1. Introduction
The placenta is a highly specialized fetal organ responsible for supporting growth and development of the fetus in utero. It forms an immune and physical barrier between the mother and fetus and provides metabolic, transport, and endocrine functions [
1]. Each of these functions exact a metabolic cost so it comes with no surprise that the placenta has an extraordinarily high metabolic rate, accounting for approximately 40% of the total oxygen consumed by the fetus and placenta combined [
2]. The chorionic villi of the placenta, which contain fetal capillaries, are bathed with maternal blood and it is here on its outer surface where oxygen and nutrient uptake and transfer between maternal and fetal circulations occurs across the syncytiotrophoblast (ST) cell layer, a multinuclear syncytium 13 m
2 in area at term. The ST also synthesizes and secretes large amounts of peptide and steroid hormones [
3]. The ST cell layer is formed from underlying mononucleated villous cytotrophoblast (CT) cells which constantly proliferate in vivo and fuse into the fully differentiated multinucleated ST. Formation of ST can be recapitulated in vitro by culture of isolated CT cells which, although they cannot proliferate in-vitro, spontaneously fuse, and differentiate into ST [
4]. As the majority of energy requiring placental functions (nutrient/waste transfer and hormone production) are carried out by the ST layer, it has traditionally been assumed that the ST is more metabolically active than CT cells, although recent studies suggest a more complex metabolic status [
5].
Trophoblast cells, like other cells, produce chemical energy in the form of adenosine triphosphate (ATP) mainly via oxidative phosphorylation. Glycolysis, the TCA cycle, and fatty acid oxidation all result in formation of energy-rich NADH and FADH2 which donate their electrons into the electron transport chain (ETC) for shuttling down a chain of protein complexes while protons are pumped out of the mitochondrial matrix into the intermembrane space, creating a proton gradient across the inner mitochondrial membrane. In the final step of oxidative phosphorylation, protons travel down their concentration gradient through complex 5 (ATP synthase) and phosphorylate adenosine diphosphate (ADP), creating ATP. In most cells, breakdown of glucose via glycolysis and formation of acetyl CoA is the primary pathway that provides metabolites for oxidative phosphorylation. However, cells can switch to other metabolites in either the absence of glucose or excess of fatty acids. This ability to switch metabolite substrates depending on nutrient availability is called metabolic flexibility and is a crucial cell survival mechanism when faced with sub-optimal metabolic conditions. We recently showed that in addition to glucose, trophoblast cells can also utilize amino acids, e.g., glutamine, and fatty acids for generation of ATP via the ETC and that the proportions of each used can change with metabolic condition, e.g., obesity or gestational diabetes [
6]. Since, the proliferative CT and differentiated ST have different role in terms of transport, metabolism, and steroid and peptide hormone production, we hypothesized that they might differ in their use of fuel sources and metabolic flexibility.
Cytotrophoblast cells share many similarities with cancer cells which proliferate, migrate, and invade tissues to establish a continuous nutrient supply to support the development of a tumor. In-vivo, CT proliferate, migrate, and invade (as extravillous trophoblast) endometrial tissue to establish a nutrient supply but also as villous cytotrophoblast undergo fusion to form ST [
7]. Otto Warburg described a phenomenon, the Warburg effect, where cancer cells preferentially utilize glycolysis in the presence of oxygen (aerobic glycolysis) to produce the bulk of their ATP requirement, unlike normal body cells that generate ATP through mitochondrial respiration using metabolites from glycolysis, the TCA cycle and β-oxidation of fatty acids [
8,
9,
10]. Based on the similarities between CT cells and cancer cells, we therefore hypothesized that CT might have higher glycolytic function, compared to ST cells.
There is now an overwhelming body of data indicating a sexual dimorphism exists in placental physiology underpinned by a sex-dependent difference in placental gene expression [
11,
12,
13,
14]. This may be linked to the different fetal growth and survival strategies where male fetuses grow larger than female fetuses but are therefore at a greater risk of suffering from adverse pregnancy outcomes if maternal nutrition and placental function are not optimal [
15,
16,
17]. We have previously reported maternal obesity, preeclampsia, and gestational diabetes mellitus to be associated with sexually dimorphic effects on energetics and autophagy in the placenta, and have also shown sexual dimorphism in placental antioxidant enzyme activity [
6,
18,
19,
20]. In this study we also investigated if fetal sex had effects on glycolytic and mitochondrial metabolism in CT vs. ST cells from women of a healthy weight.
3. Discussion
Cell differentiation and differentiated functions are highly energy-consuming processes [
26]. Several studies have reported significant modifications in cellular bioenergetics as progenitor cells differentiate [
27,
28]. However, the shifts in mitochondrial and cellular bioenergetic pathways during ST differentiation are not well understood. Additionally, while sexual dimorphism in placental function has been reported, the effect of fetal sex on CT and ST bioenergetics and mitochondrial function has been largely unexplored.
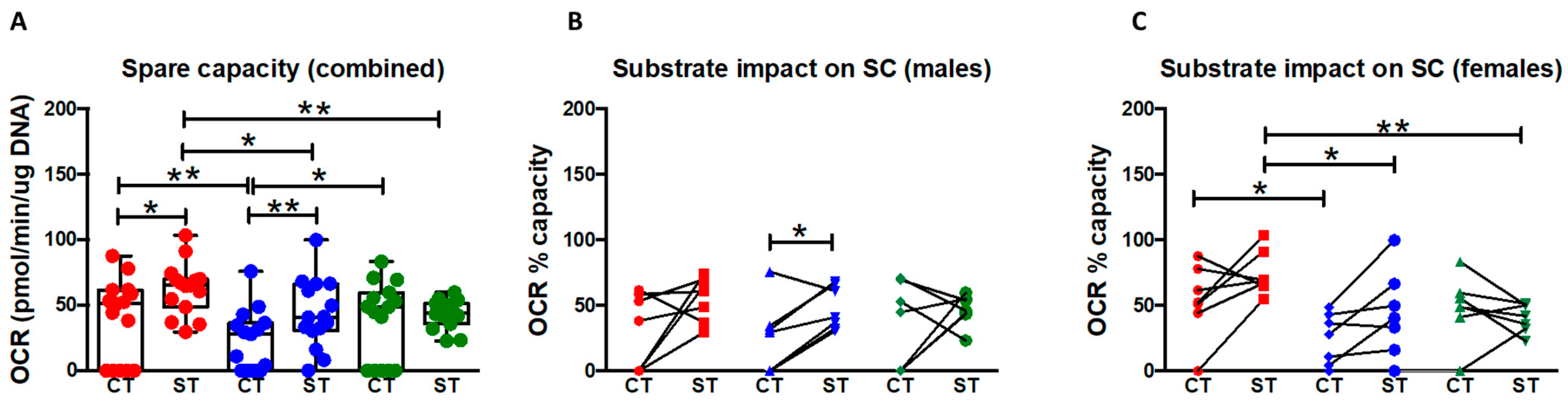
The present study provides several lines of evidence that CT and ST differ significantly in their metabolic phenotypes. CT have equivalent basal glycolysis but a higher glycolytic capacity and reserve than ST whereas ST have a higher mitochondrial respiratory function than CT under both basal conditions and conditions mimicking physiological stress and increased energy demand. ST also appear to utilize glucose and glutamine more efficiently than CT whereas the two cell types show no difference in their ability to use fatty acids to generate energy. Further, both CT and ST show a distinct sexual dimorphism in their energy metabolism with male ST having lower glycolytic capacity and reserve compared to their CT and with female ST having comparable glycolytic capacity, but lower reserve than their CT. On the other hand, both male and female ST have higher mitochondrial respiration (compared to their respective CT) for all parameters except basal respiration which is not different in male ST vs. CT and proton leak which is not different in female ST vs. CT.
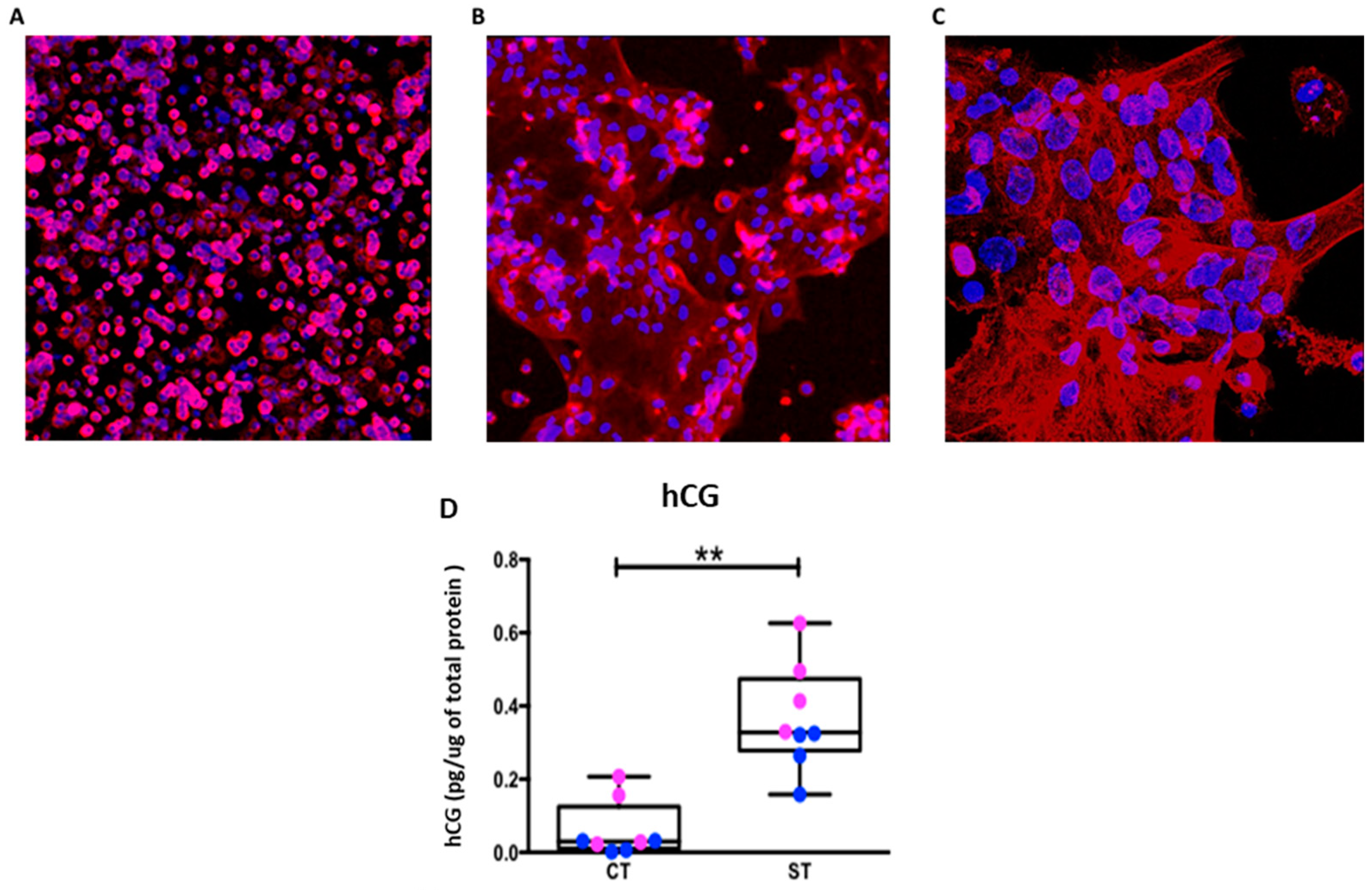
In the current study, we used isolated term CT cells cultured for 24 h and 96 h representing progenitor CT cells and syncytialized ST, respectively. Syncytialization over this timeframe was confirmed by staining for the trophoblast marker CK-7 and for nuclear aggregates and measuring hCG secretion as shown in
Figure 1. We then assessed glycolytic function and mitochondrial respiration in both CT and ST using the Seahorse assay. The assay measures the rate of depletion of O
2 from the media, “oxygen consumption rate” (OCR) and protons released into the media, “extracellular acidification rate” (ECAR) as indicators of mitochondrial oxidative phosphorylation and glycolytic function, respectively.
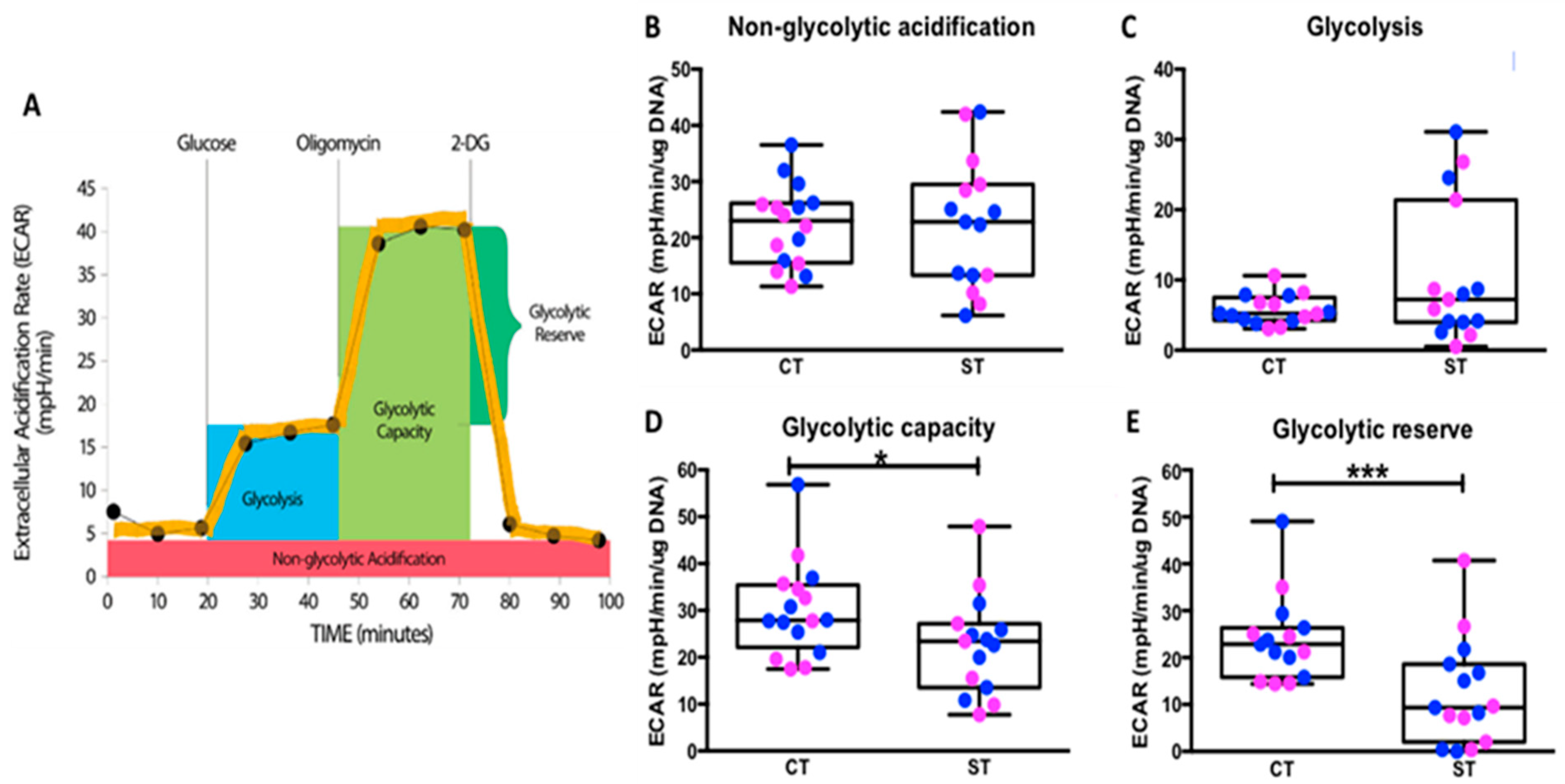
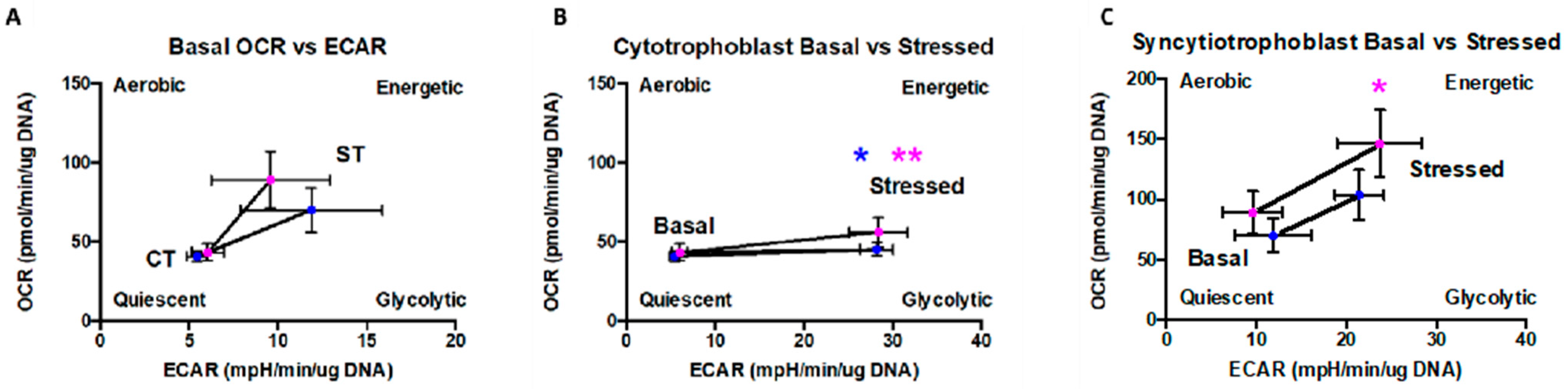
Although, there was no statistical difference in the basal rate of glycolysis between CT and ST, we observed that CT had a significantly higher glycolytic capacity and reserve capacity than ST (
Figure 2). Kolahi et al. previously reported significantly higher basal glycolysis rate in CT but no difference in the glycolytic reserve. However, their study was performed with media containing pyruvate, a product in the glycolysis pathway which upon breakdown releases lactate and proton measured as ECAR in the Seahorse assay. The presence of pyruvate would thus affect the baseline measurements performed in the study and may account for the differences seen in this study. Higher glycolytic capacity and reserve in CT suggests that under physiologically energy demanding conditions, CT but not ST could rapidly increase their glycolytic function to survive. From a bioenergetic perspective, glycolysis is not as efficient as mitochondrial respiration for ATP production with 2 vs. 36 ATP molecules being generated per glucose molecule respectively. However, it is generally accepted that proliferating progenitor cells, such as cytotrophoblast, are glycolytic in nature [
29,
30,
31,
32] and it is the preferred way to generate ATP in cancer cells described as the Warburg effect [
8].
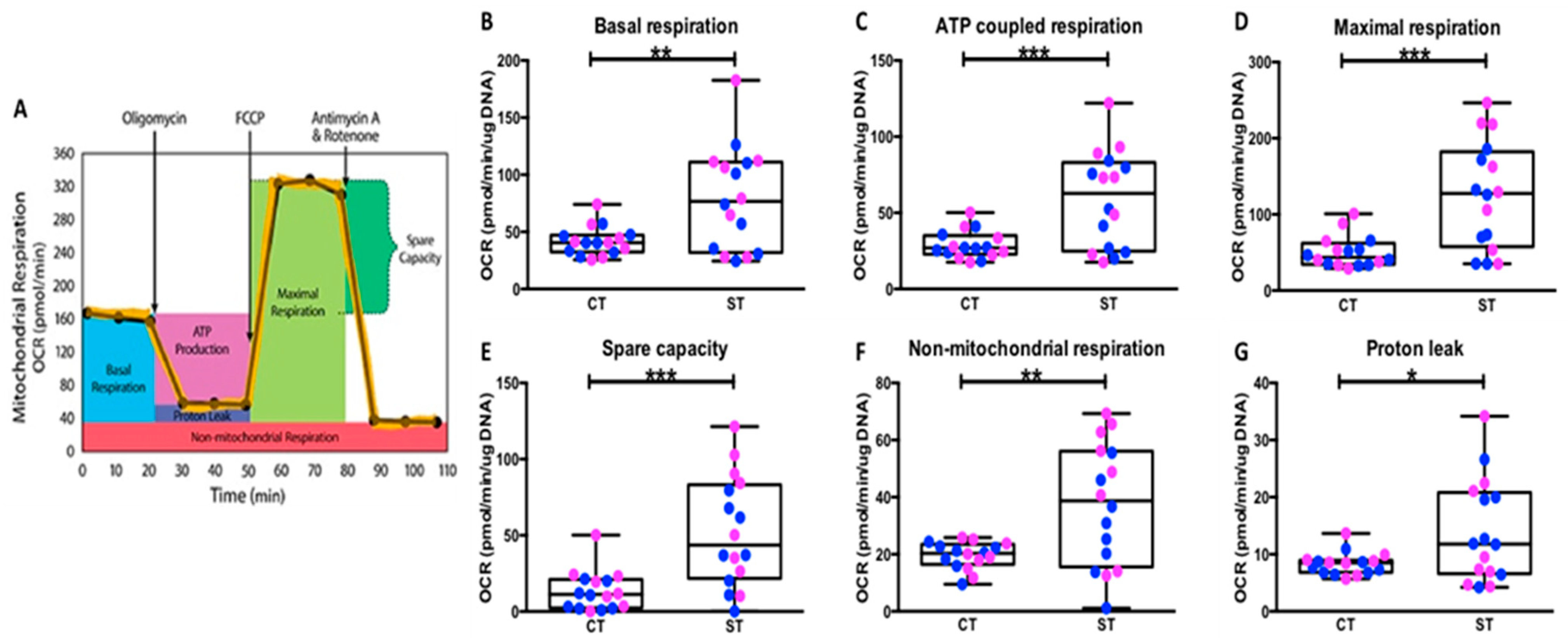
We also observed that the differentiated ST have significantly higher levels of both basal mitochondrial respiration and higher reserve capacity (
Figure 3). ST were also more flexible in their fuel dependency and were able to better utilize glucose and glutamine for energy generation under conditions mimicking physiological stress and energy demand (
Figure 5). Studies assessing bioenergetics of neuronal, osteogenic, and erythroid differentiation also made similar observations where differentiation was accompanied by, and required, a shift towards mitochondrial respiration [
27,
28,
32,
33]. Interestingly, previous studies on mitochondrial function in human placenta have reported observations contradictory to ours. Fisher et al. reported reduced oxygen consumption, mitochondrial respiration, and ATP production in mitochondria of ST vs. CT [
34]. However, these studies use intact mitochondria isolated from whole placental tissue by homogenization followed by density gradient purification. This separates bigger mitochondria from small mitochondria which the authors refer to as cyto–mito and syncytio–mito, respectively, based on previous studies that reported large circular mitochondria in cytotrophoblast and small irregular shaped ones in syncytiotrophoblast [
35]. As prepared this “cyto–mito” fraction will also contain normal sized mitochondria found in other cell types of the placenta such as stromal and endothelial cells and hence does not only represent cytotrophoblast mitochondrial respiration. Our use of intact cells (individual or syncytialized) better mimics the physiological milieu which may have impact on availability of substrates, membrane potential and proton gradients all crucial for mitochondrial function. Similarly, Kolahi et al. reported reduced OCR in ST contradictory to our observations. However, their study was focused on fatty acid metabolism and assays contained high concentrations of saturated long chain and monounsaturated fatty acids which could account for the observed differences in OCR seen.
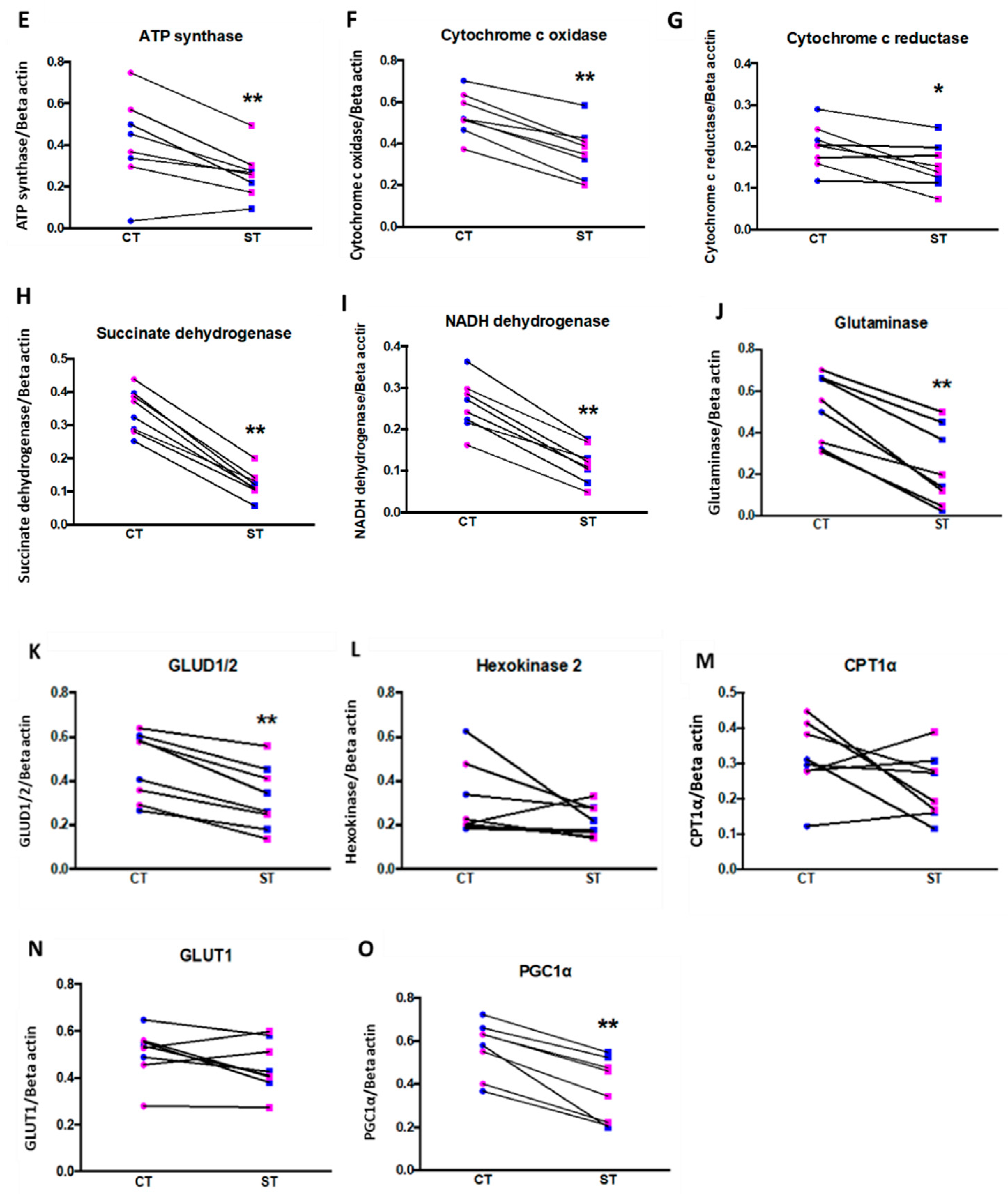
The differentiated functions of syncytiotrophoblast means their mitochondria have several functions distinct from those of the proliferative cytotrophoblast, particularly steroidogenesis. Martinez et al. reported that ST mitochondria have significantly increased cytochrome P450 expression an enzyme responsible for catalyzing the first step in steroidogenesis, highlighting the role of syncytiotrophoblast in hormone synthesis [
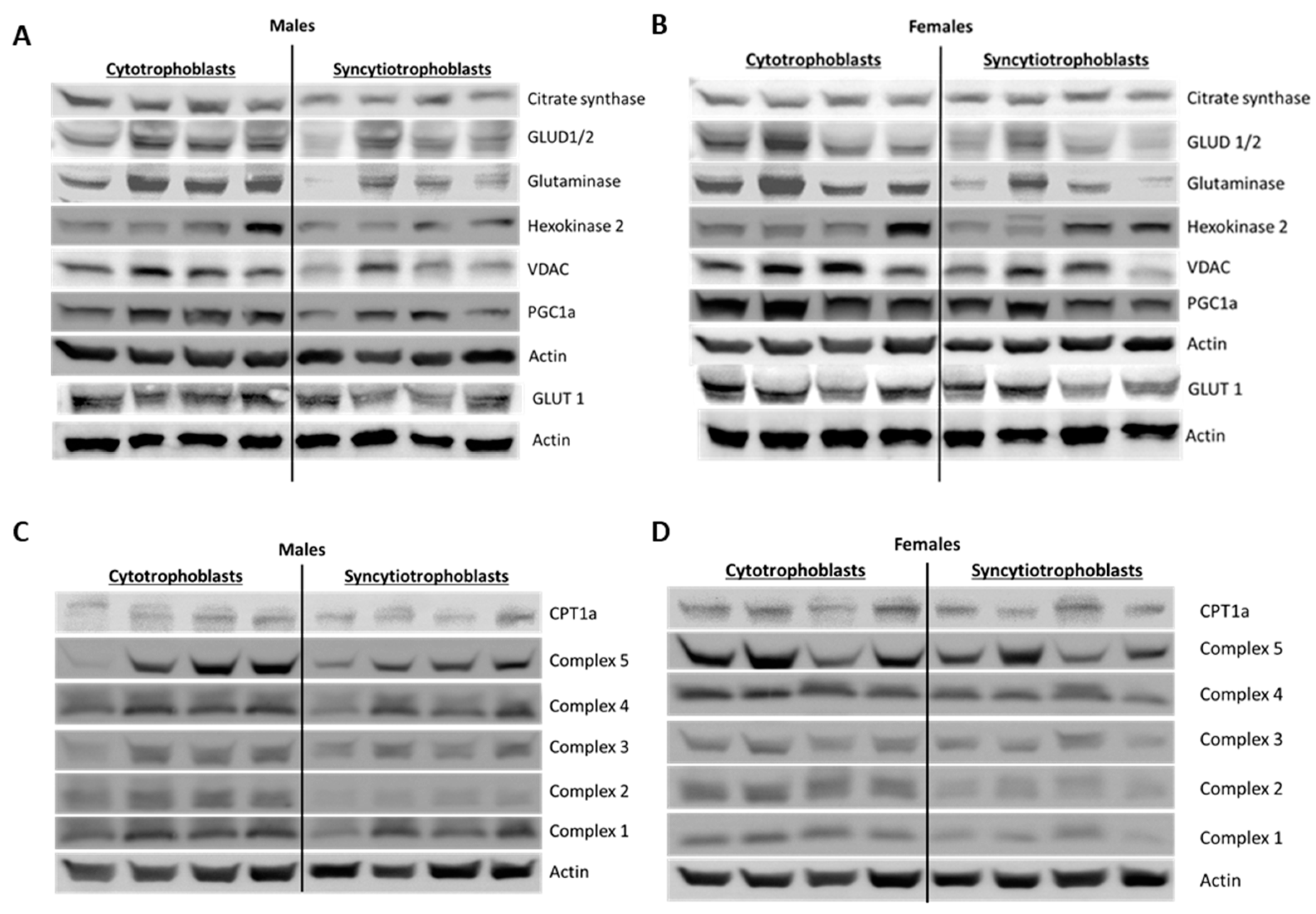
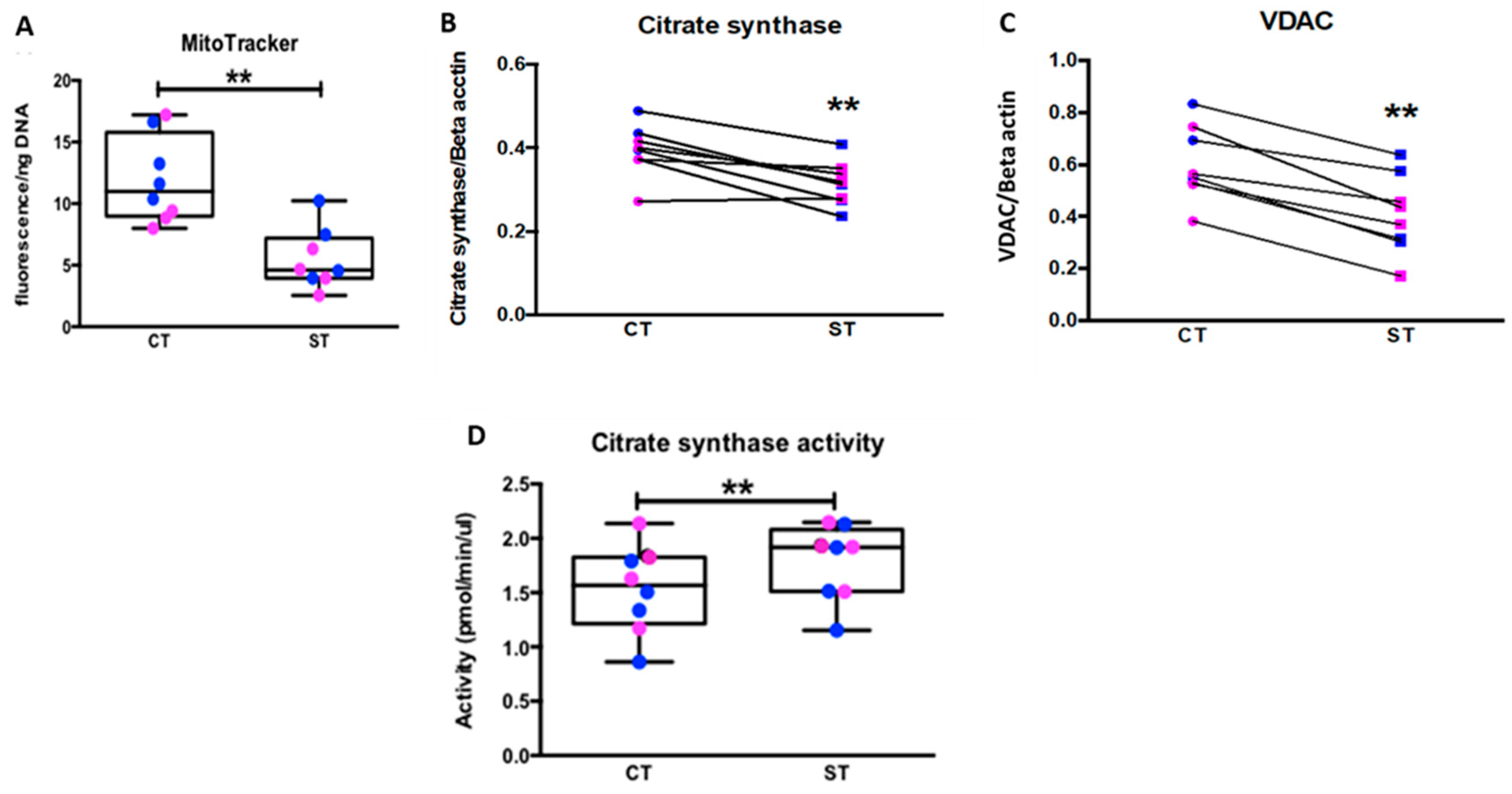
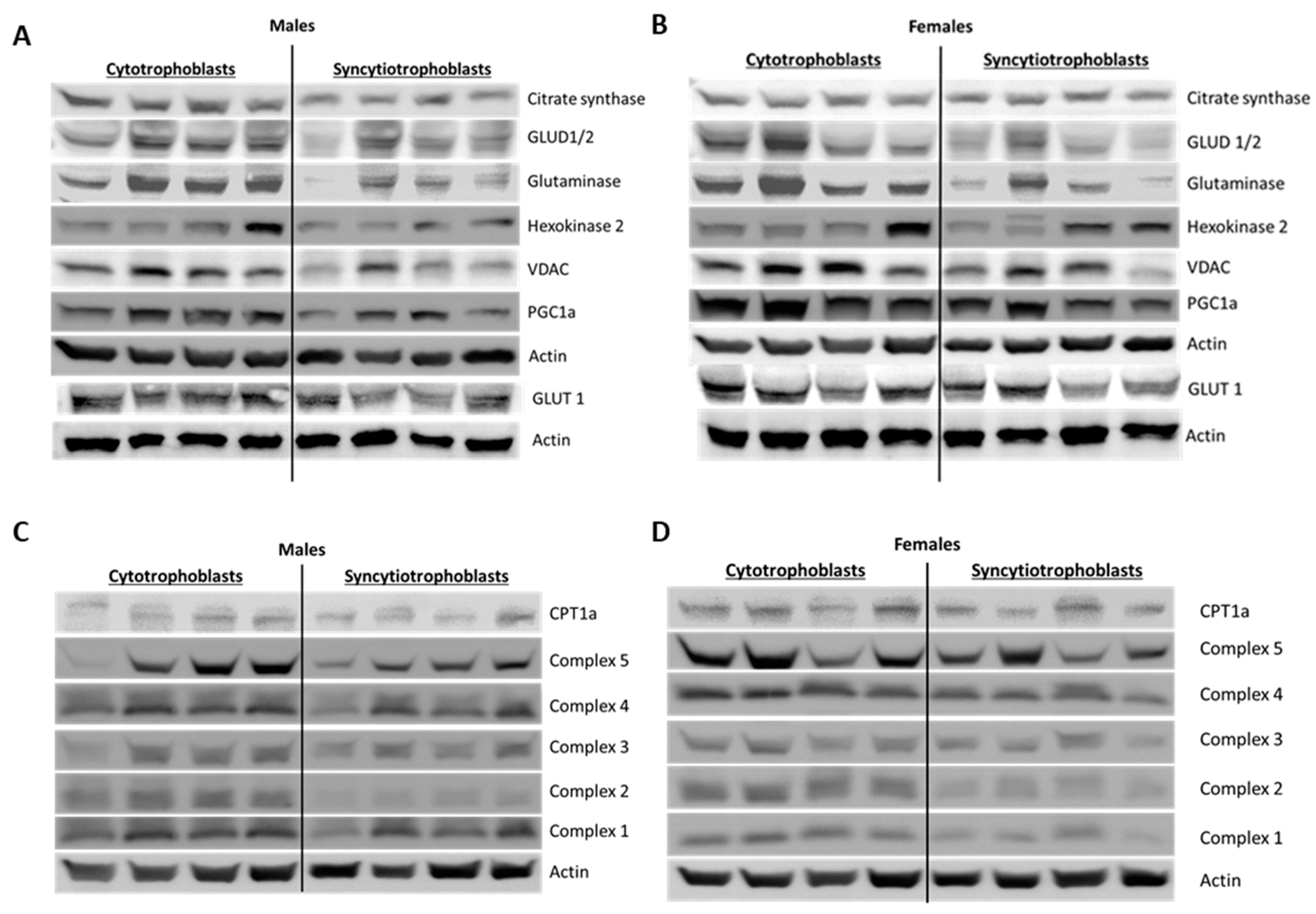
35]. Similarly, Fisher et al. reported increased CYP11A1 expression and increased progesterone production in ST mitochondria. While we did not assess steroidogenesis, we measured citrate synthase activity as a marker for mitochondrial activity. Like the above studies, we found that ST had higher citrate synthase activity, again implying greater mitochondrial function. However, our assessment showed significantly reduced mitochondrial content, as well as decreased protein expression of citrate synthase, VDAC, mitochondrial ETC complexes and other enzymes involved in mitochondrial respiration seemingly counter-intuitive to our results on mitochondrial function. (
Figure 6 and
Figure 7,
Supplementary Figures S4 and S5). We assessed mitochondrial content using the specific dye MitoTracker
TM deep red and normalizing it to the nuclear DNA content determined by the Hoechst DNA stain. The dye accumulates in active mitochondria and is used for mitochondrial tracking in live cells. Its correlation to mitochondrial mass is, however, not clearly defined. Complementing our results with additional assays such as quantifying cardiolipin content or ratio of mitochondrial DNA to nuclear DNA might provide a better idea of mitochondrial mass in ST [
36].
Several studies have also highlighted how mitochondrial ultrastructure and cristae organization play a critical role in its function (comprehensive review in [
37]). Detailed ultrastructure studies using cryo-electron tomography have suggested that ATP synthase dimers preferentially localize in (and even aid in formation of) the curved regions of the cristae, such as the tips, whereas the ETC complexes are in less curved regions, such as the stalks [
38,
39,
40,
41]. These observations suggest that the cristae structure is finely tuned to support the energetic needs of the respective cells. Increased number of cristae could improve mitochondrial function but substantially reduce the available matrix space for metabolic enzymes [
37] which would explain the reduced expression but increased function observed in our study. Recently, Cagiliati et al. showed that cristae structure drives the assembly of respiratory chain super complexes (RCS) (consisting of ATP synthase and ETC enzymes) on their surface and therefore affect the efficiency of mitochondrial respiration [
42]. They further reported that mitochondrial fusion protein OPA1 (Optic Atrophy Protein 1) was crucial for cristae organization and structure, assembly of the RCS, and respiratory function. Increased expression of another fusion protein mitochondrial fusion protein-2 (Mfn2) has also been correlated to increased mitochondrial function further emphasizing the correlation between mitochondrial ultrastructure, function and ‘mitochondria-shaping’ proteins that regulate the organelle’s fission and fusion [
43]. We propose that a detailed analysis of ST and CT mitochondrial cristae structure and studying expression of mitochondrial shaping proteins might provide further insights into the above results.
An important aim for the study was to assess sexual dimorphism, if any, in placental mitochondrial function. Sexual dimorphism in fetal and placental development as well as placental gene expression has been reported before [
14,
44]. Male fetuses are known to be bigger and heavier than females with equivalent placental weight as observed in our study [
21,
22,
45] and are therefore considered more efficient, but vulnerable to gestational stressors. Placental responses to environmental stress, such as hyperlipidemia and asthma, are influenced by fetal sex wherein female fetus growth is limited increasing the chances of survival, but male fetuses continue growing normally, increasing their chances of a poor outcome in case of acute exacerbation of the stressors [
11,
16,
46]. We have previously shown that indeed male and female syncytiotrophoblast show differences in metabolic flexibility in use of glucose, glutamine, or fatty acids when they are exposed to different intrauterine environments, i.e., from normal weight, obese, or type A2 gestational diabetes, with male trophoblasts being more severely affected [
6,
14,
47]. To the best of our knowledge, this is the first study assessing sexual dimorphism in basal mitochondrial function and response to stressors as CT from normal pregnancies differentiate to ST. We report that when CT differentiate into ST, they reduce their glycolytic capacity with a more pronounced reduction in male ST. On the other hand, while ST from both sexes have an efficient and higher rate of mitochondrial respiration over their respective CT, this is more pronounced in female ST. The reduced capability of male trophoblasts (ST) to shift to the more efficient mitochondrial oxidation suggests that they might not be equipped at coping with situations that require an increase in energy production. This is further evident in their reduced metabolic flexibility in using either glucose, glutamine, or fatty acids as substrates. Our results thus provide evidence for sexual dimorphism on the cellular, metabolic, and functional level in placental trophoblast. Collectively, our results fortify the notion that male placentas function at near their maximum limit, and if presented with a stress, may not be able to increase energy production and are at a higher risk of suffering from adverse pregnancy outcomes [
16,
46].
4. Materials and Methods
4.1. Ethical Approval of the Study
Placentae were collected from the Labor and Delivery Unit at Oregon Health and Science University into a tissue repository under a protocol approved by the Institutional Review Board with informed consent from the patients. Fetal weight was recorded. All tissues and clinical data were de-identified before being made available to the investigative team.
4.2. Collection of Placental Tissues
Placentae were collected and weighed immediately following Cesarean section from uncomplicated pregnancies at term in the absence of labor from patients with either a male or a female fetus and a pre-pregnancy BMI in the normal weight range (NW, BMI = 18.5–24.9, n = 8 male, 8 female). Exclusion criteria included overweight or obesity, multifetal gestation, gestational diabetes mellitus, preeclampsia, chronic inflammatory diseases, use of tobacco/illicit drugs, and recent bariatric surgery. Five random samples of tissue (~80 g) were collected from each placenta and placed in PBS to be transported back to the lab.
4.3. Primary Cell Isolation and Culture
The chorionic plate and decidua were removed from each randomly isolated placental sample, leaving only villous tissue, which was thoroughly rinsed in PBS to remove excess blood. Primary cytotrophoblast were isolated from villous tissue using a protocol adapted from Eis et al. [
48] using trypsin/DNAse digestion followed by density gradient purification. Isolated cytotrophoblast cells were then frozen in freezing media (10% DMSO in FBS) and stored in liquid nitrogen until usage.
Cytotrophoblast cells were rapidly thawed in a 37 °C water bath and immediately diluted in Iscove’s modified Dulbecco’s medium (25 mM glucose, 4mM glutamine, and 1 mM pyruvate (ThermoFisher Scientific, Waltham, MA, Cat. #12440053) supplemented with 10% FBS and 1% penicillin/streptomycin (complete media) (ThermoFisher Scientific, Cat. #MT35010CV, #15140 respectively). Cells were centrifuged at 1000 × RCF for 10 min and re-suspended in fresh complete media. Trophoblast cells were plated in a 96-well Seahorse plate (100,000 cells/well) in 100µL of complete media for glycolysis and respiration measurements or plated in a 6-well plate (4 million cells/well) in 2 mL complete media for protein expression studies. The following day, additional complete media was added to each well. All studies were performed at two time points—24 hrs (labelled as cytotrophoblast/CT) and 96 hrs to allow fusion and formation of syncytiotrophoblast (ST). ST formation was confirmed by staining the cells for the trophoblast marker Cytokeratin-7.
4.4. Immunocytochemistry
CT cells were plated on circular coverslips at a cell density of 1.5 million cells/mL in a volume of 0.3 mL. CT (24 h) and ST (96 h) were fixed in ice-cold methanol for 10 min at −20 °C and washed three times with cold PBS. Cells were then blocked in 3% BSA diluted in PBS + 0.1% Tween 20 (PBST) for 2 hrs at room temperature. Cytokeratin-7 primary antibody (1:100) (ThermoFisher Scientific, Waltham, MA, Cat. #MA1-06315) was incubated overnight at 4 °C. Following primary antibody incubation, cells were washed three times in PBST and incubated with anti-mouse Alexa fluor 555 secondary antibodies (1:1000) (Thermofisher Scientific, Cat. #A31570) for 3 hrs at room temperature. Cells were then washed three times in PBST followed by Hoechst 33342 (1:10,000) counterstain for 30 s. Cells were washed three more times with PBST and mounted on slides using SlowFade Diamond Antifade Mountant (Thermofisher Scientific, Cat. #S36972). After allowing to set for 24 hr, cover-slips were sealed in place using clear nail polish. Images were captured using a Zeiss LSM 880 confocal microscope and processed using ImageJ Software (Bethesda, Rockville, MD, USA).
4.5. Metabolic Analysis and Cellular Bioenergetics Measurements
CT and ST bioenergetics were measured using Seahorse XF Analyzer (Agilent Technologies, Santa Clara, CA, USA) assays following the manufacturer’s protocol outlined briefly below. For all assays, 100,000 cells were plated per well in a 96-well Seahorse assay plate.
4.5.1. Mitochondrial Stress Test
This was used to assess mitochondrial function parameters: basal respiration, ATP production-coupled respiration, maximal respiration, spare capacity, and non-mitochondrial respiration using the Seahorse XF Cell Mito Stress Test (Agilent Technologies, Cat # 103010). One hr prior to running the mitochondrial stress test, complete media was exchanged with basal Seahorse media supplemented with glucose, glutamine, and pyruvate to match culture conditions. The cells were then allowed to equilibrate in a non-CO
2 37 °C incubator for 1 hr before the first rate measurement, called ‘Basal respiration rate’, and is defined as the initial oxygen consumption rate (OCR). This represents the total mitochondrial respiration rate. After measuring the baseline, 75 µL of oligomycin (ATP synthase inhibitor), FCCP (protonophore), and a combination of rotenone (NADH dehydrogenase inhibitor) and antimycin A (cytochrome c reductase inhibitor) solutions were sequentially added to each well at a 1 µM working concentration to determine the ATP coupled respiration, maximum respiration, and non-mitochondrial oxygen consumption rates, respectively. The ATP coupled response is defined the rate of oxygen consumption linked to ATP production and is calculated as the difference between the basal OCR and the OCR after oligomycin injection. Maximal respiratory rate was calculated as the difference between the OCR after uncoupled addition (FCCP) and the lowest OCR reached after oligomycin addition. Spare (reserve) capacity is calculated as the difference between OCR after FCCP and basal respiration and represents the spare metabolic potential thought to guard against stressful conditions (
Figure 3A) [
49].
4.5.2. Modified Mitochondrial Stress Test
An adapted version of the mitochondrial stress test described above that was used to examine substrate impact on spare capacity by determining the rate of oxidation of a single substrate (glucose, glutamine, or long-chain fatty acids) while the other two substrate pathways are blocked. The pathway inhibitors used were 2 µM UK5099 (inhibitor of glucose oxidation, blocks action of mitochondrial pyruvate carrier (MPC), which converts glucose to pyruvate), 3 µM BPTES (inhibitor of glutamine oxidation, blocks glutaminase (GSL1), which converts glutamine to glutamate) and 4 µM Etomoxir (inhibitor of long-chain fatty acid oxidation, which blocks carnitine palmitoyltransferase 1 alpha (CPT1α). The cells were treated with either a combination of two pathway inhibitors or a combination of all three pathway inhibitors followed by the mitochondrial stress test ETC inhibitors to calculate the capacity of each pathway using the following formula.
4.5.3. Glycolysis Stress Test
This was used to assess glycolytic function parameters: glycolysis, glycolytic capacity, glycolytic reserve, and non-glycolytic acidification using the Seahorse XF Glycolysis Stress kit (Agilent Technologies, Cat # 103020). One hr prior to running the glycolysis stress test, the cell culture medium was exchanged with basal Seahorse media supplemented with glutamine (excluding glucose and pyruvate) to match culture conditions. The cells were then allowed to equilibrate in a non-CO2 37 °C incubator for 1 hr before the first rate measurement called ‘Non-glycolytic acidification’ and is defined as the extracellular acidification rate (ECAR) that is not attributed to glycolysis. After measuring Non-glycolytic acidification rate, 75 µL of glucose (converted to pyruvate through glycolysis), Oligomycin (ATP synthase inhibitor), and 2-deoxyglucose-glucose (competitive inhibitor of hexokinase, the first enzyme in the glycolysis pathway) solutions were sequentially added to each well at a 10 mM glucose, 1 µM Oligomycin and 50 mM 2-deoxy-glucose working concentration to determine the rate of glycolysis under basal conditions, maximum glycolytic capacity and to confirm the initial ECAR measured is due to glycolysis, respectively. Glycolysis is defined as the glucose-induced increase in ECAR and is calculated by subtracting non-glycolytic acidification from the highest ECAR measurement following the addition of glucose. Maximum glycolytic capacity was calculated as the difference between the highest ECAR measurement during non-glycolytic acidification and the highest ECAR measurement after the addition of Oligomycin. Glycolytic reserve was calculated as the difference between ECAR after glucose and after oligomycin.
Data from all Seahorse assays were normalized to cellular DNA content measured immediately after the assay was finished. Hoescht 33342 dye (Thermofisher Scientific, Cat. #H1399) was added to each well (1:1000 final concentration) and incubated for 30 min at 37 °C with constant shaking. Fluorescence was measured using a plate reader (excitation 350 nm emission 461 nm).
4.6. Protein Extraction and Western Blotting
Proteins were extracted from cultured trophoblast cells (after 24 hrs for CT fraction and after 96 hrs for ST fraction). Briefly, media was collected and frozen for ELISA analysis. To isolate protein, cells were washed in PBS followed by lysing in 100 uL RIPA buffer with added protease/phosphatase inhibitors (ThermoFisher Scientific, Cat. #89901 #A32959 respectively). Cells were then scraped, and the cell lysate transferred to a sterile 1.5 mL tube and placed on ice. Cell debris was removed by centrifuging the cell lysate at 1000 × RCF for 10 min at 4 °C and storing the supernatant at −80 °C. Total protein was quantified using the Pierce BCA Protein Assay Kit (ThermoFisher Scientific, Cat. #23225). Approximately 20 µg of protein was separated on 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) hand-cast gels for approximately 30 min at 30V followed by 2 hrs at 100 V and transferred for 1 hr at 100 V onto polyvinylidene difluoride (PVDF) membranes using Mini-PROTEAN tetra cell electrophoresis chamber (BioRad, Hercules, CA, Cat. # 1658004). Membranes were blocked in 5% (w/v) nonfat milk in TBS + 0.1% Tween 20 (TBST) for 1 hr and incubated with primary antibody overnight at 4 °C. On the next day, membranes were washed three times in TBST for 5 min each and incubated with HRP-conjugated secondary antibodies. Membranes were washed and incubated in Supersignal West Pico Plus ECL Substrate (ThermoFisher Scientific, Cat. #34578) for 5 min and imaged using the GBOX system (Syngene, Frederick, MD, USA). All samples were normalized to β-Actin and analyzed using Genetools software (Syngene).
The following primary antibodies were used for western blotting: Citrate Synthase (Cell Signaling Technology, Danvers, MA, USA, Cat# 14309, RRID:AB_2665545), glutamate dehydrogenase GLUD1/GLUD2 (Abcam, Cambridge, UK, Cat# ab154027), Glutaminase (Abcam, Cat# ab93434, RRID:AB_10561964), Hexokinase 2 (Cell Signaling Technology, Cat# 2867, RRID:AB_2232946), VDAC (Cell Signaling Technology, Cat# 4661, RRID:AB_10557420), PGC1α (Novus Biologicals, Littleton, CO, USA, Cat# NBP1-04676SS, RRID: AB_1522119), CPT1α (Cell Signaling Technology, Cat# 12252, RRID:AB_2797857), OXPHOS (Abcam, Cat# ab110411, RRID:AB_2756818) and β actin (Sigma-Aldrich, St. Louis, MO, USA, Cat# A2228, RRID:AB_476697). The following HRP conjugated secondary antibodies were used: goat anti-rabbit (Cell Signaling Technology, Cat# 7074, RRID:AB_2099233) and horse anti-mouse (Cell Signaling Technology, Cat# 7076, RRID:AB_330924).
4.7. Enzyme Linked Immunosorbent (ELISA) Assay
The levels of human chorionic gonadotropin (hCG) hormone were measured in media collected from CT and ST cells using an ELISA based assay (R&D Systems, Minneapolis, MN, Cat. #DY9034-05) following manufacturer instructions. Data were then normalized to cellular protein measured using the Pierce BCA Protein Assay Kit (ThermoFisher Scientific, Cat. #23225).
4.8. Citrate Synthase Activity
Citrate synthase activity was measured using the citrate synthase activity kit (Millipore Sigma, St. Louis, MO, USA, Cat. #MAK193) following manufacturer instructions. Briefly, 2 × 106 cells/well were plated in 12-well tissue-culture plates. At 24 hrs and 96 hrs cells were lysed using 90 µL ice cold CS Assay Buffer. The total protein in the lysate was determined using Pierce BCA Protein Assay Kit (ThermoFisher Scientific, Cat. #23225) and all samples were adjusted to 40 μg of protein/50 μL using the CS assay buffer. 50 μL of the lysate was transferred to a 96-well reaction plate along with the standards supplied in the kit. 50 μL Reaction buffer was added to each well and an initial absorbance was measured at 412 nm. The plate was incubated at 25 °C for a total of 10 min before the final measurement was taken. The CS activity was calculated as Sa/(Reaction Time) × Sv; where Sa = Amount of GSH (nmole) generated in unknown sample well between Tinitial and Tfinal from standard curve, Reaction Time = Tfinal − Tinitial (minutes) and Sv = sample volume (mL) added to well. CS activity is reported as pmole/min/μL = microunit/μL.
4.9. Quantitation of Mitochondrial Content
To quantitate mitochondrial number CT cells were plated in a 96-well tissue-culture dish at a cell density of 1 million cells/mL in a volume of 0.1 mL/well for 24 hrs (CT) or 96 hrs (ST). Cells were then incubated with 200 nM MitoTrackerTM Deep Red (Thermo Fisher Scientific, Cat. #M22426) diluted in HBSS for 30 min at 37 °C. Cells were washed three times in HBSS and MitoTrackerTM fluorescence (excitation 644 nm/emission 665 nm). MitoTrackerTM Deep Red specifically stains the mitochondria, and the OD data was normalized to DNA content measured using Quant-it Pico Green dsDNA Reagent (Thermo Fisher Scientific, Cat. #P7581).
4.10. Statistical Analysis
Data are reported as box-and-whisker plots (min to max with mean) with individual data points. Data separated by fetal sex are reported as individual symbols and lines. Statistical significance between groups was calculated using the Friedman test, Wilcoxon test or paired t-test where appropriate. * p < 0.05, ** p < 0.01, and *** p < 0.001 are reported as statistically significant. Graphpad Prism was used to perform all statistical analyses and to generate all graphs.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}