
Evolution of Electronic State and Properties of Silver Nanoparticles during Their Formation in Aqueous Solution



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
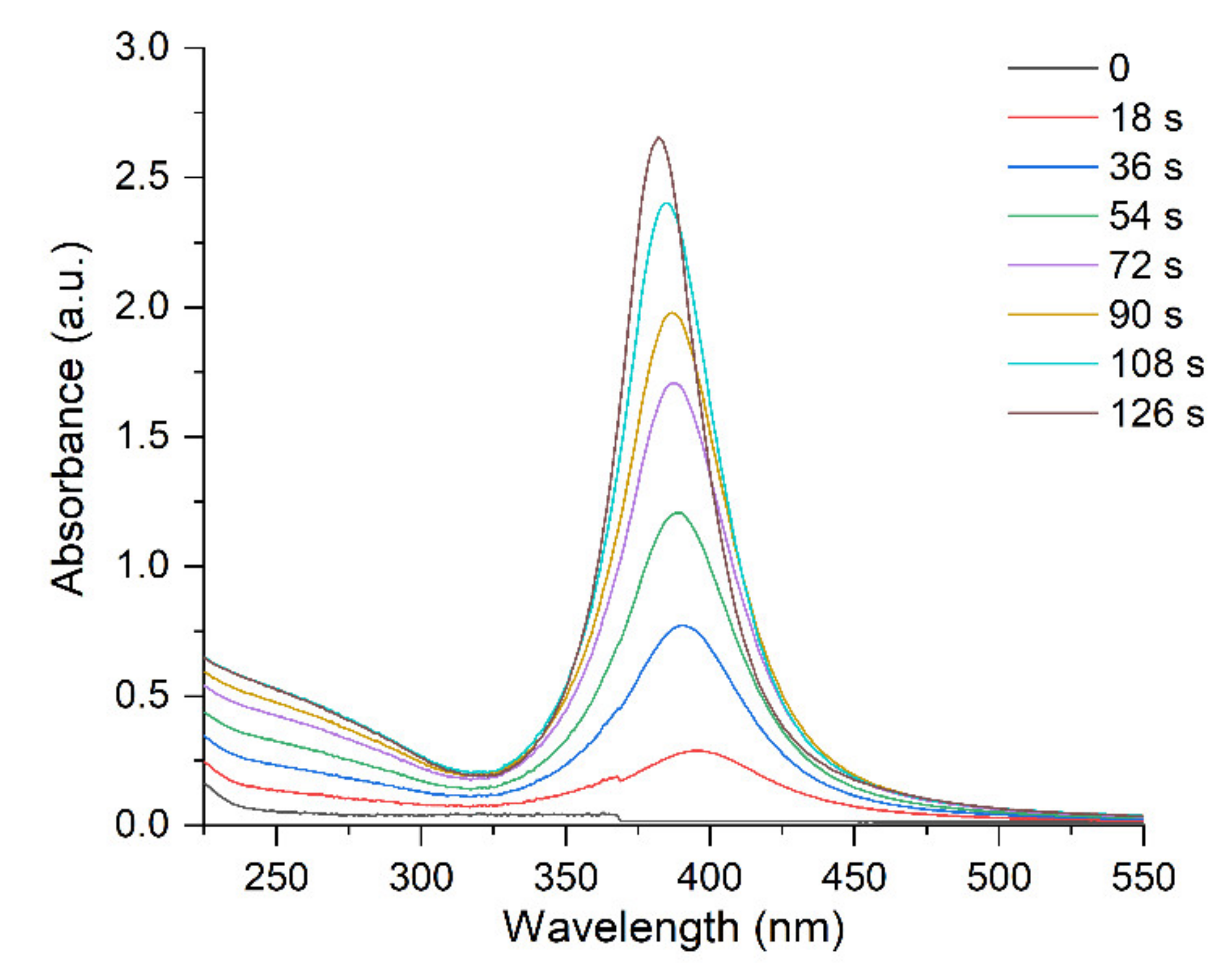
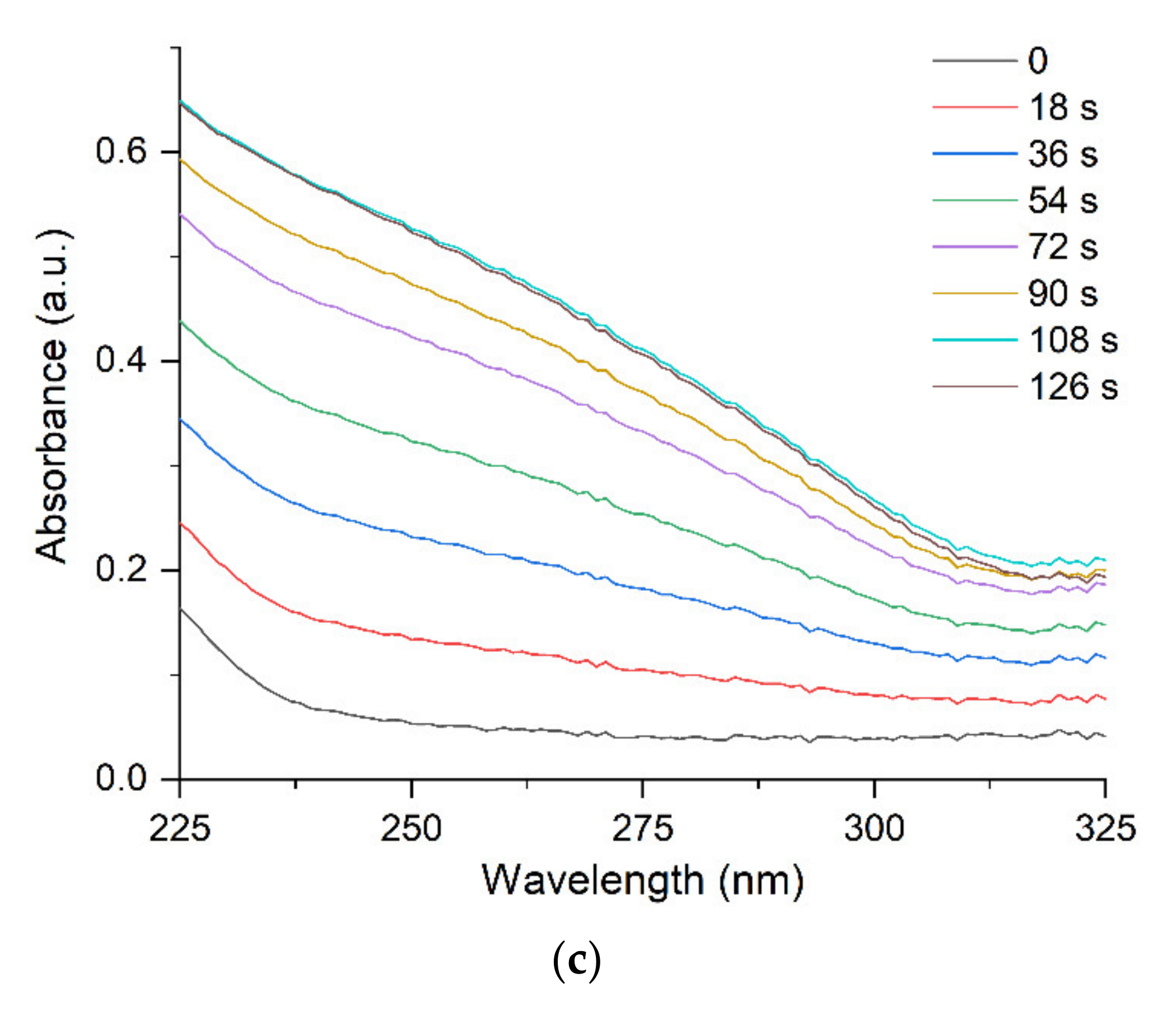
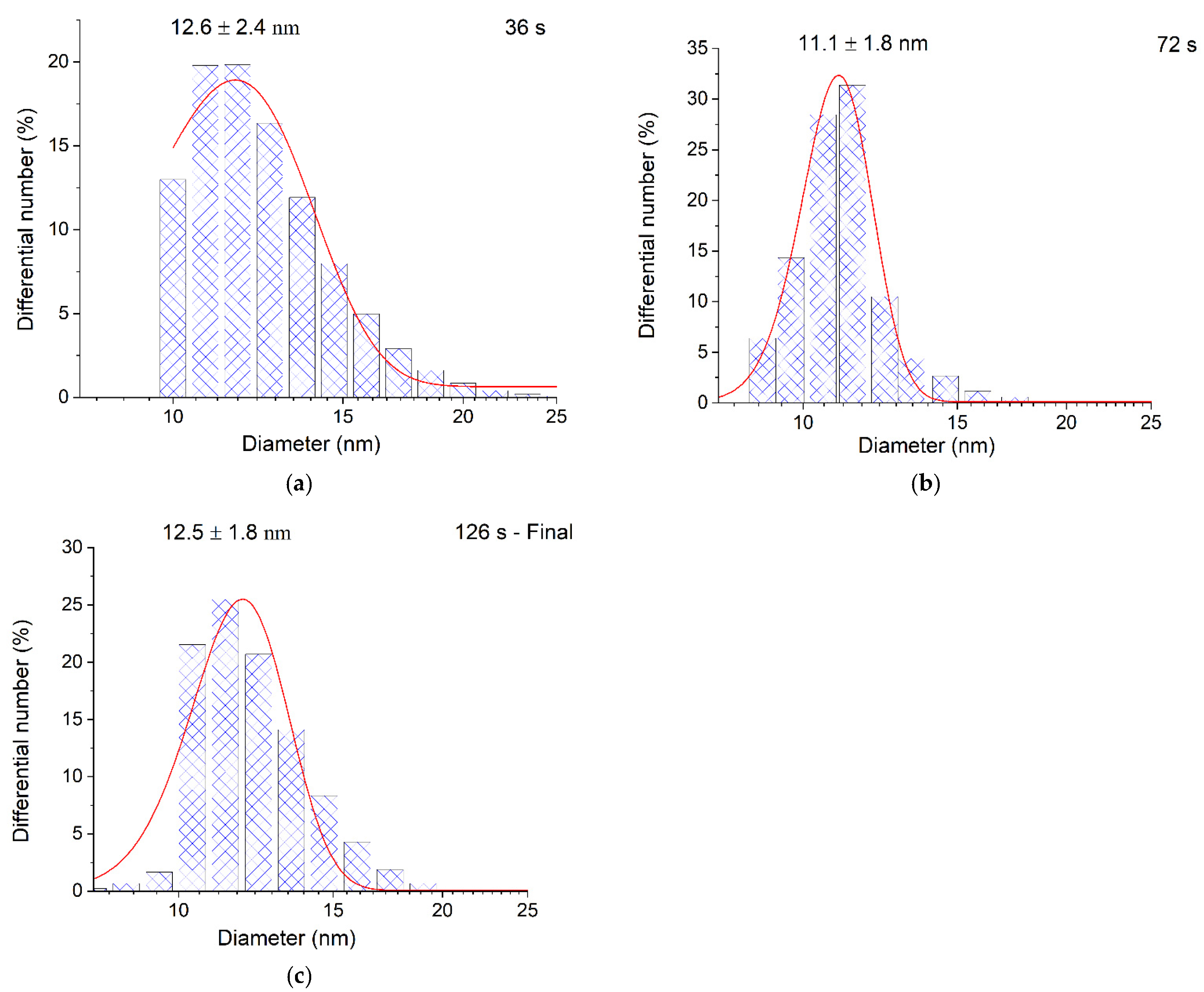
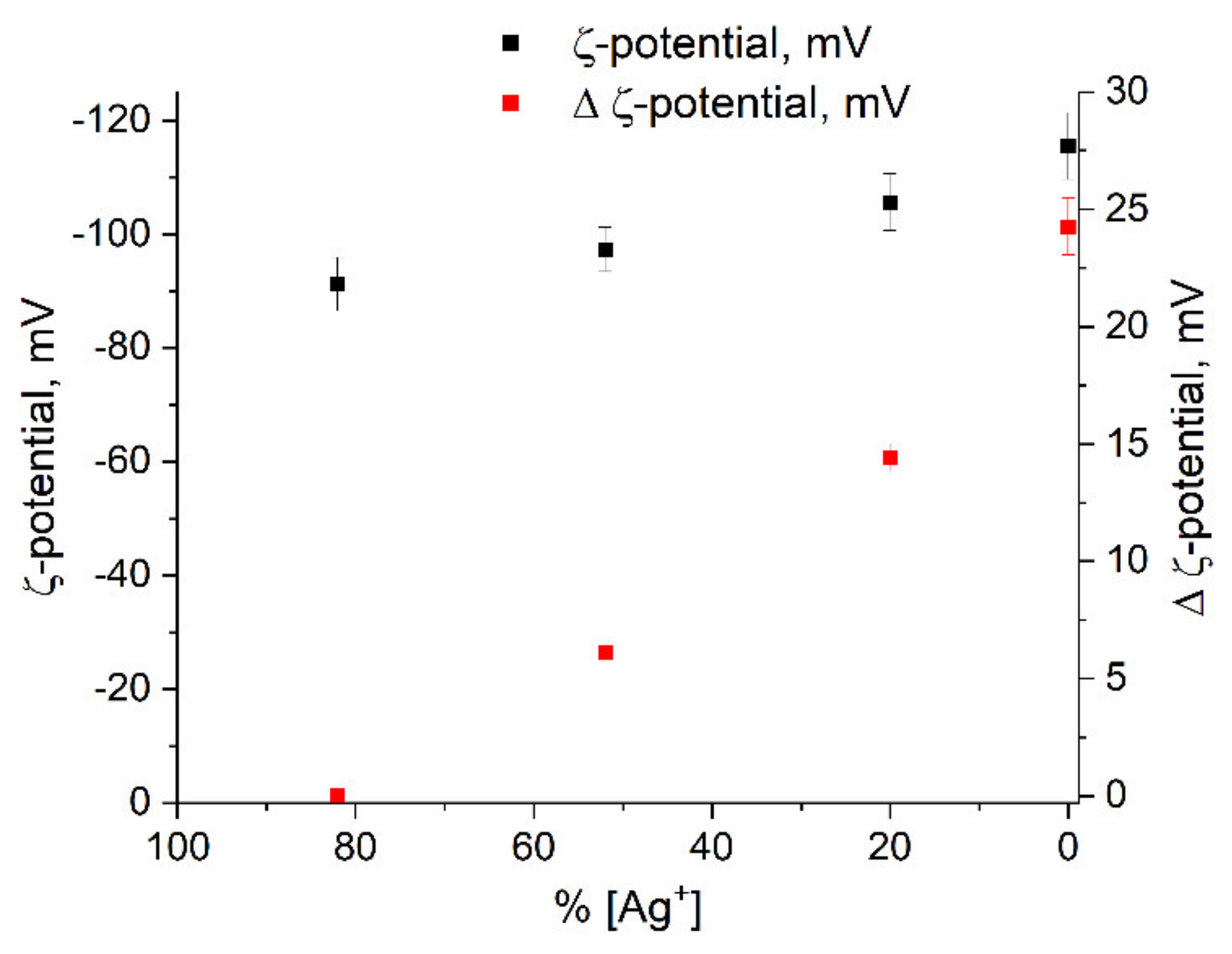
 discharging of the hydrosol of a constant composition on the structure of its optical absorption. For this purpose, charged hydrosol (corresponding to absorption after 126 s of UV irradiation in Figure 1) was kept in the dark at a constant room temperature for a long period in which spontaneous discharging took place. Figure 6 shows the variation in the spectrum of the hydrosol 3 days later. It can be seen that the LSPR band decreased by about 10% in intensity, was broadened, and shifted by about 5 nm to longer wavelengths (red shift). The intensity and position of the IBM band remained unchanged, which indicated that the number of silver atoms in the hydrosol was retained. The hydrosol was then exposed to UV light (~20 s). The absorption of the hydrosol returned to nearly the initial state. In other words, the LSPR band increased in intensity, narrowed, and shifted to the blue region. The described charging discharging procedure could be repeated. When the hydrosol was stored, nanoparticles lost the excess charge. A decrease in the charge can be caused both by a decrease in the concentration of free electrons and by the partial ionization of adatoms on the surface of a silver particle. The repeated photochemical charging of the hydrosol with electrons from the donor ( radical ion) restored the nanoparticle charge, which resulted in the restoration of the former position and shape of the LSPR band. In these experiments, we used a hydrosol with an invariable composition of silver nanoparticles (size and size distribution).
discharging of the hydrosol of a constant composition on the structure of its optical absorption. For this purpose, charged hydrosol (corresponding to absorption after 126 s of UV irradiation in Figure 1) was kept in the dark at a constant room temperature for a long period in which spontaneous discharging took place. Figure 6 shows the variation in the spectrum of the hydrosol 3 days later. It can be seen that the LSPR band decreased by about 10% in intensity, was broadened, and shifted by about 5 nm to longer wavelengths (red shift). The intensity and position of the IBM band remained unchanged, which indicated that the number of silver atoms in the hydrosol was retained. The hydrosol was then exposed to UV light (~20 s). The absorption of the hydrosol returned to nearly the initial state. In other words, the LSPR band increased in intensity, narrowed, and shifted to the blue region. The described charging discharging procedure could be repeated. When the hydrosol was stored, nanoparticles lost the excess charge. A decrease in the charge can be caused both by a decrease in the concentration of free electrons and by the partial ionization of adatoms on the surface of a silver particle. The repeated photochemical charging of the hydrosol with electrons from the donor ( radical ion) restored the nanoparticle charge, which resulted in the restoration of the former position and shape of the LSPR band. In these experiments, we used a hydrosol with an invariable composition of silver nanoparticles (size and size distribution).3. Materials and Methods
3.1. Chemicals and Materials
3.2. Synthesis Procedure
3.3. Instrumentation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Chen, F.; Guo, L. Catalytic Activity of Palladium-doped Silver Dilute Nanoalloys for Formate Oxidation from a Theoretical Perspective. Phys. Chem. Chem. Phys. 2019, 21, 22598–22610. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Zeng, C.; Zhou, M.; Chen, Y. Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities. Chem. Rev. 2016, 116, 10346–10413. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.Y.; Zhou, J.; Zou, H.Z.; Li, Y.F.; Gao, P.F.; Huang, C.Z. In situ investigating the size-dependent scattering signatures and sensing sensitivity of single silver nanocube through a multi-model approach. J. Colloid Interface Sci. 2021, 584, 253–262. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, M.; Xiong, Y.; Lim, B.; Xia, Y. Shape-Controlled Synthesis of Pd Nanocrystals and Their Catalytic Applications. Acc. Chem. Res. 2013, 46, 1783–1794. [Google Scholar] [CrossRef]
- Song, J.; Guan, R.; Liu, X.; Jiang, C.; Hou, G. Tuning Local Surface Plasmon Resonance of Silver and Photoluminescence Intensity Enhancement by Adding Copper. J. Phys. Chem. Solids 2018, 120, 44–51. [Google Scholar] [CrossRef]
- Shalabney, A.; Abdulhalim, I. Sensitivity-Enhancement Methods for Surface Plasmon Sensors. Laser Photonics Rev. 2011, 5, 571–606. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, E. Metal Nanoclusters: New Fluorescent Probes for Sensors and Bioimaging. Nano Today 2014, 9, 132–157. [Google Scholar] [CrossRef]
- Tao, Y.; Li, M.; Ren, J.; Qu, X. Metal Nanoclusters: Novel Probes for Diagnostic and Therapeutic Applications. Chem. Soc. Rev. 2015, 44, 8636–8663. [Google Scholar] [CrossRef]
- Webb, J.; Bardhan, R. Emerging Advances in Nanomedicine with Engineered Gold Nanostructures. Nanoscale 2014, 6, 2502–2530. [Google Scholar] [CrossRef]
- Somlyai-Sipos, L.; Baumli, P.; Sycheva, A.; Kaptay, G.; Szőri-Dorogházi, E.; Kristály, F.; Mikó, T.; Janovszky, D. Development of Ag Nanoparticles on the Surface of Ti Powders by Chemical Reduction Method and Investigation of Their Antibacterial Properties. Appl. Surf. Sci. 2020, 533, 147494. [Google Scholar] [CrossRef]
- Hassanzadeh, P. Nanotheranostics against COVID-19: From Multivalent to Immune-Targeted Materials. J. Control Release 2020, 328, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.A.; Biji, P.; Panthalingal, M.K.; Murali, C.; Kulkarni, A.; Rajkumar, S.; Joshi, D.S.; Natarajan, S. Development of Integrated Microfluidic Platform Coupled with Surface-Enhanced Raman Spectroscopy for Diagnosis of COVID-19. Med. Hypotheses 2021, 146, 110356. [Google Scholar] [CrossRef]
- Teengam, P.; Siangproh, W.; Tuantranont, A.; Vilaivan, T.; Chailapakul, O.; Henry, C.S. Multiplex Paper-Based Colorimetric DNA Sensor Using Pyrrolidinyl Peptide Nucleic Acid-Induced AgNPs Aggregation for Detecting MERS-CoV, MTB, and HPV Oligonucleotides. Anal. Chem. 2017, 89, 5428–5435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mie, G. Beiträge zur Optik trüber Medien, speziell kolloidaler Metallösungen. Ann. Phys. 1908, 330, 377–445. [Google Scholar] [CrossRef]
- Van de Hulst, H.C. Light Scattering by Small Particles; Chapman and Hall: London, UK, 1957. [Google Scholar]
- Kerker, M. The Scattering of Light and Other Electromagnetic Radiation; Academic Press: New York, NY, USA, 1969. [Google Scholar]
- Kreibig, U.; Vollmer, M. Optical Properties of Metal Clusters; Springer: Berlin, Germany, 1995. [Google Scholar]
- Qu, Y.; Cheng, R.; Su, Q.; Duan, X. Plasmonic Enhancements of Photocatalytic Activity of Pt/n-Si/Ag Photodiodes Using Au/Ag Core/Shell Nanorods. J. Am. Chem. Soc. 2011, 133, 16730–16733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herne, T.M.; Ahern, A.M.; Garrell, R.L. Surface-Enhanced Raman Spectroscopy of Peptides: Preferential N-Terminal Adsorption on Colloidal Silver. J. Am. Chem. Soc. 1991, 113, 846–854. [Google Scholar] [CrossRef]
- Henglein, A. Colloidal Silver Nanoparticles: Photochemical Preparation and Interaction with O2, CCl4, and Some Metal Ions. Chem. Mater. 1998, 10, 444–450. [Google Scholar] [CrossRef]
- Ershov, B.G.; Abkhalimov, E.V.; Roldughin, V.I.; Rudoy, V.M.; Dement’eva, O.V.; Solovov, R.D. Adsorption of Ozone and Plasmonic Properties of Gold Hydrosol: The Effect of the Nanoparticle Size. Phys. Chem. Chem. Phys. 2015, 17, 18431–18436. [Google Scholar] [CrossRef]
- Muhammed, M.A.H.; Döblinger, M.; Rodríguez-Fernández, J. Switching Plasmons: Gold Nanorod-Copper Chalcogenide Core-Shell Nanoparticle Clusters with Selectable Metal/Semiconductor NIR Plasmon Resonances. J. Am. Chem. Soc. 2015, 137, 11666–11677. [Google Scholar] [CrossRef]
- Yuan, K.; Qin, R.; Yu, J.; Li, X.; Li, L.; Yang, X.; Yu, X.; Lu, Z.; Zhang, X.; Liu, H. Effects of Localized Surface Plasmon Resonance of Ag Nanoparticles on Luminescence of Carbon Dots with Blue, Green and Yellow Emission. Appl. Surf. Sci. 2020, 502, 144277. [Google Scholar] [CrossRef]
- Patel, M.; Pataniya, P.M.; Late, D.J.; Sumesh, C.K. Plasmon-Enhanced Photoresponse in Ag-WS2/Si Heterojunction. Appl. Surf. Sci. 2020, 538, 148121. [Google Scholar] [CrossRef]
- Celebrano, M.; Kukura, P.; Renn, A.; Sandoghdar, V. Single-molecule imaging by optical absorption. Nat. Photonics 2011, 5, 95–98. [Google Scholar] [CrossRef]
- Ringe, E.; Sharma, B.; Henry, A.-I.; Marks, L.D.; Van Duyne, R. Single nanoparticle plasmonics. Phys. Chem. Chem. Phys. 2013, 15, 4110–4129. [Google Scholar] [CrossRef]
- Murali, G.; Vattikuti, S.V.P.; Kshetri, Y.K.; Lee, H.; Modigunta, J.K.R.; Reddy, C.S.; Park, S.; Lee, S.; Poornaprakash, B.; Lee, H.; et al. Near-infrared-activated Z-scheme NaYF4:Yb/Tm@Ag3PO4/Ag@g-C3N4 photocatalyst for enhanced H2 evolution under simulated solar light irradiation. Chem. Eng. J. 2021, 421, 129687. [Google Scholar] [CrossRef]
- Vattikuti, S.V.P.; Nagajyothi, P.C.; Devarayapalli, K.C.; Yoo, K.; Nam, N.D.; Shim, J. Hybrid Ag/MoS2 nanosheets for efficient electrocatalytic oxygen reduction. Appl. Surf. Sci. 2020, 526, 146751. [Google Scholar] [CrossRef]
- Nagajyothi, P.C.; Reddy, L.V.; Devarayapalli, K.C.; Vattikuti, S.V.P.; Wee, Y.J.; Shim, J. Environmentally Friendly Synthesis: Photocatalytic Dye Degradation and Bacteria Inactivation Using Ag/f-MWCNTs Composite. J. Clust. Sci. 2021, 32, 711–718. [Google Scholar] [CrossRef]
- Abkhalimov, E.V.; Ershov, V.A.; Ershov, B.G. “Pure” Silver Hydrosol: Nanoparticles and Stabilizing Carbonate Ions. J. Nanopart. Res. 2019, 21, 93. [Google Scholar] [CrossRef]
- Kai, T.; Zhou, M.; Johnson, S.; Ahn, H.S.; Bard, A.J. Direct Observation of C2O4•− and CO2•− by Oxidation of Oxalate within Nanogap of Scanning Electrochemical Microscope. J. Am. Chem. Soc. 2018, 140, 16178–16183. [Google Scholar] [CrossRef]
- Anderson, G.K.; Lumetta, G.J.; Siria, J.W. Photochemical Reactions of Diphosphineplatinum(II) Oxalate Complexes. J. Organomet. Chem. 1992, 434, 253–259. [Google Scholar] [CrossRef]
- Wardman, P. Reduction potentials of one-electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef] [Green Version]
- Henglein, A. Remarks on the electrochemical potential of small silver clusters in aqueous solution. Ber. Bunsenges. Phys. Chem. 1990, 94, 600–603. [Google Scholar] [CrossRef]
- Gupta, S.; Prakash, R. Photochemically Assisted Formation of Silver Nanoparticles by Dithizone, and Its Application in Amperometric Sensing of Cefotaxime. J. Mater. Chem. C 2014, 2, 6859–6866. [Google Scholar] [CrossRef]
- Ershov, B.G.; Janata, E.; Henglein, A.; Fojtic, A. Silver Atoms and Clusters in Aqueous Solution: Absorption Spectra and The Particles Growth in Absence of Stabilizing Ag+ Ions. J. Phys. Chem. 1993, 97, 4589–4594. [Google Scholar] [CrossRef]
- Ershov, B.G. Short-Lived Metal Clusters in Aqueous Solutions: Formation, Identification, and Properties. Russ. Chem. Bull. 1999, 48, 1–15. [Google Scholar] [CrossRef]
- Belloni, J.; Marignier, J.-L.; Mostafavi, M. Mechanisms of Metal Nanoparticles Nucleation and Growth Studied by Radiolysis. Radiat. Phys. Chem. 2020, 169, 107952. [Google Scholar] [CrossRef]
- Russel, W.B.; Saville, D.A.; Schowalter, W.R. Colloidal Dispersions, 1st ed.; Cambridge University Press: Cambridge, UK, 1989; pp. 258–309. [Google Scholar] [CrossRef]
- Sánchez-Iglesias, A.; Claes, N.; Solís, D.M.; Taboada, J.M.; Bals, S.; Liz-Marzán, L.M.; Grzelczak, M. Reversible Clustering of Gold Nanoparticles under Confinement. Angew. Chem. Int. Ed. Agew. Chem. 2018, 57, 3183–3186. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Takezako, T.; Yamamoto, K.; Serizawa, T.; Akashi, M. Poly(N-vinylisobutyramide)-Stabilized Platinum Nanoparticles: Synthesis and Temperature-Responsive Behavior in Aqueous Solution. Colloids. Surf. A Physicochem. Eng. Asp. 2000, 169, 107–116. [Google Scholar] [CrossRef]
- Mavroyannis, C. The interaction of neutral molecules with dielectric surfaces. Mol. Phys. 1963, 6, 593–600. [Google Scholar] [CrossRef]
- Yagodovskii, V.D. Modifying the Catalytic and Adsorption Properties of Metals and Oxides. Russ. J. Phys. Chem. A 2015, 89, 1758–1767. [Google Scholar] [CrossRef]
- Von Vol’kenshtein, F.F. The electronic theory of catalysis on semiconductors. Angew. Chem. 1964, 76, 696. [Google Scholar] [CrossRef]
- Liao, G.; Fang, J.; Li, Q.; Li, S.; Xu, Z.; Fang, B. Ag-Based Nanocomposites: Synthesis and Applications in Catalysis. Nanoscale 2019, 11, 7062–7096. [Google Scholar] [CrossRef]



 discharging» procedure: 1—after formation; 2—after aging for 3 days; 3—after UV irradiation for 20 s. Solution: Ag+ (3 × 10−4 mol L−1) and (5 × 10−4 mol L−1). Optical length is 5 mm.
discharging» procedure: 1—after formation; 2—after aging for 3 days; 3—after UV irradiation for 20 s. Solution: Ag+ (3 × 10−4 mol L−1) and (5 × 10−4 mol L−1). Optical length is 5 mm.
discharging» procedure: 1—after formation; 2—after aging for 3 days; 3—after UV irradiation for 20 s. Solution: Ag+ (3 × 10−4 mol L−1) and (5 × 10−4 mol L−1). Optical length is 5 mm.
discharging» procedure: 1—after formation; 2—after aging for 3 days; 3—after UV irradiation for 20 s. Solution: Ag+ (3 × 10−4 mol L−1) and (5 × 10−4 mol L−1). Optical length is 5 mm.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ershov, V.; Tarasova, N.; Ershov, B. Evolution of Electronic State and Properties of Silver Nanoparticles during Their Formation in Aqueous Solution. Int. J. Mol. Sci. 2021, 22, 10673. https://doi.org/10.3390/ijms221910673
Ershov V, Tarasova N, Ershov B. Evolution of Electronic State and Properties of Silver Nanoparticles during Their Formation in Aqueous Solution. International Journal of Molecular Sciences. 2021; 22(19):10673. https://doi.org/10.3390/ijms221910673
Chicago/Turabian StyleErshov, Vadim, Natalia Tarasova, and Boris Ershov. 2021. "Evolution of Electronic State and Properties of Silver Nanoparticles during Their Formation in Aqueous Solution" International Journal of Molecular Sciences 22, no. 19: 10673. https://doi.org/10.3390/ijms221910673
APA StyleErshov, V., Tarasova, N., & Ershov, B. (2021). Evolution of Electronic State and Properties of Silver Nanoparticles during Their Formation in Aqueous Solution. International Journal of Molecular Sciences, 22(19), 10673. https://doi.org/10.3390/ijms221910673