
MS-275 (Entinostat) Promotes Radio-Sensitivity in PAX3-FOXO1 Rhabdomyosarcoma Cells

 ,
,  ,
,  , , , , ,
, , , , ,  ,
,  , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
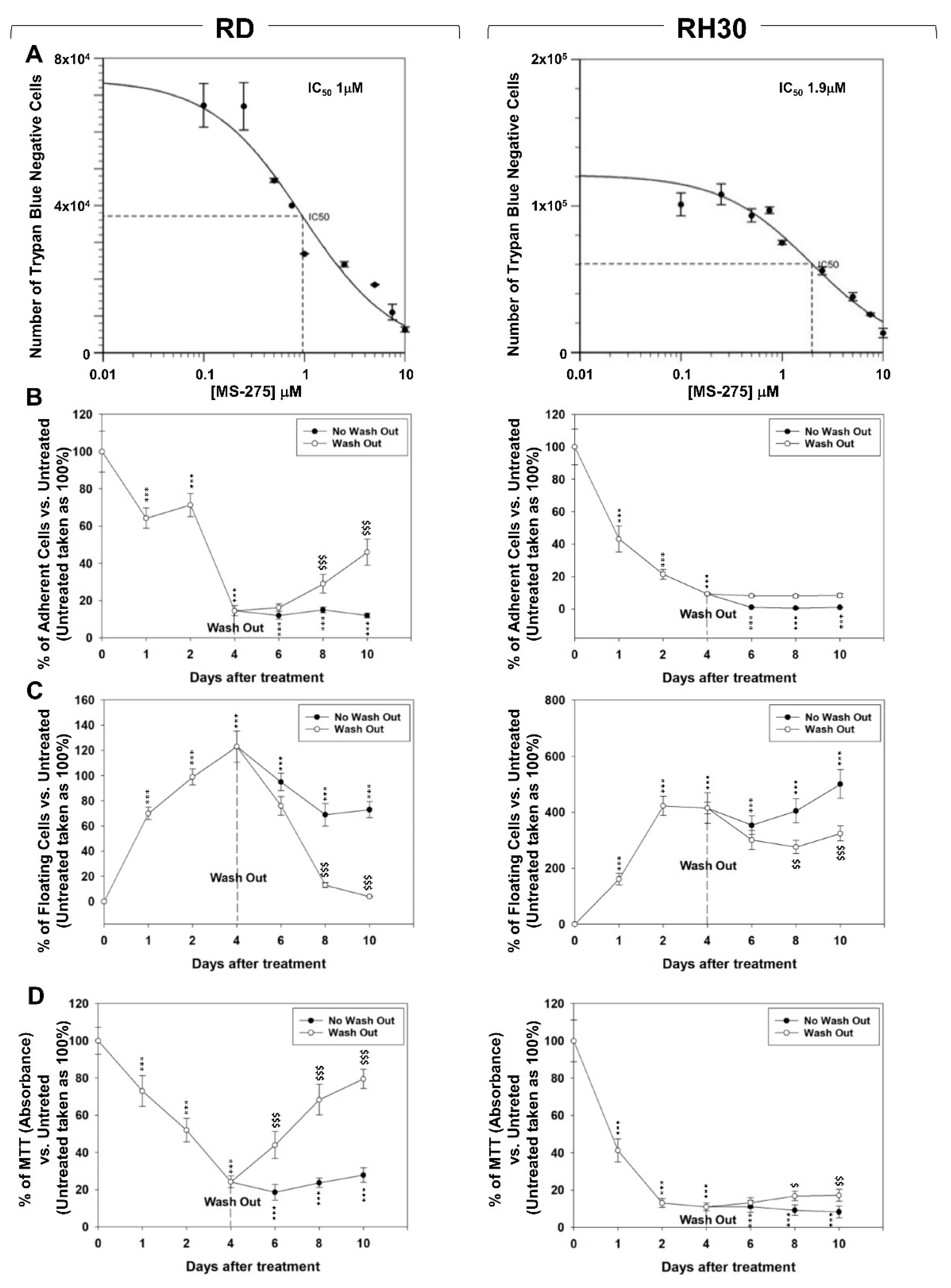
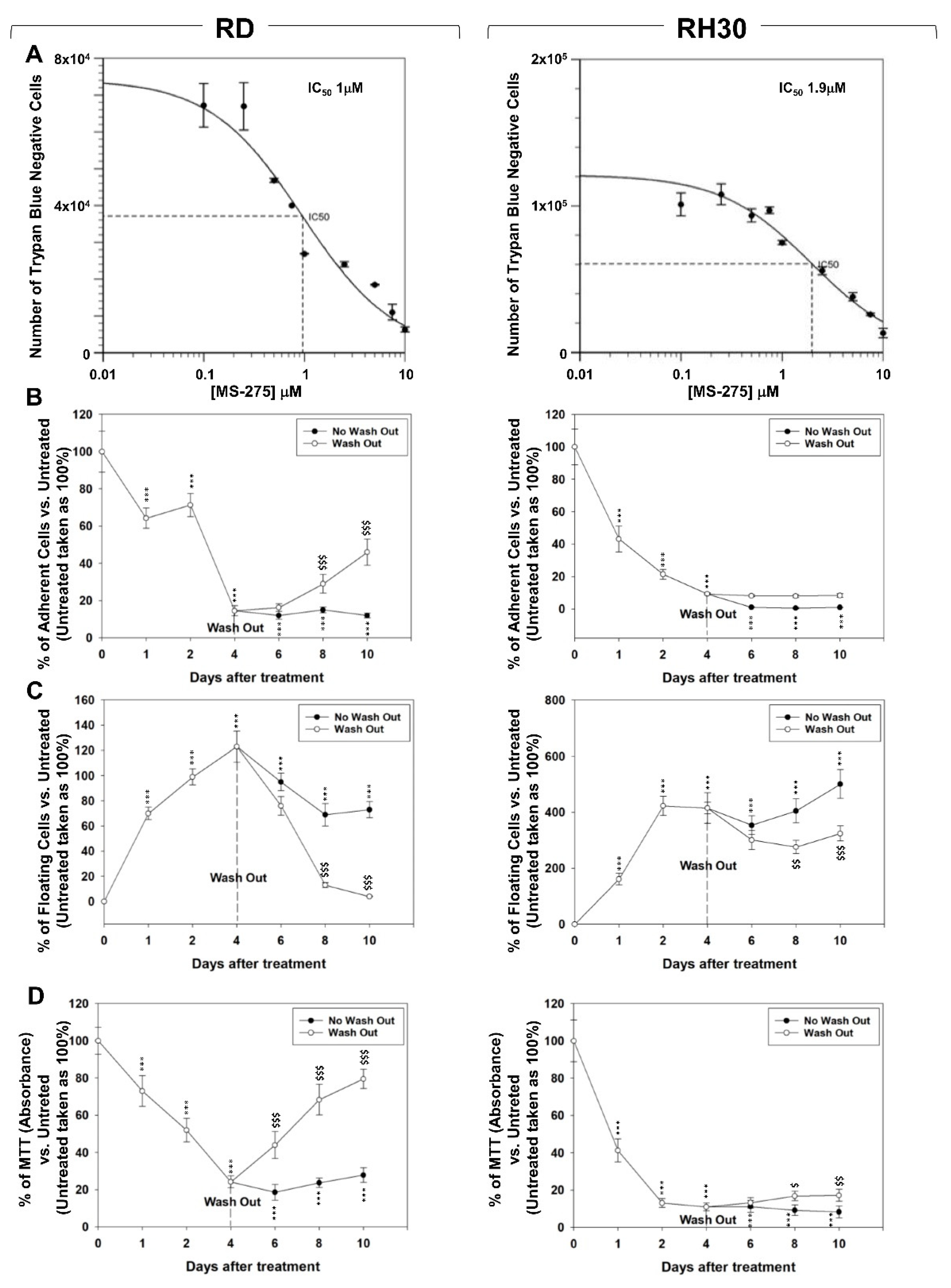
2.1. In Vitro, MS-275 Induces Growth Arrest and Cell Death of Human FN-RMS and FP-RMS
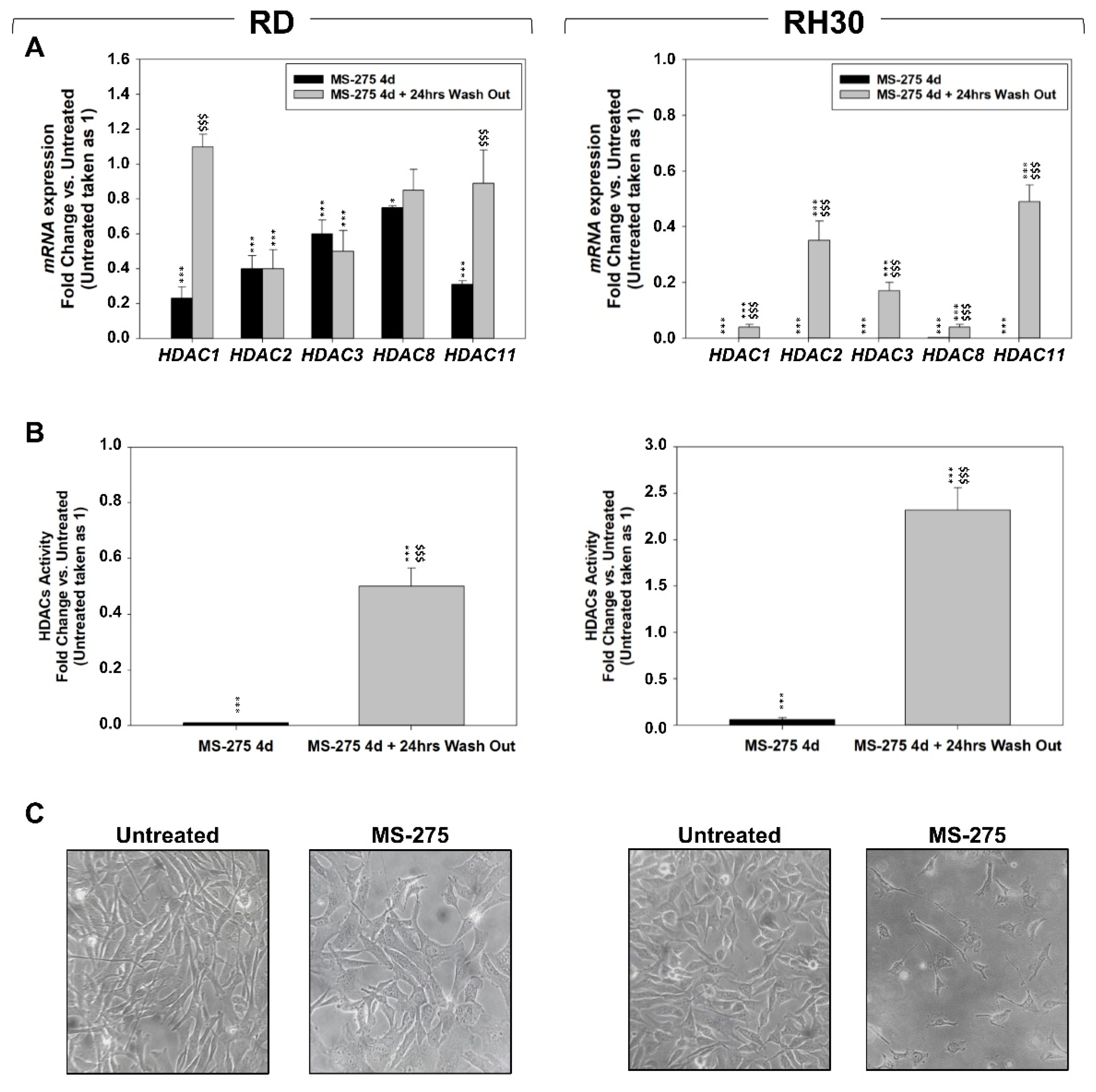
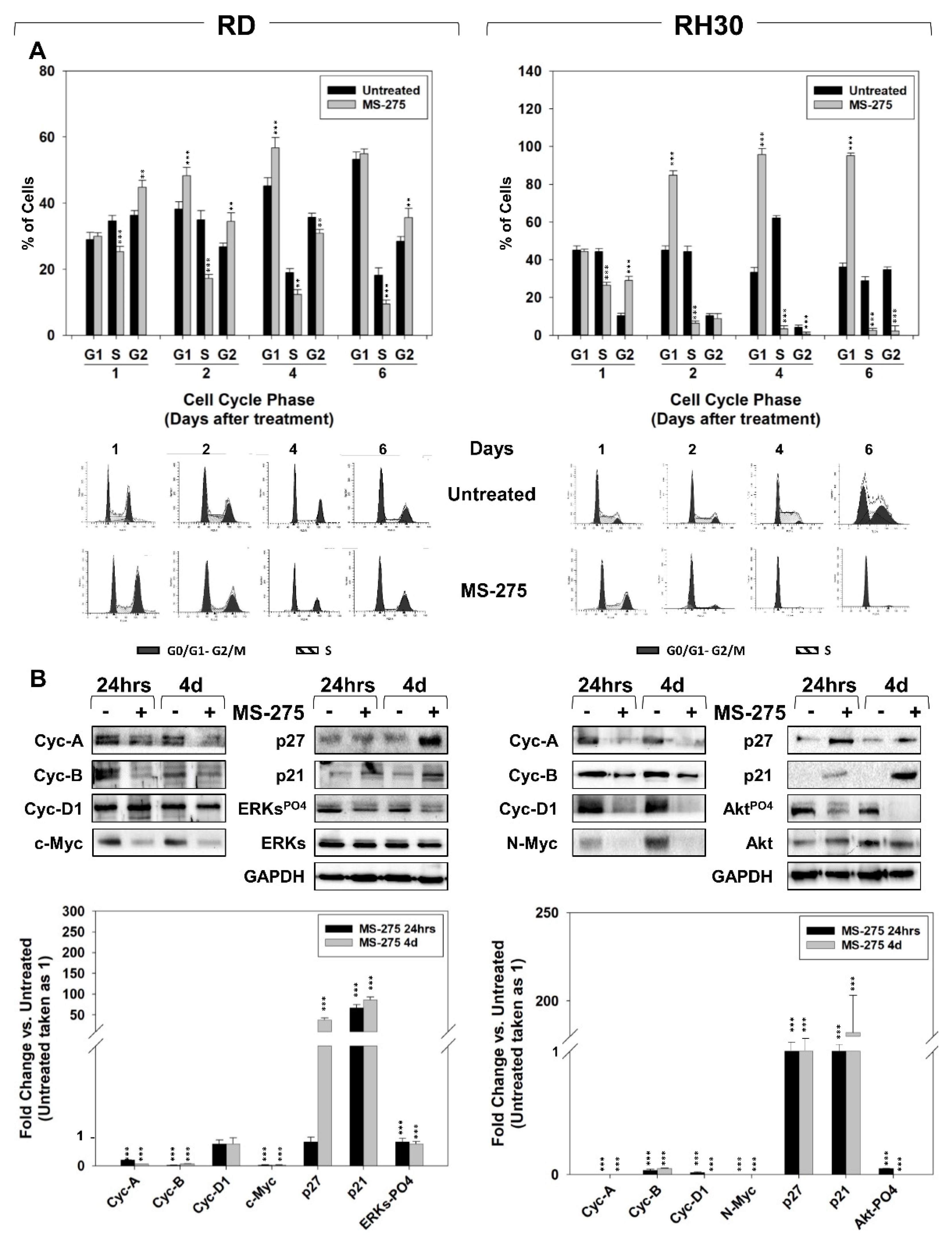
2.2. MS-275 Affected Cell Cycle Distribution and Related Molecular Signature in RD and RH30 Cells
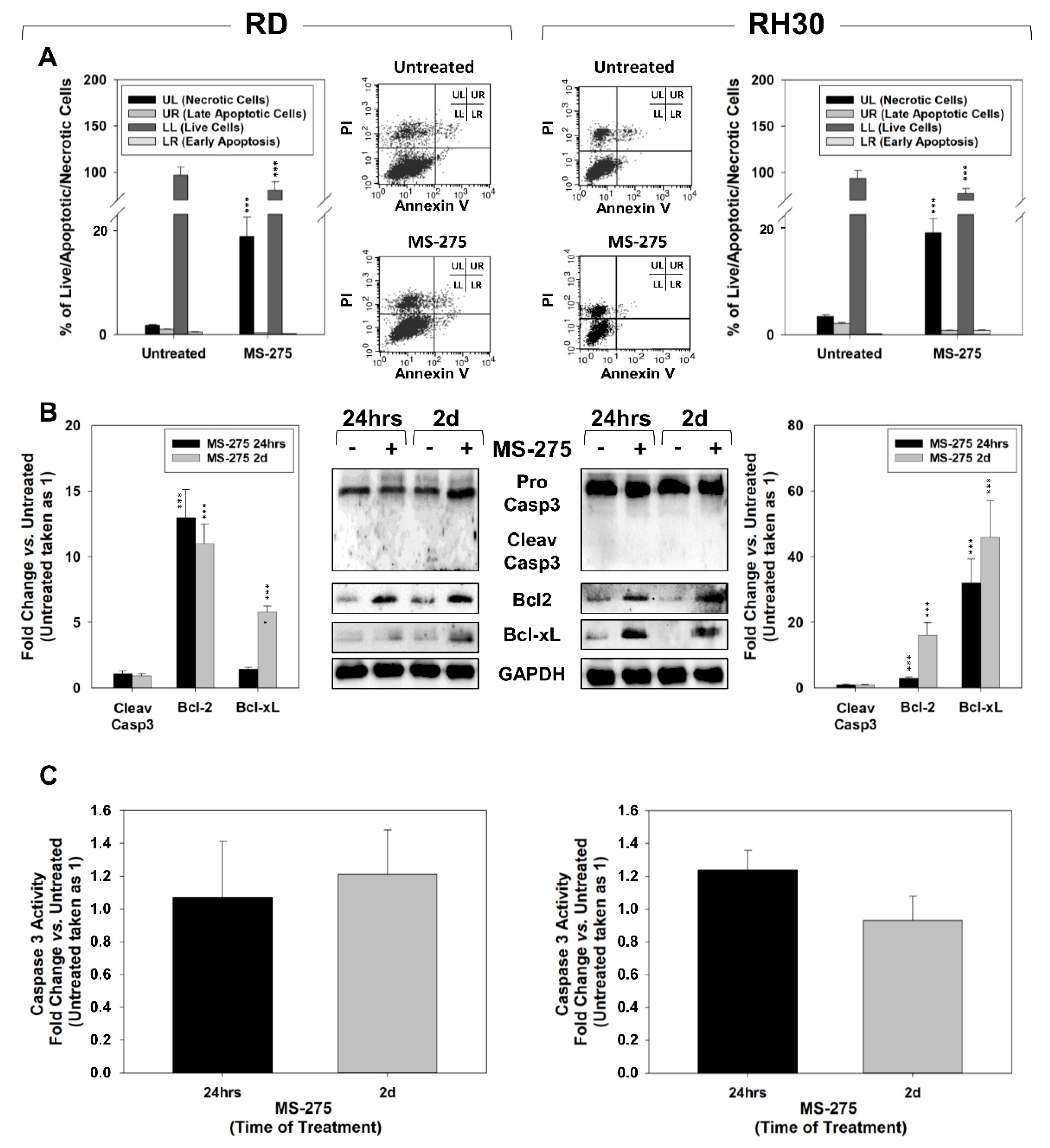
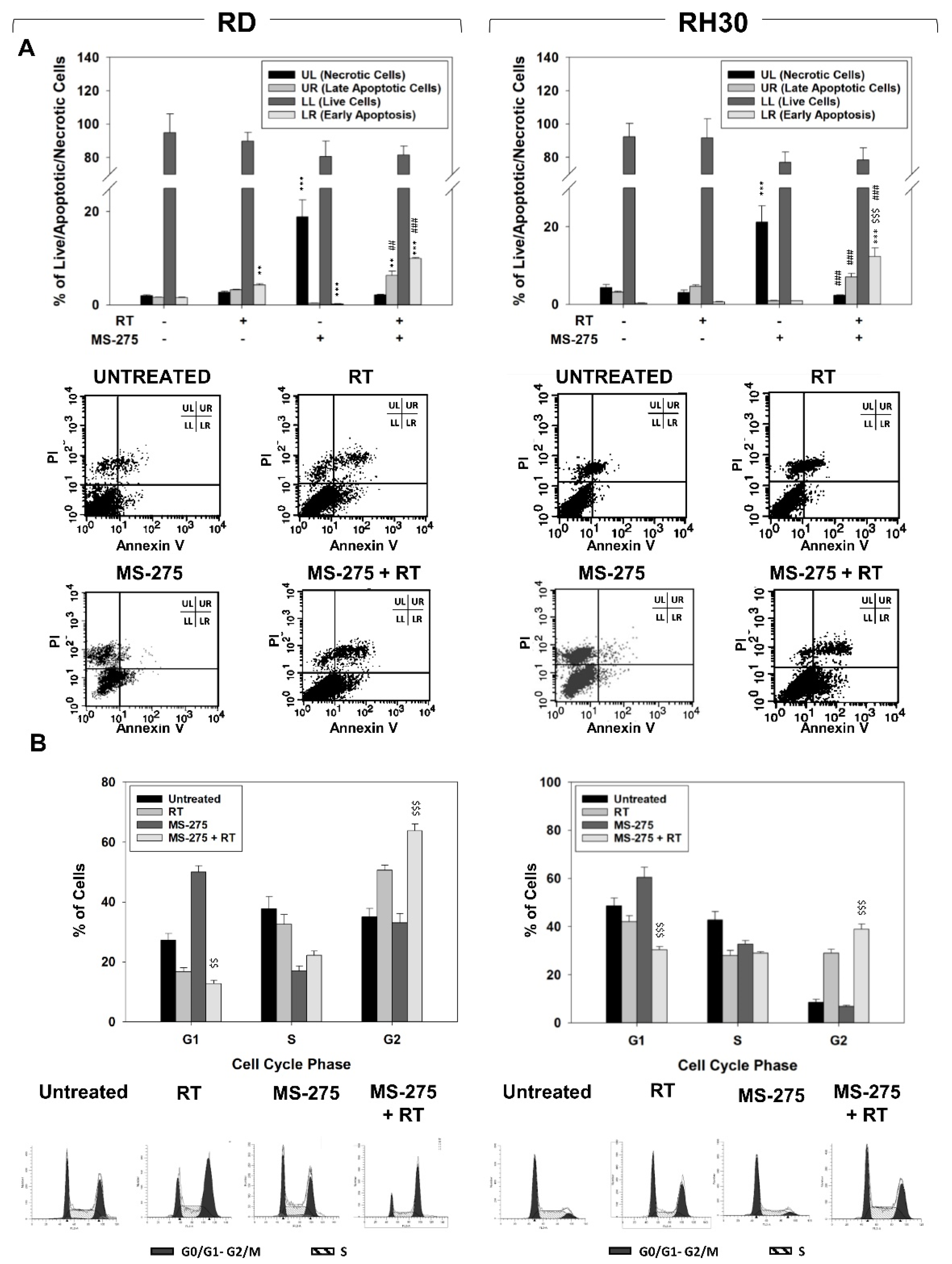
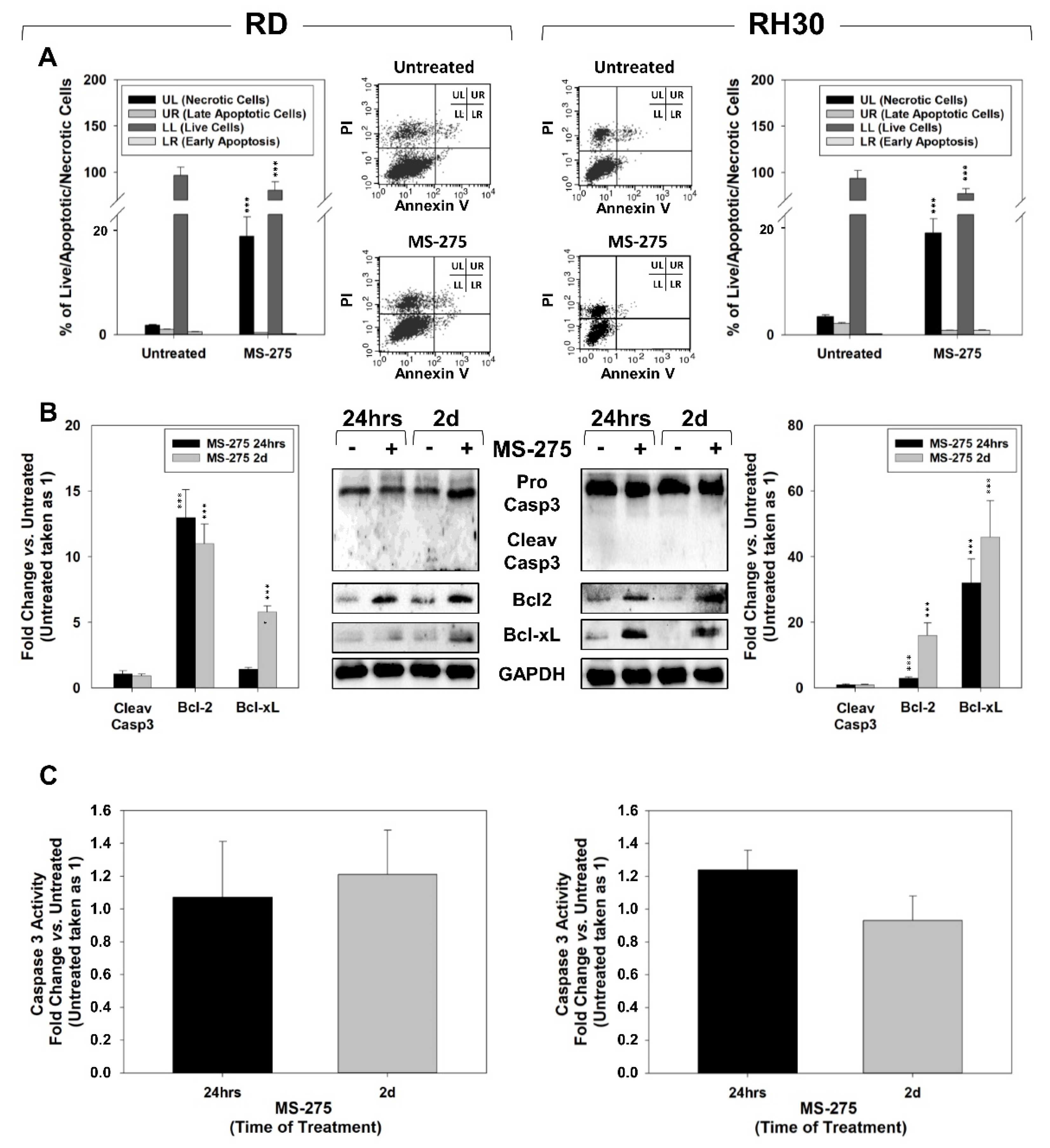
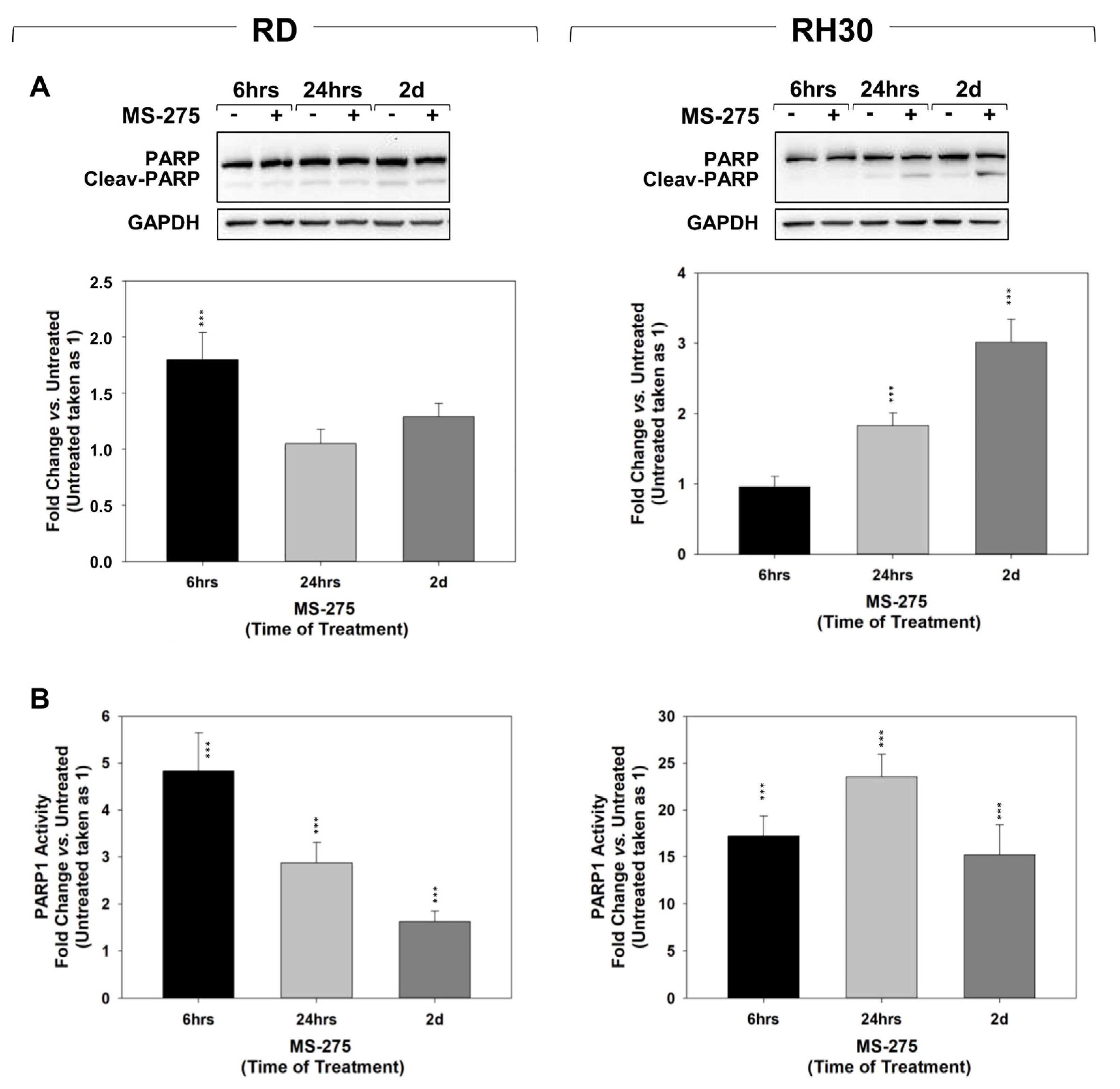
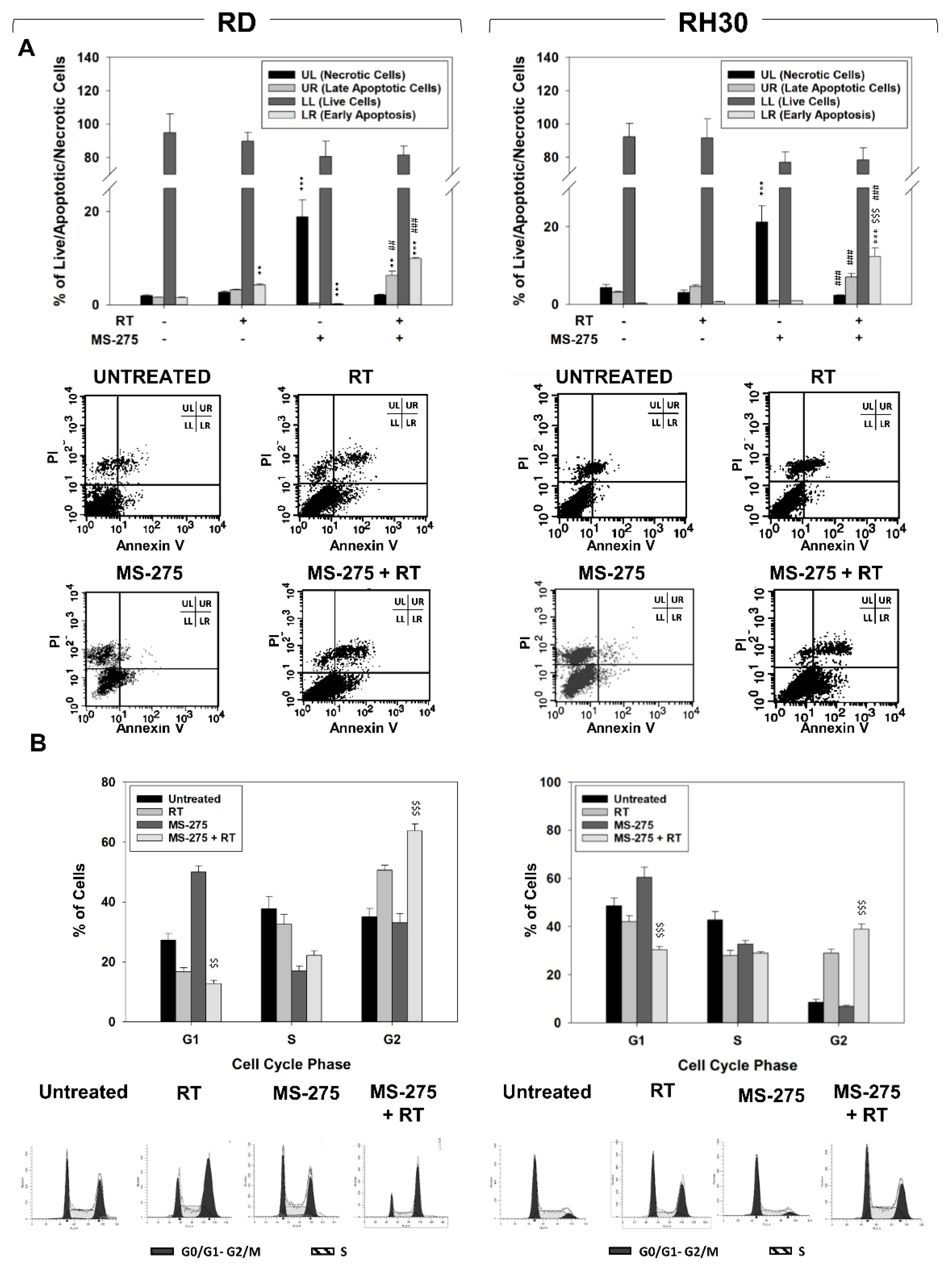
2.3. MS-275 Induced Non-Apoptotic Cell Death
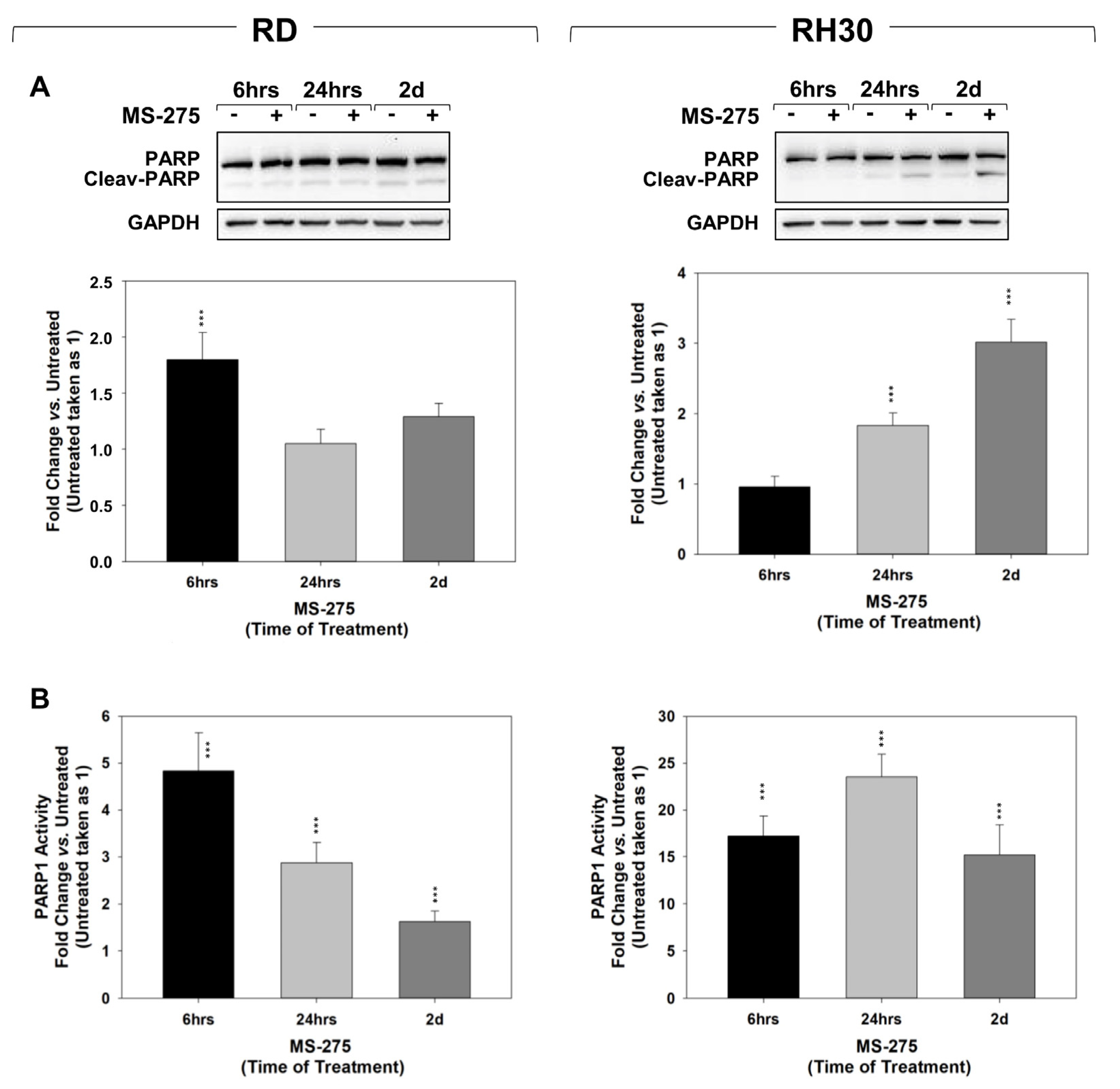
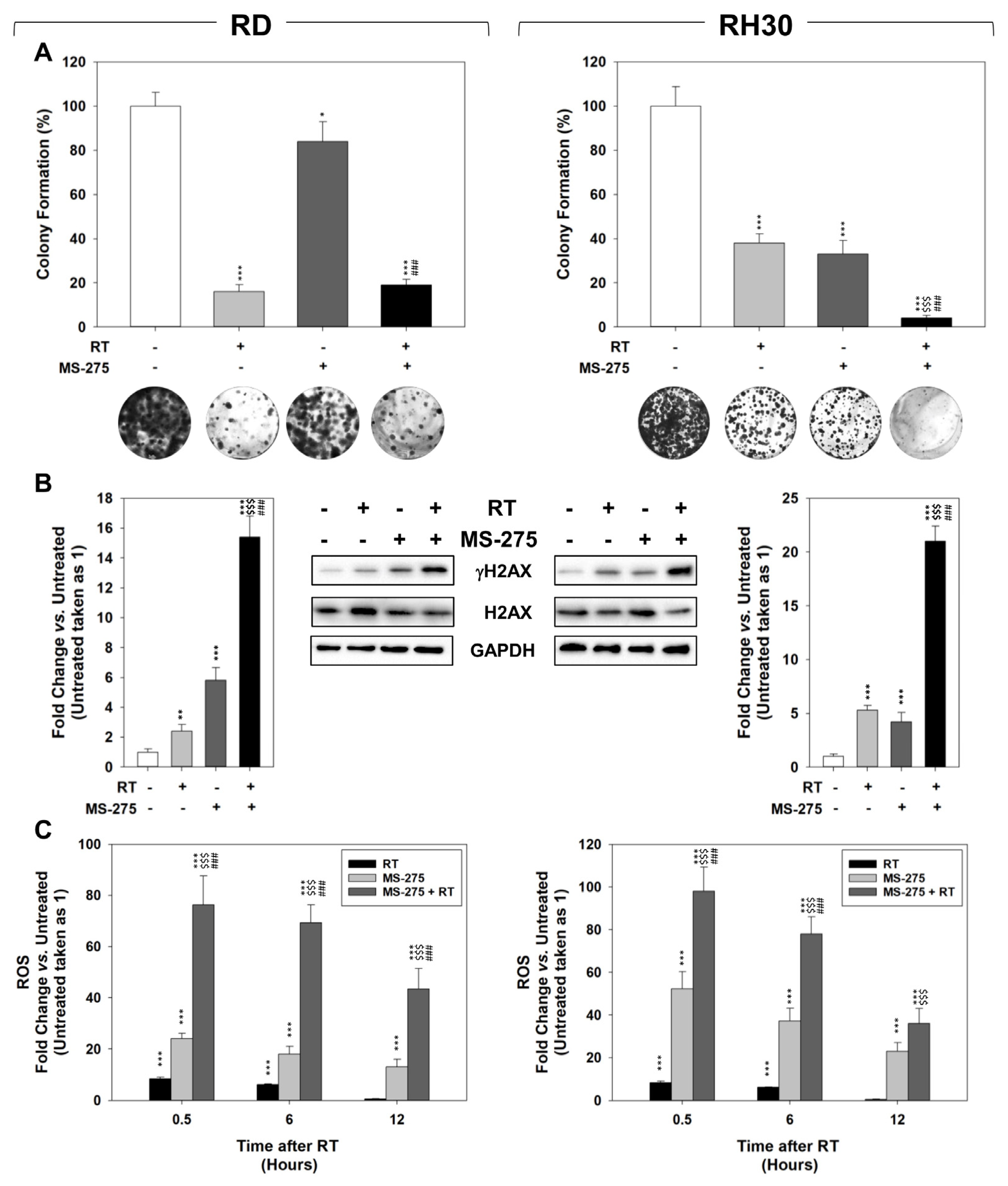
2.4. MS-275 Radiosensitizes FP-RMS Cells In Vitro
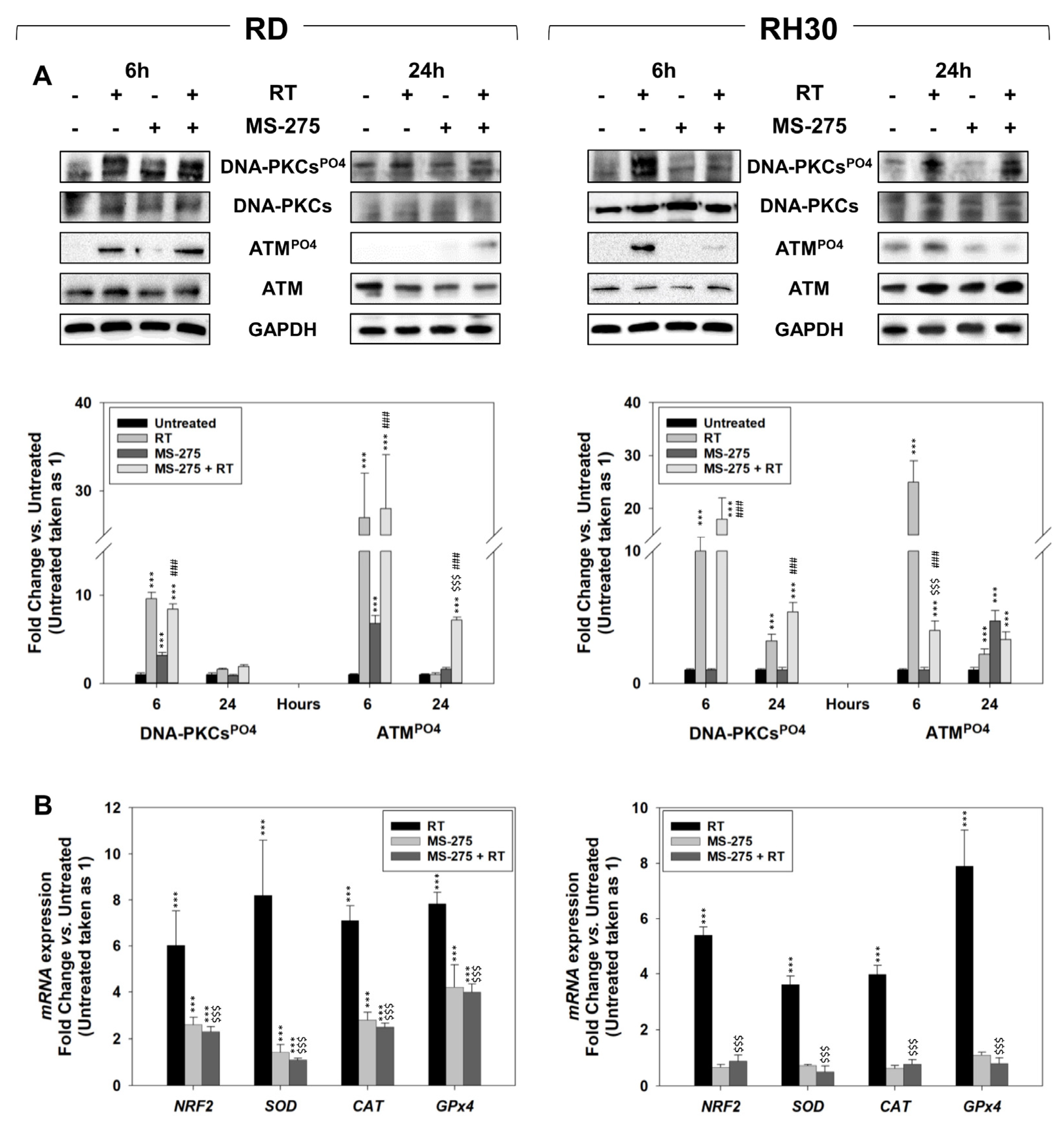
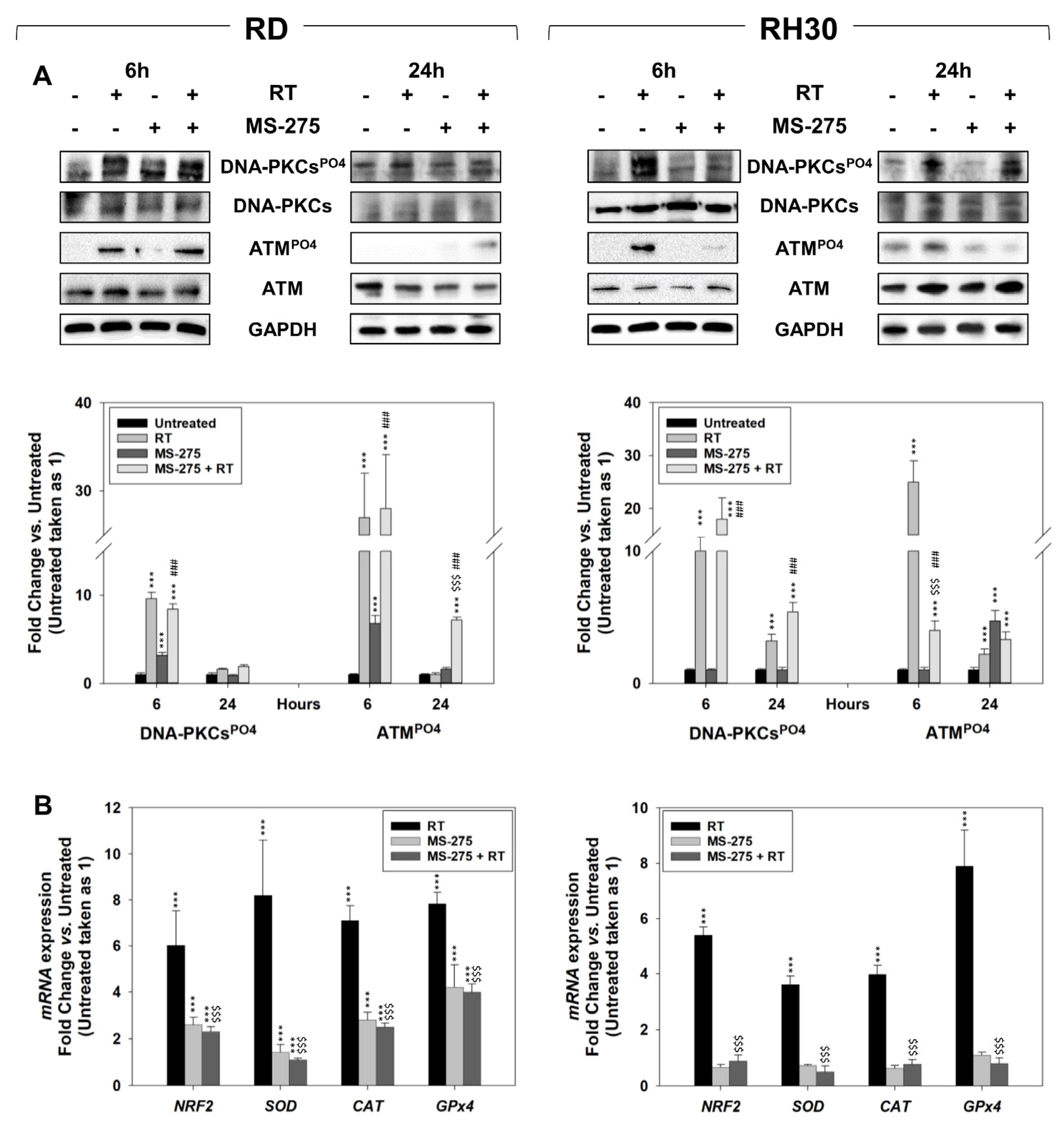
2.5. In Vitro, MS-275 Counteracts the Ability of FP-RMS to Repair DNA Damage and Detoxify from ROS Accumulation Induced by RT
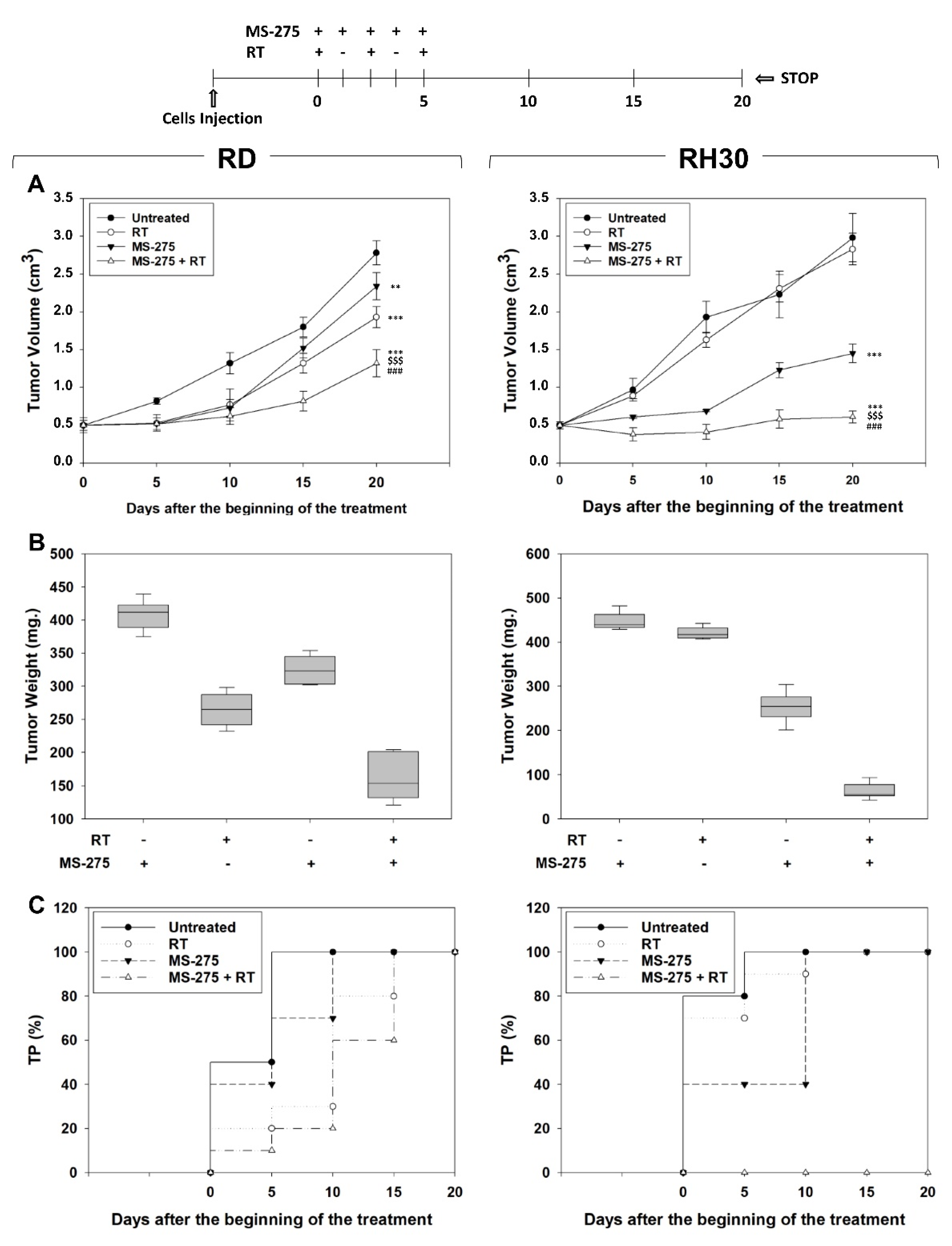
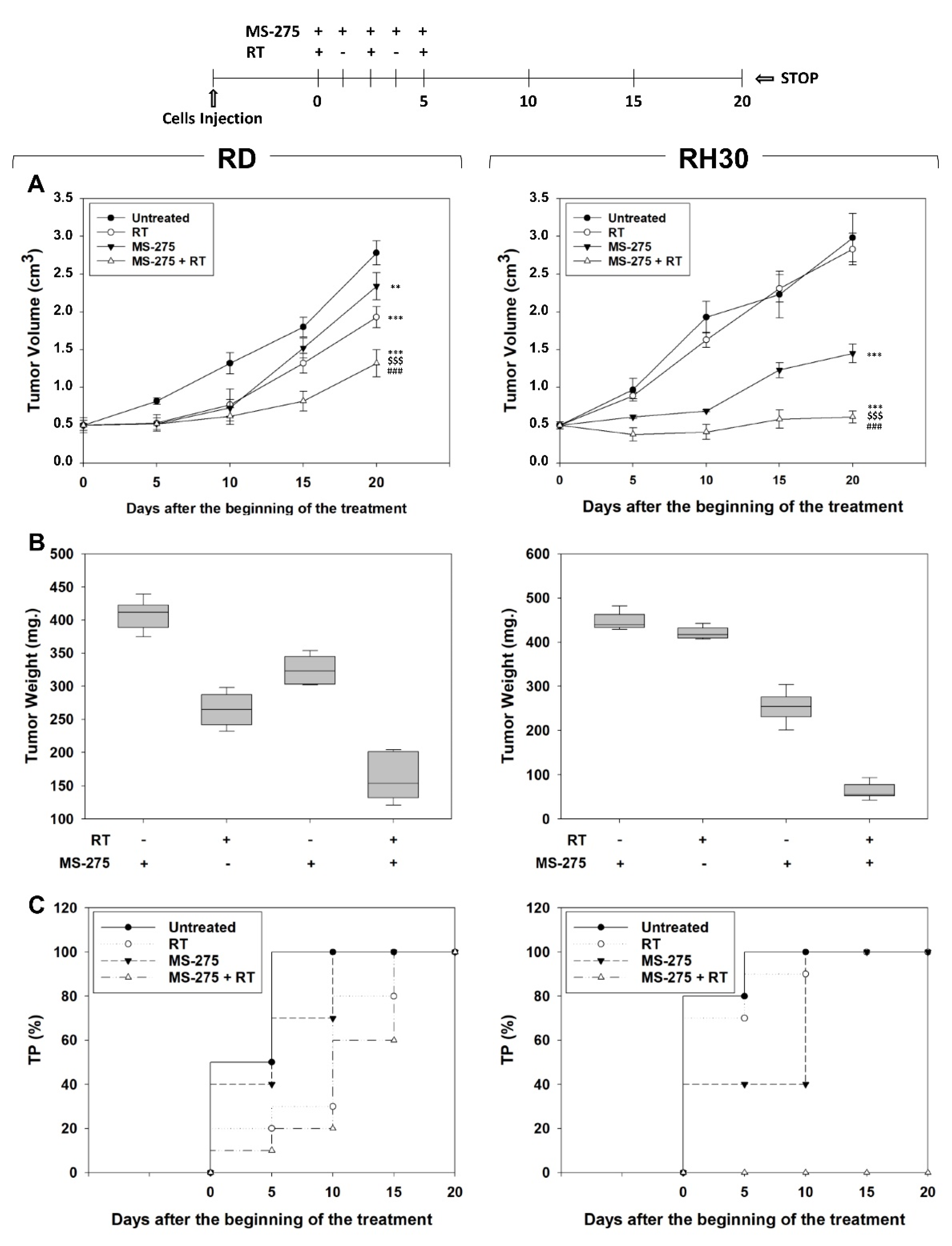
2.6. MS-275 Radiosensitizes FP-RMS Cells In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Pharmacological Treatment
4.2. Cell Viability Assay
4.3. Cell Cycle Analysis by Flow Cytometry
4.4. HDACs and PARP1 Activity Assays
4.5. Apoptosis Assay
4.6. RNA Isolation and qRT-PCR
4.7. Mitochondrial Superoxide Anion (·O2−) Production Assessment
4.8. Radiation Exposure and Clonogenic Assay
4.9. Protein Extraction and Western Blot
4.10. Animal Research Ethics Statement and In Vivo Xenograft Experiments
4.11. Evaluation of Treatment Response In Vivo
4.12. Statistical Analysis and Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F. Epigenetics in rhabdomyosarcoma: Cues to new biomarkers and targeted therapies. EBioMedicine 2020, 52, 102673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryder, B.E.; Pomella, S.; Sayers, C.; Wu, X.S.; Song, Y.; Chiarella, A.M.; Bagchi, S.; Chou, H.C.; Sinniah, R.S.; Walton, A.; et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat. Genet. 2019, 51, 1714–1722. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.Q.; Robinson, K.; Xu, L.; Pavlova, M.N.; Skapek, S.X.; Chen, E.Y. HDAC6 promotes growth, migration/invasion, and self-renewal of rhabdomyosarcoma. Oncogene 2021, 40, 578–591. [Google Scholar] [CrossRef]
- Phelps, M.P.; Bailey, J.N.; Vleeshouwer-Neumann, T.; Chen, E.Y. CRISPR screen identifies the NCOR/HDAC3 complex as a major suppressor of differentiation in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 15090–15095. [Google Scholar] [CrossRef] [Green Version]
- Monje, M. Open questions: Why are babies rarely born with cancer? BMC Biol. 2018, 16, 1–2. [Google Scholar] [CrossRef]
- Gryder, B.E.; Wachtel, M.; Chang, K.; El Demerdash, O.; Aboreden, N.G.; Mohammed, W.; Ewert, W.; Pomella, S.; Rota, R.; Wei, J.S.; et al. Miswired Enhancer Logic Drives a Cancer of the Muscle Lineage. iScience 2020, 23, 101103. [Google Scholar] [CrossRef]
- Bergeron, C.; Jenney, M.; De Corti, F.; Gallego, S.; Merks, H.; Glosli, H.; Ferrari, A.; Ranchère-Vince, D.; De Salvo, G.L.; Zanetti, I.; et al. Embryonal rhabdomyosarcoma completely resected at diagnosis: The European paediatric Soft tissue sarcoma Study Group RMS2005 experience. Eur. J. Cancer 2021, 146, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin. Second in review on chromatin dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef]
- Sadoul, K.; Wang, J.; Diagouraga, B.; Khochbin, S. The tale of protein lysine acetylation in the cytoplasm. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef]
- Khan, S.N.; Khan, A.U. Role of histone acetylation in cell physiology and diseases: An update. Clin. Chim. Acta 2010, 411, 1401–1411. [Google Scholar] [CrossRef]
- Di Martile, M.; Del Bufalo, D.; Trisciuoglio, D. The multifaceted role of lysine acetylation in cancer: Prognostic biomarker and therapeutic target. Oncotarget 2016, 7, 55789–55810. [Google Scholar] [CrossRef] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagella, L.; Federico, M. Histone deacetylase inhibitors in the treatment of hematological malignancies and solid tumors. J. Biomed. Biotechnol. 2011, 2011, 475641. [Google Scholar]
- Fantin, V.R.; Richon, V.M. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin. Cancer Res. 2007, 13, 7237–7242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [Green Version]
- Bieliauskas, A.V.; Pflum, M.K.H. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402–1413. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Sharda, A.; Rashid, M.; Shah, S.G.; Sharma, A.K.; Singh, S.R.; Gera, P.; Chilkapati, M.K.; Gupta, S. Elevated HDAC activity and altered histone phospho-acetylation confer acquired radio-resistant phenotype to breast cancer cells. Clin. Epigenet. 2020, 12, 4. [Google Scholar] [CrossRef]
- Jonsson, M.; Ragnum, H.B.; Julin, C.H.; Yeramian, A.; Clancy, T.; Frikstad, K.A.M.; Seierstad, T.; Stokke, T.; Matias-Guiu, X.; Ree, A.H.; et al. Hypoxia-independent gene expression signature associated with radiosensitisation of prostate cancer cell lines by histone deacetylase inhibition. Br. J. Cancer 2016, 115, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Wu, W.; Zhang, Q.; Sun, Y.; Cui, Y.; Wu, F.; Wei, X.; Qi, G.; Liang, X.; Tang, F.; et al. Inhibition of tribbles protein-1 attenuates radioresistance in human glioma cells. Sci. Rep. 2015, 5, 15961. [Google Scholar] [CrossRef] [Green Version]
- Frame, F.M.; Pellacani, D.; Collins, A.T.; Simms, M.S.; Mann, V.M.; Jones, G.; Meuth, M.; Bristow, R.G.; Maitland, N.J. HDAC inhibitor confers radiosensitivity to prostate stem-like cells. Br. J. Cancer 2013, 109, 3023–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blattmann, C.; Thiemann, M.; Stenzinger, A.; Christmann, A.; Roth, E.; Ehemann, V.; Debus, J.; Kulozik, A.E.; Weichert, W.; Huber, P.E.; et al. Radiosensitization by histone deacetylase inhibition in an osteosarcoma mouse model. Strahlenther. Onkol. 2013, 189, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.; Tucker, C.; Creech, J.; Favours, E.; Billups, C.A.; Liu, T.; Fouladi, M.; Freeman, B.B.; Stewart, C.F.; Houghton, P.J. Evaluation of the antitumor efficacy, pharmacokinetics, and pharmacodynamics of the histone deacetylase inhibitor depsipeptide in childhood cancer models in vivo. Clin. Cancer Res. 2006, 12, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutko, M.C.; Glick, R.D.; Butler, L.M.; Coffey, D.C.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; Laquaglia, M.P. Histone Deacetylase Inhibitors Induce Growth Suppression and Cell Death in Human Rhabdomyosarcoma in Vitro. Clin. Cancer Res. 2003, 9, 5749–5755. [Google Scholar] [PubMed]
- Hedrick, E.; Crose, L.; Linardic, C.M.; Safe, S. Histone deacetylase inhibitors inhibit rhabdomyosarcoma by reactive oxygen species-dependent targeting of specificity protein transcription factors. Mol. Cancer Ther. 2015, 14, 2143–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vleeshouwer-Neumann, T.; Phelps, M.; Bammler, T.K.; MacDonald, J.W.; Jenkins, I.; Chen, E.Y. Histone deacetylase inhibitors antagonize distinct pathways to suppress tumorigenesis of embryonal rhabdomyosarcoma. PLoS ONE 2015, 10, e0144320. [Google Scholar] [CrossRef] [Green Version]
- Ghayad, S.E.; Rammal, G.; Sarkis, O.; Basma, H.; Ghamloush, F.; Fahs, A.; Karam, M.; Harajli, M.; Rabeh, W.; Mouawad, J.E.; et al. The histone deacetylase inhibitor Suberoylanilide Hydroxamic Acid (SAHA) as a therapeutic agent in rhabdomyosarcoma. Cancer Biol. Ther. 2019, 20, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Tomoyasu, C.; Kikuchi, K.; Kaneda, D.; Yagyu, S.; Miyachi, M.; Tsuchiya, K.; Iehara, T.; Sakai, T.; Hosoi, H. OBP-801, a novel histone deacetylase inhibitor, induces M-phase arrest and apoptosis in rhabdomyosarcoma cells. Oncol. Rep. 2019, 41, 643–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Martile, M.; Desideri, M.; Tupone, M.G.; Buglioni, S.; Antoniani, B.; Mastroiorio, C.; Falcioni, R.; Ferraresi, V.; Baldini, N.; Biagini, R.; et al. Histone deacetylase inhibitor ITF2357 leads to apoptosis and enhances doxorubicin cytotoxicity in preclinical models of human sarcoma. Oncogenesis 2018, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Bharathy, N.; Berlow, N.E.; Wang, E.; Abraham, J.; Settelmeyer, T.P.; Hooper, J.E.; Svalina, M.N.; Bajwa, Z.; Goros, M.W.; Hernandez, B.S.; et al. Preclinical rationale for entinostat in embryonal rhabdomyosarcoma. Skelet. Muscle 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Kurmasheva, R.T.; Bandyopadhyay, A.; Favours, E.; Del Pozo, V.; Ghilu, S.; Phelps, D.A.; Erickson, S.W.; Peer, C.J.; Figg, W.D.; Smith, M.A.; et al. Evaluation of entinostat alone and in combination with standard-of-care cytotoxic agents against rhabdomyosarcoma xenograft models. Pediatr. Blood Cancer 2019, 66, e27820. [Google Scholar] [CrossRef] [PubMed]
- Jaboin, J.; Wild, J.; Hamidi, H.; Khanna, C.; Kim, C.J.; Robey, R.; Bates, S.E.; Thiele, C.J. MS-27-275, an Inhibitor of Histone Deacetylase, Has Marked in Vitro and in Vivo Antitumor Activity against Pediatric Solid Tumors. Cancer Res. 2002, 62, 6108–6115. [Google Scholar] [PubMed]
- Marampon, F.; Di Nisio, V.; Pietrantoni, I.; Petragnano, F.; Fasciani, I.; Scicchitano, B.M.; Ciccarelli, C.; Gravina, G.L.; Festuccia, C.; Del Fattore, A.; et al. Pro-differentiating and radiosensitizing effects of inhibiting HDACs by PXD-101 (Belinostat) in in vitro and in vivo models of human rhabdomyosarcoma cell lines. Cancer Lett. 2019, 461, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, A.; Petragnano, F.; Milazzo, L.; Vulcano, F.; Macioce, G.; Codenotti, S.; Cassandri, M.; Pomella, S.; Cicchetti, F.; Fasciani, I.; et al. Romidepsin (FK228) fails in counteracting the transformed phenotype of rhabdomyosarcoma cells but efficiently radiosensitizes, in vitro and in vivo, the alveolar phenotype subtype. Int. J. Radiat. Biol. 2021, 97, 943–957. [Google Scholar] [CrossRef]
- Bukowinski, A.; Chang, B.; Reid, J.M.; Liu, X.; Minard, C.G.; Trepel, J.B.; Lee, M.J.; Fox, E.; Weigel, B.J. A phase 1 study of entinostat in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1513, Pediatric Early Phase-Clinical Trial Network (PEP-CTN). Pediatr. Blood Cancer 2021, 68, e28892. [Google Scholar] [CrossRef]
- Chu, Q.S.C.; Nielsen, T.O.; Alcindor, T.; Gupta, A.; Endo, M.; Goytain, A.; Xu, H.; Verma, S.; Tozer, R.; Knowling, M.; et al. A phase II study of SB939, a novel pan-histone deacetylase inhibitor, in patients with translocationassociated recurrent/metastatic sarcomas-NCIC-CTG IND 200. Ann. Oncol. 2015, 26, 973–981. [Google Scholar] [CrossRef]
- Choy, E.; Flamand, Y.; Balasubramanian, S.; Butrynski, J.E.; Harmon, D.C.; George, S.; Cote, G.M.; Wagner, A.J.; Morgan, J.A.; Sirisawad, M.; et al. Phase 1 study of oral abexinostat, a histone deacetylase inhibitor, in combination with doxorubicin in patients with metastatic sarcoma. Cancer 2015, 121, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Vitfell-Rasmussen, J.; Judson, I.; Safwat, A.; Jones, R.L.; Rossen, P.B.; Lind-Hansen, M.; Knoblauch, P.; Krarup-Hansen, A. A phase I/II clinical trial of belinostat (PXD101) in combination with doxorubicin in patients with soft tissue sarcomas. Sarcoma 2016, 2016, 2090271. [Google Scholar] [CrossRef] [Green Version]
- Cassier, P.A.; Lefranc, A.; Y Amela, E.; Chevreau, C.; Bui, B.N.; Lecesne, A.; Ray-Coquard, I.; Chabaud, S.; Penel, N.; Berge, Y.; et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. Br. J. Cancer 2013, 109, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yamashita, T.; Mariko, Y.; Nosaka, Y.; Tsuchiya, K.; Ando, T.; Suzuki, T.; Tsuruo, T.; Nakanishi, O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 4592–4597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Perales, S.; Martínez-Ramírez, A.; De Andrés, S.A.; Valle, L.; Urioste, M.; Benítez, J.; Cigudosa, J.C. Molecular cytogenetic characterization of rhabdomyosarcoma cell lines. Cancer Genet. Cytogenet. 2004, 148, 35–43. [Google Scholar] [CrossRef]
- Gross, A.; Katz, S.G. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [CrossRef]
- Robert, C.; Rassool, F.V. HDAC Inhibitors: Roles of DNA damage and repair. In Advances in Cancer Research; Academic Press Inc.: Cambridge, MA, USA, 2012; Volume 116, pp. 87–129. [Google Scholar]
- Nias, A.H.W. An Introduction to Radiobiology, 2nd ed.; Wiley: Hoboken, NJ, USA, 1998; Available online: https://www.wiley.com/en-us/An+Introduction+to+Radiobiology%2C+2nd+Edition-p-9780471975908 (accessed on 22 June 2021).
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [Green Version]
- Gryder, B.E.; Yohe, M.E.; Chou, H.C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, R.; McIntyre, A.; Camerin, C.; Walters, Z.S.; Di Leo, K.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor activity of sustained N-myc reduction in rhabdomyosarcomas and transcriptional block by antigene therapy. Clin. Cancer Res. 2012, 18, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Camero, S.; Ceccarelli, S.; De Felice, F.; Marampon, F.; Mannarino, O.; Camicia, L.; Vescarelli, E.; Pontecorvi, P.; Pizer, B.; Shukla, R.; et al. PARP inhibitors affect growth, survival and radiation susceptibility of human alveolar and embryonal rhabdomyosarcoma cell lines. J. Cancer Res. Clin. Oncol. 2019, 145, 137–152. [Google Scholar] [CrossRef] [Green Version]
- Marampon, F.; Ciccarelli, C.; Zani, B.M. Down-regulation of c-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol. Cancer 2006, 145, 137–152. [Google Scholar] [CrossRef] [Green Version]
- Gravina, G.L.; Festuccia, C.; Popov, V.M.; Di Rocco, A.; Colapietro, A.; Sanità, P.; Delle Monache, S.; Musio, D.; De Felice, F.; Di Cesare, E.; et al. C-Myc Sustains Transformed Phenotype and Promotes Radioresistance of Embryonal Rhabdomyosarcoma Cell Lines. Radiat. Res. 2016, 185, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Dutta, C.; Day, T.; Kopp, N.; Van Bodegom, D.; Davids, M.S.; Ryan, J.; Bird, L.; Kommajosyula, N.; Weigert, O.; Yoda, A.; et al. BCL2 suppresses PARP1 function and nonapoptotic cell death. Cancer Res. 2012, 72, 4193–4203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oike, T.; Ohno, T. Molecular mechanisms underlying radioresistance: Data compiled from isogenic cell experiments. Ann. Transl. Med. 2020, 8, 273. [Google Scholar] [CrossRef]
- Toulany, M. Targeting DNA double-strand break repair pathways to improve radiotherapy response. Genes 2019, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 2010, 31, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Tarnawski, R.; Fowler, J.; Skladowski, K.; Wierniak, A.; Suwiński, R.; Maciejewski, B.; Wygoda, A. How fast is repopulation of tumor cells during the treatment gap? Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 229–236. [Google Scholar] [CrossRef]
- Trott, K.R. Cell repopulation and overall treatment time. Int. J. Radiat. Oncol. Biol. Phys. 1990, 19, 1071–1075. [Google Scholar] [CrossRef]
- Maciejewski, B.; Withers, H.R.; Taylor, J.M.G.; Hliniak, A. Dose fractionation and regeneration in radiotherapy for cancer of the oral cavity and oropharynx. Part 2. Normal tissue responses: Acute and late effects. Int. J. Radiat. Oncol. Biol. Phys. 1990, 18, 101–111. [Google Scholar] [CrossRef]
- Withers, H.R.; Maciejewski, B.; Taylor, J.M.; Hliniak, A. Accelerated repopulation in head and neck cancer. Front. Radiat. Ther. Oncol. 1988, 22, 105–110. [Google Scholar] [CrossRef]
- Harriss-Phillips, W.M.; Bezak, E.; Yeoh, E.K. Monte Carlo radiotherapy simulations of accelerated repopulation and reoxygenation for hypoxic head and neck cancer. Br. J. Radiol. 2011, 84, 903–918. [Google Scholar] [CrossRef] [Green Version]
- Marcu, L.; van Doorn, T.; Olver, I. Modelling of post-irradiation accelerated repopulation in squamous cell carcinomas. Phys. Med. Biol. 2004, 49, 3767. [Google Scholar] [CrossRef]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 239–253. [Google Scholar] [CrossRef]
- Boustani, J.; Grapin, M.; Laurent, P.A.; Apetoh, L.; Mirjolet, C. The 6th R of radiobiology: Reactivation of anti-tumor immune response. Cancers 2019, 11, 860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Withers, H.R. The Four R’s of Radiotherapy. In Advances in Radiation Biology; Elsevier: Amsterdam, The Netherlands, 1975. [Google Scholar]
- McDonald, J.T.; Kim, K.; Norris, A.J.; Vlashi, E.; Phillips, T.M.; Lagadec, C.; Della Donna, L.; Ratikan, J.; Szelag, H.; Hlatky, L.; et al. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010, 70, 8886–8895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marampon, F.; Codenotti, S.; Megiorni, F.; Del Fattore, A.; Camero, S.; Gravina, G.L.; Festuccia, C.; Musio, D.; De Felice, F.; Nardone, V.; et al. NRF2 orchestrates the redox regulation induced by radiation therapy, sustaining embryonal and alveolar rhabdomyosarcoma cells radioresistance. J. Cancer Res. Clin. Oncol. 2019, 145, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Hammers, H.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009, 280, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Wu, F.; Jin, Y.M.; Chang, W.Q.; Xu, T.M. HDAC11: A rising star in epigenetics. Biomed. Pharmacother. 2020, 131, 110607. [Google Scholar] [CrossRef]
- Zhang, F.; Shao, C.; Chen, Z.; Li, Y.; Jing, X.; Huang, Q. Low Dose of Trichostatin A Improves Radiation Resistance by Activating Akt/Nrf2-Dependent Antioxidation Pathway in Cancer Cells. Radiat. Res. 2021, 195, 366–377. [Google Scholar] [CrossRef]
- Deubzer, H.E.; Schier, M.C.; Oehme, I.; Lodrini, M.; Haendler, B.; Sommer, A.; Witt, O. HDAC11 is a novel drug target in carcinomas. Int. J. Cancer 2013, 103, 2200–2208. [Google Scholar] [CrossRef]
- Megiorni, F.; Gravina, G.L.; Camero, S.; Ceccarelli, S.; Del Fattore, A.; Desiderio, V.; Papaccio, F.; McDowell, H.P.; Shukla, R.; Pizzuti, A.; et al. Pharmacological targeting of the ephrin receptor kinase signalling by GLPG1790 in vitro and in vivo reverts oncophenotype, induces myogenic differentiation and radiosensitizes embryonal rhabdomyosarcoma cells. J. Hematol. Oncol. 2017, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Pomella, S.; Sreenivas, P.; Gryder, B.E.; Wang, L.; Milewski, D.; Cassandri, M.; Baxi, K.; Hensch, N.R.; Carcarino, E.; Song, Y.; et al. Interaction between SNAI2 and MYOD enhances oncogenesis and suppresses differentiation in Fusion Negative Rhabdomyosarcoma. Nat. Commun. 2021, 12, 192. [Google Scholar] [CrossRef]
- Vulcano, F.; Milazzo, L.; Ciccarelli, C.; Eramo, A.; Sette, G.; Mauro, A.; Macioce, G.; Martinelli, A.; La Torre, R.; Casalbore, P.; et al. Wharton’s jelly mesenchymal stromal cells have contrasting effects on proliferation and phenotype of cancer stem cells from different subtypes of lung cancer. Exp. Cell Res. 2016, 345, 190–198. [Google Scholar] [CrossRef]
- Camero, S.; Camicia, L.; Marampon, F.; Ceccarelli, S.; Shukla, R.; Mannarino, O.; Pizer, B.; Schiavetti, A.; Pizzuti, A.; Tombolini, V.; et al. BET inhibition therapy counteracts cancer cell survival, clonogenic potential and radioresistance mechanisms in rhabdomyosarcoma cells. Cancer Lett. 2020, 479, 71–88. [Google Scholar] [CrossRef]
- Petragnano, F.; Pietrantoni, I.; Camero, S.; Codenotti, S.; Milazzo, L.; Vulcano, F.; MacIoce, G.; Giordani, I.; Tini, P.; Cheleschi, S.; et al. Clinically relevant radioresistant rhabdomyosarcoma cell lines: Functional, molecular and immune-related characterization. J. Biomed. Sci. 2020, 27, 90. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; Camero, S.; Ceccarelli, S.; McDowell, H.P.; Mannarino, O.; Marampon, F.; Pizer, B.; Shukla, R.; Pizzuti, A.; Marchese, C.; et al. DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget 2016, 7, 79342–79356. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassandri, M.; Pomella, S.; Rossetti, A.; Petragnano, F.; Milazzo, L.; Vulcano, F.; Camero, S.; Codenotti, S.; Cicchetti, F.; Maggio, R.; et al. MS-275 (Entinostat) Promotes Radio-Sensitivity in PAX3-FOXO1 Rhabdomyosarcoma Cells. Int. J. Mol. Sci. 2021, 22, 10671. https://doi.org/10.3390/ijms221910671
Cassandri M, Pomella S, Rossetti A, Petragnano F, Milazzo L, Vulcano F, Camero S, Codenotti S, Cicchetti F, Maggio R, et al. MS-275 (Entinostat) Promotes Radio-Sensitivity in PAX3-FOXO1 Rhabdomyosarcoma Cells. International Journal of Molecular Sciences. 2021; 22(19):10671. https://doi.org/10.3390/ijms221910671
Chicago/Turabian StyleCassandri, Matteo, Silvia Pomella, Alessandra Rossetti, Francesco Petragnano, Luisa Milazzo, Francesca Vulcano, Simona Camero, Silvia Codenotti, Francesca Cicchetti, Roberto Maggio, and et al. 2021. "MS-275 (Entinostat) Promotes Radio-Sensitivity in PAX3-FOXO1 Rhabdomyosarcoma Cells" International Journal of Molecular Sciences 22, no. 19: 10671. https://doi.org/10.3390/ijms221910671
APA StyleCassandri, M., Pomella, S., Rossetti, A., Petragnano, F., Milazzo, L., Vulcano, F., Camero, S., Codenotti, S., Cicchetti, F., Maggio, R., Festuccia, C., Gravina, G. L., Fanzani, A., Megiorni, F., Catanoso, M., Marchese, C., Tombolini, V., Locatelli, F., Rota, R., & Marampon, F. (2021). MS-275 (Entinostat) Promotes Radio-Sensitivity in PAX3-FOXO1 Rhabdomyosarcoma Cells. International Journal of Molecular Sciences, 22(19), 10671. https://doi.org/10.3390/ijms221910671