Moriarity, B.S. Engineering of primary human B cells with CRISPR/Cas9 targeted nuclease. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Voss, J.; Gonzalez-Martin, A.; Andrabi, R.; Fuller, R.P.; Murrell, B.; McCoy, L.; Porter, K.; Huang, D.; Li, W.; Sok, D.; et al. Reprogramming the antigen specificity of B cells using genome-editing technologies. eLife 2019, 8, e42995. [Google Scholar] [CrossRef] [PubMed]
- Moffett, H.F.; Harms, C.K.; Fitzpatrick, K.S.; Tooley, M.R.; Boonyaratanakornkit, J.; Taylor, J.J. B cells engineered to express pathogen-specific antibodies protect against infection. Sci. Immunol. 2019, 4, eaax0644. [Google Scholar] [CrossRef]
- Greiner, V.; Puerto, R.B.; Liu, S.; Herbel, C.; Carmona, E.M.; Goldberg, M.S. CRISPR-mediated editing of the B Cell receptor in primary human B cells. iScience 2019, 12, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Nahmad, A.D.; Raviv, Y.; Horovitz-Fried, M.; Sofer, I.; Akriv, T.; Nataf, D.; Dotan, I.; Carmi, Y.; Burstein, D.; Wine, Y.; et al. Engineered B cells expressing an anti-HIV antibody enable memory retention, isotype switching and clonal expansion. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Cheong, T.-C.; Compagno, M.; Chiarle, R. Editing of mouse and human immunoglobulin genes by CRISPR-Cas9 system. Nat. Commun. 2016, 7, 10934. [Google Scholar] [CrossRef]
- Lin, Y.; Pecetta, S.; Steichen, J.M.; Kratochvil, S.; Melzi, E.; Arnold, J.; Dougan, S.K.; Wu, L.; Kirsch, K.H.; Nair, U.; et al. One-step CRISPR/Cas9 method for the rapid generation of human antibody heavy chain knock-in mice. EMBO J. 2018, 37, e99243. [Google Scholar] [CrossRef]
- Nahmad, A.D.; Lazzarotto, C.R.; Zelikson, N.; Kustin, T.; Tenuta, M.; Huang, D.; Reuveni, I.; Horovitz-Fried, M.; Dotan, I.; Rosin-Arbesfeld, R.; et al. In vivo engineered B cells retain memory and secrete high titers of anti-HIV antibodies in mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kato, K.; Cantwell, M.J.; Sharma, S.; Kipps, T.J. Gene transfer of CD40-ligand induces autologous immune recognition of chronic lymphocytic leukemia B cells. J. Clin. Investig. 1998, 101, 1133–1141. [Google Scholar] [CrossRef]
- Stripecke, R.; Cardoso, A.A.; Pepper, K.A.; Skelton, D.C.; Yu, X.J.; Mascarenhas, L.; Weinberg, K.I.; Nadler, L.M.; Kohn, D.B. Lentiviral vectors for efficient delivery of CD80 and granulocyte-macrophage colony-stimulating factor in human acute lymphoblastic leukemia and acute myeloid leukemia cells to induce antileukemic immune responses. Blood 2000, 96, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.-A.; Cho, H.; Shin, A.-R.; Sohn, H.-J.; Kim, T.-G.; Cho, H.-I. Co-expression of CD40L with CD70 or OX40L increases B-cell viability and antitumor efficacy. Oncotarget 2016, 7, 46173–46186. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.; Meitlis, I.; Hale, M.; Chen, C.-Y.; Singh, S.; Jackson, S.W.; Miao, C.H.; Khan, I.F.; Rawlings, D.J.; James, R.G. Engineering protein-secreting plasma cells by homology-directed repair in primary human B cells. Mol. Ther. 2018, 26, 456–467. [Google Scholar] [CrossRef]
- Luo, B.; Zhan, Y.; Luo, M.; Dong, H.; Liu, J.; Lin, Y.; Zhang, J.; Wang, G.; Verhoeyen, E.; Zhang, Y.; et al. Engineering of α-PD-1 antibody-expressing long-lived plasma cells by CRISPR/Cas9-mediated targeted gene integration. Cell Death Dis. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lesch, S.; Blumenberg, V.; Stoiber, S.; Gottschlich, A.; Ogonek, J.; Cadilha, B.L.; Dantes, Z.; Rataj, F.; Dorman, K.; Lutz, J.; et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 2021, 1–15. [Google Scholar] [CrossRef]
- Roybal, K.T.; Lim, W.A. Synthetic immunology: Hacking immune cells to expand their therapeutic capabilities. Annu. Rev. Immunol. 2017, 35, 229–253. [Google Scholar] [CrossRef]
- Chabannon, C.; Bouabdallah, R.; Fürst, S.; Granata, A.; Saillard, C.; Vey, N.; Mokart, D.; Fougereau, E.; Lemarie, C.; Mfarrej, B.; et al. CAR-T cells: Lymphocytes exprimant un récepteur chimérique à l’antigène. La Rev. Médecine Interne 2019, 40, 545–552. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T cells releasing IL-18 convert to T-Bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef]
- Chassin, H.; Geering, B.; Schukur, L.; Ausländer, D.; Lang, B.M.; Fussenegger, M. Sensing and responding to allergic response cytokines through a genetically encoded circuit. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Shao, J.; Xue, S.; Yu, G.; Yu, Y.; Yang, X.; Bai, Y.; Zhu, S.; Yang, L.; Yin, J.; Wang, Y.; et al. Smartphone-controlled optogenetically engineered cells enable semiautomatic glucose homeostasis in diabetic mice. Sci. Transl. Med. 2017, 9, eaal2298. [Google Scholar] [CrossRef] [PubMed]
- Schukur, L.; Geering, B.; Hamri, G.C.-E.; Fussenegger, M. Implantable synthetic cytokine converter cells with AND-gate logic treat experimental psoriasis. Sci. Transl. Med. 2015, 7, 318ra201. [Google Scholar] [CrossRef] [PubMed]
- Qudrat, A.; Al Mosabbir, A.; Truong, K. Engineered proteins program mammalian cells to target inflammatory disease sites. Cell Chem. Biol. 2017, 24, 703.e2–711.e2. [Google Scholar] [CrossRef] [PubMed]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; Daringer, N.M.; Dolberg, T.; Leonard, J.N. Rewiring human cellular input-output using modular extracellular sensors. Nat. Chem. Biol. 2016, 13, 202–209. [Google Scholar] [CrossRef]
- Daringer, N.M.; Dudek, R.M.; Schwarz, K.A.; Leonard, J.N. Modular extracellular sensor architecture for engineering mammalian cell-based devices. ACS Synth. Biol. 2014, 3, 892–902. [Google Scholar] [CrossRef]
- Pesch, T.; Bonati, L.; Kelton, W.; Parola, C.; Ehling, R.A.; Csepregi, L.; Kitamura, D.; Reddy, S.T. Molecular design, optimization, and genomic integration of chimeric B cell receptors in murine B cells. Front. Immunol. 2019, 10, 2630. [Google Scholar] [CrossRef]
- Wu, Y.; Chang, T.; Long, Y.; Huang, H.; Kandeel, F.; Yee, J.-K. Using gene editing to establish a safeguard system for pluripotent stem-cell-based therapies. iScience 2019, 22, 409–422. [Google Scholar] [CrossRef]
- Bonfá, G.; Blazquez-Roman, J.; Tarnai, R.; Siciliano, V. Precision tools in immuno-oncology: Synthetic gene circuits for cancer immunotherapy. Vaccines 2020, 8, 732. [Google Scholar] [CrossRef]
- Muldoon, J.J.; Kandula, V.; Hong, M.; Donahue, P.S.; Boucher, J.D.; Bagheri, N.; Leonard, J.N. Model-guided design of mammalian genetic programs. Sci. Adv. 2021, 7, eabe9375. [Google Scholar] [CrossRef] [PubMed]
- Smole, A.; Lainšček, D.; Bezeljak, U.; Horvat, S.; Jerala, R. A synthetic mammalian therapeutic gene circuit for sensing and suppressing inflammation. Mol. Ther. 2017, 25, 102–119. [Google Scholar] [CrossRef]
- Weisel, F.J.; Zuccarino-Catania, G.V.; Chikina, M.; Shlomchik, M.J. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity 2016, 44, 116–130. [Google Scholar] [CrossRef]
- Pape, K.A.; Taylor, J.J.; Maul, R.; Gearhart, P.J.; Jenkins, M. Different B cell populations mediate early and late memory during an endogenous immune response. Science 2011, 331, 1203–1207. [Google Scholar] [CrossRef]
- Zhao, K.-L.; Yang, X.-J.; Jin, H.-Z.; Zhao, L.; Hu, J.-L.; Qin, W.-J. Double-edge role of B cells in tumor immunity: Potential molecular mechanism. Curr. Med. Sci. 2019, 39, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2016, 23, 2255–2266. [Google Scholar] [CrossRef]
- Brendel, C.; Rio, P.; Verhoeyen, E. Humanized mice are precious tools for evaluation of hematopoietic gene therapies and preclinical modeling to move towards a clinical trial. Biochem. Pharmacol. 2019, 174, 113711. [Google Scholar] [CrossRef]
- Pesch, T. Molecular and Cellular Engineering of B Cells for Immunotherapy Applications; ETH Zurich: Zurich, Switzerland, 2019; p. 138. [Google Scholar]
- Goodnow, C.; Crosbie, J.; Adelstein, S.; Lavoie, T.B.; Smith-Gill, S.J.; Brink, R.A.; Pritchard-Briscoe, H.; Wotherspoon, J.S.; Loblay, R.H.; Raphael, K.; et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 1988, 334, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Cascalho, M.; Ma, A.; Lee, S.; Masat, L.; Wabl, M. A quasi-monoclonal mouse. Science 1996, 272, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Dougan, S.K.; Ogata, S.; Hu, C.-C.A.; Grotenbreg, G.M.; Guillen, E.; Jaenisch, R.; Ploegh, H.L. IgG1+ ovalbumin-specific B-cell transnuclear mice show class switch recombination in rare allelically included B cells. Proc. Natl. Acad. Sci. USA 2012, 109, 13739–13744. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin of B Cells | Antigen | Number of B Cells (Total) | Route of Infusion | Treatment/Prior Activation | Cancer Model | Results | Ref. |
|---|---|---|---|---|---|---|---|
| TDLN | Endogenous | 1 × 106 | iv | IL-2 (infused in vivo) | Metastasis of 4T1 mammary tumor | Anti-4T1- antibodies CXCR4 expression by B cells Reduction of pulmonary metastasis (combined with IL2) | [13] |
| TDLN | Endogenous | 3 × 106 | iv | LPS Anti CD40 | Metastasis of 4T1 mammary tumor | Anti-4T1- antibodies Generation of T cell responses Reduction of pulmonary metastasis | [14] |
| TDLN | Endogenous | 1 × 106 to 3 × 106 | iv | LPS Anti CD40 | 3-methylcholanthrene-induced fibrosarcoma | Antitumor antigen antibodies Reduction of pulmonary metastasis and tumor size | [15] |
| Spleens and dCLNs (tumor-bearing mice) | Endogenous | 1.5 × 106 | iv | CD40 agonist IFNγ BAFF | Glioblastoma | Migration at tumor site and in the SLOs Generation of CD8+ T cell responses 80% of tumor eradication (combined with anti PD-L1, radiotherapy) Memory response | [16] |
| Spleen | Endogenous (OVA transgenic mice) or peptide loading (pulsed) | 1 × 105 | iv | CpG Anti-CD40 | Thymoma-derived EG-7 cells expressing OVA | Generation of CTL cell responses Protection against tumor growth | [17] |
| Spleen | Endogenous (frequency of OVA specific cells increased by immunization) | 0.1 × 106 to 2 × 106 | iv | Feeder cell line expressing CD40L IL4 (+ IL21; CD40L;OVA tetramers to generate plasma cells) | Panc02OVA tumor cells expressing OVA | Migration at tumor site and in the SLOs Anti-OVA antibodies Decreased of tumor growth | [18] |
| Spleen | Endogenous (frequency of antigen specific cells increase by in vitro culture) | 2 × 107 | iv | IL-4 IL-21 Feeder cell line expressing CD40L, BAFF, tumor Ag and FasL) | Melanoma metastasis | Anti-HEL antibodies Decreased of tumor growth Increased survival | [19] |
| Spleen | Tumor peptide loading | 5 × 106 | iv | CpG Anti-CD40 | Thymoma-derived EG-7 cells expressing OVA | Generation of T cell responses Regression of established tumors Increased survival | [20] |
| Spleen | Tumor peptide loading | 1 × 106 to 1 × 107 | iv ip sc | Feeder cell line expressing CD40L | Thymoma-derived EG-7 cells expressing OVA Melanoma B16F10 OVA tumor cells | Generation of T cell responses Decreased of tumor growth | [21] |
| Spleen | Electroporation of RNA encoding tumor Ag | 1 × 106 | iv sc | LPS | Colorectal cancer | Antitumor antigen antibodies Generation of CD4+ T cell responses Regression of established tumors Increased survival | [22] |
| Spleen | Viral delivered (Adenovirus) | 2 × 106 | iv | CFm40L harbored on adenovirus | E6/E7-expressing TC-1 cell line human Her-2/neu-expressing CT26 cell line murine Her-2/ neu-expressing CT26 cell line | Generation of CTL cell responses Decreased of tumor growth Increased survival | [23] |
| Spleen | Viral delivered (adenovirus) | 2 × 106 | iv | α-galactosylceramide | Her2/neu-expressing transfectoma cell line CT26-hHer2 Her2/neu-expressing SK-Br-3 human breast carcinoma | Migration in the SLOs Antitumor antigen antibodies Generation of CD8+ T cell responses | [24] |
| Spleen | Viral delivery (adenovirus) | 2 × 106 | iv | α-galactosylceramide | Her2/neu-expressing transfectoma cell line CT26-hHer2 or CT26-hp95Her2 | Antitumor antigen antibodies Generation of CTL cell responses Decreased of tumor growth Increased survival | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Page, A.; Hubert, J.; Fusil, F.; Cosset, F.-L. Exploiting B Cell Transfer for Cancer Therapy: Engineered B Cells to Eradicate Tumors. Int. J. Mol. Sci. 2021, 22, 9991. https://doi.org/10.3390/ijms22189991
Page A, Hubert J, Fusil F, Cosset F-L. Exploiting B Cell Transfer for Cancer Therapy: Engineered B Cells to Eradicate Tumors. International Journal of Molecular Sciences. 2021; 22(18):9991. https://doi.org/10.3390/ijms22189991
Chicago/Turabian StylePage, Audrey, Julie Hubert, Floriane Fusil, and François-Loïc Cosset. 2021. "Exploiting B Cell Transfer for Cancer Therapy: Engineered B Cells to Eradicate Tumors" International Journal of Molecular Sciences 22, no. 18: 9991. https://doi.org/10.3390/ijms22189991
APA StylePage, A., Hubert, J., Fusil, F., & Cosset, F.-L. (2021). Exploiting B Cell Transfer for Cancer Therapy: Engineered B Cells to Eradicate Tumors. International Journal of Molecular Sciences, 22(18), 9991. https://doi.org/10.3390/ijms22189991