Nitric Oxide and S-Nitrosylation in Cardiac Regulation: G Protein-Coupled Receptor Kinase-2 and β-Arrestins as Targets


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. NO: An Essential Signaling Molecule
1.1.1. Endothelial Nitric Oxide Synthase
1.1.2. Inducible Nitric Oxide Synthase
1.1.3. Neuronal Nitric Oxide Synthase
1.1.4. Nitric Oxide Signaling
1.2. NO-Mediated Signaling: S-nitrosylation
1.3. Role of S-Nitrosylation in the HEART
1.4. Role of S-Nitrosylation in GPCR Signaling
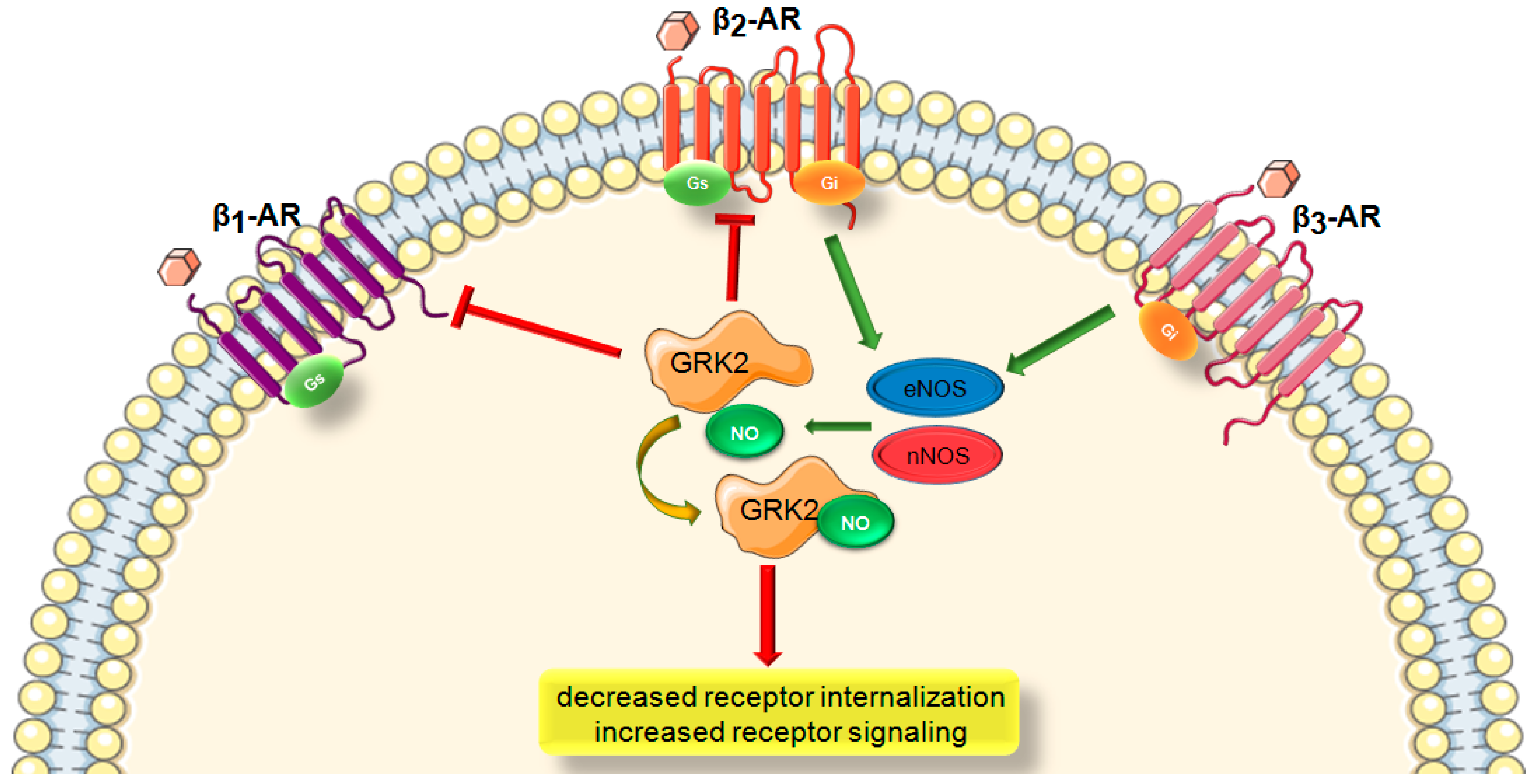
1.5. Role of S-Nitrosylation in GRK Signaling
1.6. Role of S-Nitrosylation in β-Arrestin Signaling
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GPCR | G protein-coupled receptor |
| GRK | G protein-coupled receptor kinase |
| β-Arr | β-Arrestin |
| NO | Nitric oxide |
| eNOS | Endothelial nitric oxide synthase |
| nNOS | Neuronal nitric oxide synthase |
| β-AR | β-Adrenergic receptor |
| SNO | S-nitrosylation |
| ROS | Reactive oxygen species |
References
- Pfleger, J.; Gresham, K.; Koch, W.J. G protein-coupled receptor kinases as therapeutic targets in the heart. Nat. Rev. Cardiol. 2019, 16, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein–Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; Schumacher, S.M.; Tilley, D.G.; Koch, W.J. Designer Approaches for G Protein–Coupled Receptor Modulation for Cardiovascular Disease. JACC Basic Transl. Sci. 2018, 3, 550–562. [Google Scholar] [CrossRef]
- Sato, P.Y.; Chuprun, J.K.; Schwartz, M.; Koch, W.J. The Evolving Impact of G Protein-Coupled Receptor Kinases in Cardiac Health and Disease. Physiol. Rev. 2015, 95, 377–404. [Google Scholar] [CrossRef]
- Farah, C.; Michel, L.Y.M.; Balligand, J.-L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018, 15, 292–316. [Google Scholar] [CrossRef]
- Cannavo, A.; Koch, W.J. GRK2 as negative modulator of NO bioavailability: Implications for cardiovascular disease. Cell. Signal. 2017, 41, 33–40. [Google Scholar] [CrossRef]
- Haldar, S.M.; Stamler, J.S. S-nitrosylation: Integrator of cardiovascular performance and oxygen delivery. J. Clin. Investig. 2013, 123, 101–110. [Google Scholar] [CrossRef]
- Lima, B.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. S-Nitrosylation in Cardiovascular Signaling. Circ. Res. 2010, 106, 633–646. [Google Scholar] [CrossRef]
- Sun, J.; Murphy, E. Protein S -Nitrosylation and Cardioprotection. Circ. Res. 2010, 106, 285–296. [Google Scholar] [CrossRef]
- Hayashi, H.; Hess, D.T.; Zhang, R.; Sugi, K.; Gao, H.; Tan, B.L.; Bowles, D.E.; Milano, C.A.; Jain, M.K.; Koch, W.J.; et al. S-Nitrosylation of β-Arrestins Biases Receptor Signaling and Confers Ligand Independence. Mol. Cell 2018, 70, 473–487.e6. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.M.; Gao, E.; Chuprun, J.K.; Koch, W.J. GRK2 in the Heart: A GPCR Kinase and Beyond. Antioxidants Redox Signal. 2014, 21, 2032–2043. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Whalen, E.J.; Nelson, C.D.; Mu, Y.; Hess, D.T.; Lefkowitz, R.J.; Stamler, J.S. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol. Cell 2008, 31, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Whalen, E.J.; Foster, M.W.; Matsumoto, A.; Ozawa, K.; Violin, J.D.; Que, L.G.; Nelson, C.D.; Moran, B.; Keys, J.R.; Rockman, H.A.; et al. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell 2007, 129, 511–522. [Google Scholar] [CrossRef]
- Loscalzo, J. The Identification of Nitric Oxide as Endothelium-Derived Relaxing Factor. Circ. Res. 2013, 113, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Singel, D.J.; Loscalzo, J. Biochemistry of nitric oxide and its redox-activated forms. Science 1992, 258, 1898–1902. [Google Scholar] [CrossRef]
- Stamler, J.S.; Toone, E.J.; Lipton, S.A.; Sucher, N.J. (S)NO Signals: Translocation, Regulation, and a Consensus Motif. Neuron 1997, 18, 691–696. [Google Scholar] [CrossRef]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzellie, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free. Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef]
- Daaka, Y. S-nitrosylation-regulated GPCR signaling. Biochim. Biophys. Acta BBA Gen. Subj. 2012, 1820, 743–751. [Google Scholar] [CrossRef]
- Vidanapathirana, A.K.; Psaltis, P.J.; Bursill, C.A.; Abell, A.D.; Nicholls, S.J. Cardiovascular bioimaging of nitric oxide: Achievements, challenges, and the future. Med. Res. Rev. 2021, 41, 435–463. [Google Scholar] [CrossRef]
- Webb, A.; Bond, R.; McLean, P.; Uppal, R.; Benjamin, N.; Ahluwalia, A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc. Natl. Acad. Sci. USA 2004, 101, 13683–13688. [Google Scholar] [CrossRef] [PubMed]
- Bredt, D.S.; Snyder, S.H. Nitric oxide: A physiologic messenger molecule. Annu. Rev. Biochem. 1994, 63, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2011, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Damy, T.; Ratajczak, P.; Shah, A.M.; Camors, E.; Marty, I.; Hasenfuss, G.; Marotte, F.; Samuel, J.-L.; Heymes, C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 2004, 363, 1365–1367. [Google Scholar] [CrossRef]
- Stuehr, D.J.; Santolini, J.; Wang, Z.-Q.; Wei, C.-C.; Adak, S. Update on Mechanism and Catalytic Regulation in the NO Synthases. J. Biol. Chem. 2004, 279, 36167–36170. [Google Scholar] [CrossRef] [PubMed]
- Abu-Soud, H.M.; Stuehr, D.J. Nitric oxide synthases reveal a role for calmodulin in controlling electron transfer. Proc. Natl. Acad. Sci. USA 1993, 90, 10769–10772. [Google Scholar] [CrossRef]
- Barouch, L.A.; Harrison, R.W.; Skaf, M.W.; Rosas, G.O.; Cappola, T.P.; Kobeissi, Z.A.; Hobai, I.A.; Lemmon, C.A.; Burnett, A.L.; O’Rourke, B.; et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nat. Cell Biol. 2002, 416, 337–339. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Siamwala, J.H.; Chatterjee, S. eNOS phosphorylation in health and disease. Biochimie 2010, 92, 1186–1198. [Google Scholar] [CrossRef]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Fulton, D.; Gratton, J.P.; Sessa, W.C. Post-translational control of endothelial nitric oxide synthase: Why isn’t calcium/calmodulin enough? J. Pharmacol. Exp. Ther. 2001, 299, 818–824. [Google Scholar] [PubMed]
- Qian, J.; Fulton, D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol 2013, 4, 347. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Kohr, M.J.; Nguyen, T.; Aponte, A.M.; Connelly, P.S.; Esfahani, S.G.; Gucek, M.; Daniels, M.P.; Steenbergen, C.; Murphy, E. Disruption of Caveolae Blocks Ischemic Preconditioning-Mediated S-Nitrosylation of Mitochondrial Proteins. Antioxidants Redox Signal. 2012, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2019, 40, 158–189. [Google Scholar] [CrossRef]
- Soskic, S.S. Regulation of Inducible Nitric Oxide Synthase (iNOS) and its Potential Role in Insulin Resistance, Diabetes and Heart Failure. Open Cardiovasc. Med. J. 2011, 5, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H. Nitric oxide signalling and neuronal nitric oxide synthase in the heart under stress. F1000Research 2017, 6, 742. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, P.M.; Kleinert, H.; Förstermann, U. Potential functional significance of brain-type and muscle-type nitric oxide synthase I expressed in adventitia and media of rat aorta. Arter. Thromb. Vasc. Biol. 1999, 19, 2584–2590. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.D.; Rezende, B.A.; Cortes, S.F.; Lemos, V.S. Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Front. Physiol. 2016, 7, 206. [Google Scholar] [CrossRef]
- Weissman, B.A.; Jones, C.L.; Liu, Q.; Gross, S.S. Activation and inactivation of neuronal nitric oxide synthase: Characterization of Ca2+-dependent [125I]Calmodulin binding. Eur. J. Pharmacol. 2002, 435, 9–18. [Google Scholar] [CrossRef]
- Liu, V.W.; Huang, P.L. WITHDRAWN: Cardiovascular roles of nitric oxide: A review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc. Res. 2007, 77, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Feil, R.; Mülsch, A.; Lohmann, S.M.; Hofmann, F.; Walter, U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3′,5′-cyclic monophosphate-dependent protein kinase. Circulation 2003, 108, 2172–2183. [Google Scholar] [CrossRef] [PubMed]
- Massion, P.B.; Balligand, J.-L. Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): Lessons from genetically modified mice. J. Physiol. 2003, 546, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Carnicer, R.; Crabtree, M.J.; Sivakumaran, V.; Casadei, B.; Kass, D.A. Nitric Oxide Synthases in Heart Failure. Antioxid. Redox Signal. 2013, 18, 1078–1099. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E. NO generation from inorganic nitrate and nitrite: Role in physiology, nutrition and therapeutics. Arch. Pharm. Res. 2009, 32, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Seth, D.; Hess, D.T.; Hausladen, A.; Wang, L.; Wang, Y.-J.; Stamler, J.S. A Multiplex Enzymatic Machinery for Cellular Protein S-nitrosylation. Mol. Cell 2018, 69, 451–464.e6. [Google Scholar] [CrossRef]
- Zhou, H.-L.; Stomberski, C.T.; Stamler, J.S. Cross Talk Between S -Nitrosylation and Phosphorylation Involving Kinases and Nitrosylases. Circ. Res. 2018, 122, 1485–1487. [Google Scholar] [CrossRef]
- Murphy, E.; Kohr, M.; Menazza, S.; Nguyen, T.; Evangelista, A.; Sun, J.; Steenbergen, C. Signaling by S-nitrosylation in the heart. J. Mol. Cell. Cardiol. 2014, 73, 18–25. [Google Scholar] [CrossRef]
- Stamler, J.S. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994, 78, 931–936. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.-O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Gonzalez, D.R.; Beigi, F.; Treuer, A.V.; Hare, J.M. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 20612–20617. [Google Scholar] [CrossRef] [PubMed]
- Erwin, P.A.; Lin, A.J.; Golan, D.E.; Michel, T. Receptor-regulated DynamicS-Nitrosylation of Endothelial Nitric-oxide Synthase in Vascular Endothelial Cells. J. Biol. Chem. 2005, 280, 19888–19894. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Baskaran, P.; Ma, X.; Akker, F.V.D.; Beuve, A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc. Natl. Acad. Sci. USA 2007, 104, 12312–12317. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Lu, Q.; Ding, Y.; Wang, Q.; Xiao, L.; Song, P.; Zou, M.-H. Endothelial Nitric Oxide Synthase–Derived Nitric Oxide Prevents Dihydrofolate Reductase Degradation via Promoting S-Nitrosylation. Arter. Thromb. Vasc. Biol. 2015, 35, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Doctor, A.; Platt, R.; Sheram, M.L.; Eischeid, A.; McMahon, T.; Maxey, T.; Doherty, J.; Axelrod, M.; Kline, J.; Gurka, M.; et al. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc. Natl. Acad. Sci. USA 2005, 102, 5709–5714. [Google Scholar] [CrossRef]
- Liu, L.; Yan, Y.; Zeng, M.; Zhang, J.; Hanes, M.A.; Ahearn, G.; McMahon, T.J.; Dickfeld, T.; E Marshall, H.; Que, L.G.; et al. Essential Roles of S-Nitrosothiols in Vascular Homeostasis and Endotoxic Shock. Cell 2004, 116, 617–628. [Google Scholar] [CrossRef]
- Jia, J.; Arif, A.; Terenzi, F.; Willard, B.; Plow, E.F.; Hazen, S.L.; Fox, P.L. Target-Selective Protein S-Nitrosylation by Sequence Motif Recognition. Cell 2014, 159, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y. S-nitrosylation of proteins: A new insight into endothelial cell function regulated by eNOS-derived NO. Nitric Oxide 2011, 25, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Kavoussi, P.; Smith, R.; Oliver, J.L.; Costabile, R.A.; Steers, W.D.; Brown-Steinke, K.; De Ronde, K.; Lysiak, J.J.; Palmer, L.A. S-nitrosylation of endothelial nitric oxide synthase impacts erectile function. Int. J. Impot. Res. 2018, 31, 31–38. [Google Scholar] [CrossRef]
- Montagna, C.; Rizza, S.; Cirotti, C.; Maiani, E.; Muscaritoli, M.; Musarò, A.; Carrí, M.T.; Ferraro, E.; Cecconi, F.; Filomeni, G. nNOS/GSNOR interaction contributes to skeletal muscle differentiation and homeostasis. Cell Death Dis. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Eu, J.P.; Liu, L.; Zeng, M.; Stamler, J.S. An apoptotic model for nitrosative stress. Biochemistry 2000, 39, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Calay, E.S.; Fan, J.; Arduini, A.; Kunz, R.C.; Gygi, S.P.; Yalcin, A.; Fu, S.; Hotamisligil, G.S. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 2015, 349, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rivera, E.; Jayaraman, P.; Parikh, F.; Davies, M.A.; Ekmekcioglu, S.; Izadmehr, S.; Milton, D.R.; Chipuk, J.E.; Grimm, E.A.; Estrada, Y.; et al. Inducible Nitric Oxide Synthase Drives mTOR Pathway Activation and Proliferation of Human Melanoma by Reversible Nitrosylation of TSC2. Cancer Res. 2014, 74, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Choi, Y.-B.; Takahashi, H.; Zhang, D.; Li, W.; Godzik, A.; Bankston, L.A. Cysteine regulation of protein function—As exemplified by NMDA-receptor modulation. Trends Neurosci. 2002, 25, 474–480. [Google Scholar] [CrossRef]
- Fang, M.; Jaffrey, S.R.; Sawa, A.; Ye, K.; Luo, X.; Snyder, S.H. Dexras1: A G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron 2000, 28, 183–193. [Google Scholar] [CrossRef]
- Beigi, F.; Gonzalez, D.R.; Minhas, K.M.; Sun, Q.-A.; Foster, M.W.; Khan, S.A.; Treuer, A.V.; Dulce, R.A.; Harrison, R.W.; Saraiva, R.M.; et al. Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc. Natl. Acad. Sci. USA 2012, 109, 4314–4319. [Google Scholar] [CrossRef] [PubMed]
- Hatzistergos, K.E.; Paulino, E.C.; Dulce, R.A.; Takeuchi, L.M.; Bellio, M.A.; Kulandavelu, S.; Cao, Y.; Balkan, W.; Kanashiro-Takeuchi, R.M.; Hare, J.M.; et al. S -Nitrosoglutathione Reductase Deficiency Enhances the Proliferative Expansion of Adult Heart Progenitors and Myocytes Post Myocardial Infarction. J. Am. Heart Assoc. 2015, 4, e001974. [Google Scholar] [CrossRef] [PubMed]
- Sips, P.Y.; Irie, T.; Zou, L.; Shinozaki, S.; Sakai, M.; Shimizu, N. Reduction of cardiomyocyte S-nitrosylation by S-nitrosoglutathione reductase protects against sepsis-induced myocardial depression. Am. J. Physiol Heart Circ. Physiol 2013, 304, H1134–H1146. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Sips, P.Y.; Kai, S.; Kida, K.; Ikeda, K.; Hirai, S.; Moazzami, K.; Jiramongkolchai, P.; Bloch, D.B.; Doulias, P.-T.; et al. S-Nitrosylation of Calcium-Handling Proteins in Cardiac Adrenergic Signaling and Hypertrophy. Circ. Res. 2015, 117, 793–803. [Google Scholar] [CrossRef]
- Sun, J.; Yamaguchi, N.; Xu, L.; Eu, J.P.; Stamler, J.S.; Meissner, G. Regulation of the Cardiac Muscle Ryanodine Receptor by O2Tension andS-Nitrosoglutathione. Biochemistry 2008, 47, 13985–13990. [Google Scholar] [CrossRef]
- Sun, J.; Morgan, M.; Shen, R.-F.; Steenbergen, C.; Murphy, E. Preconditioning Results in S -Nitrosylation of Proteins Involved in Regulation of Mitochondrial Energetics and Calcium Transport. Circ. Res. 2007, 101, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Picht, E.; Ginburg, K.S.; Bers, D.; Steenbergen, C.; Murphy, E. Hypercontractile Female Hearts Exhibit Increased S -Nitrosylation of the L-Type Ca 2+ Channel α1 Subunit and Reduced Ischemia/Reperfusion Injury. Circ. Res. 2006, 98, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.M.; Stamler, J.S. NO/redox disequilibrium in the failing heart and cardiovascular system. J. Clin. Investig. 2005, 115, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.E.; Lu, X.; Lei, M.; Xiang, F.-L.; Hammoud, L.; Jiang, M.; Wang, H.; Jones, D.L.; Sims, S.M.; Feng, Q. Neuronal Nitric Oxide Synthase Protects Against Myocardial Infarction-Induced Ventricular Arrhythmia and Mortality in Mice. Circulation 2009, 120, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Asada, K.; Kurokawa, J.; Furukawa, T. Redox- and Calmodulin-dependentS-Nitrosylation of the KCNQ1 Channel. J. Biol. Chem. 2009, 284, 6014–6020. [Google Scholar] [CrossRef] [PubMed]
- Núñez, L.; Vaquero, L.M.; Gómez, R.; Caballero, R.; Mateos-Cáceres, P.; Macaya, C.; Iriepa, I.; Gálvez, E.; López-Farré, A.J.; Tamargo, J. Nitric oxide blocks hKv1.5 channels by S-nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc. Res. 2006, 72, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Gómez, R.; Núñez, L.; Vaquero, L.M.; Amorós, I.; Barana, A.; De Prada, T.; Macaya, C.; Maroto, L.; Rodríguez, E.; Caballero, R.; et al. Nitric oxide inhibits Kv4.3 and human cardiac transient outward potassium current (Ito1). Cardiovasc. Res. 2008, 80, 375–384. [Google Scholar] [CrossRef]
- Gonzalez, D.R.; Treuer, A.; Sun, Q.-A.; Stamler, J.S.; Hare, J.M. S-Nitrosylation of Cardiac Ion Channels. J. Cardiovasc. Pharmacol. 2009, 54, 188–195. [Google Scholar] [CrossRef]
- Figueiredo-Freitas, C.; Dulce, R.A.; Foster, M.W.; Liang, J.; Yamashita, A.M.; Lima-Rosa, F.L.; Thompson, J.W.; Moseley, M.A.; Hare, J.M.; Nogueira, L.; et al. S-Nitrosylation of Sarcomeric Proteins Depresses Myofilament Ca2+ Sensitivity in Intact Cardiomyocytes. Antioxidants Redox Signal. 2015, 23, 1017–1034. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, B.; Li, L.; Block, E.R.; Patel, J.M. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am. J. Physiol. Physiol. 2005, 288, C840–C849. [Google Scholar] [CrossRef]
- Lima, B.; Lam, G.K.W.; Xie, L.; Diesen, D.L.; Villamizar, N.; Nienaber, J.; Messina, E.; Bowles, D.; Kontos, C.D.; Hare, J.M.; et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc. Natl. Acad. Sci. USA 2009, 106, 6297–6302. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, O.; Treuer, A.V.; Gonzalez, D.R. Cardioprotective Effects of S-Nitrosothiols in Ischemia- Reperfusion: Role for Mitochondria and Calcium Channels. In Free Radicals Antioxidants and Diseases; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Ozawa, K.; Komatsubara, A.T.; Nishimura, Y.; Sawada, T.; Kawafune, H.; Tsumoto, H.; Tsuji, Y.; Zhao, J.; Kyotani, Y.; Tanaka, T.; et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 2013, 3, srep02202. [Google Scholar] [CrossRef] [PubMed]
- Radcliffe, E.; Sun, J.; Aponte, A.; Eisner, D.; Murphy, E.; Trafford, A. 198 Heart failure increases mitochondrial s-nitrosylation. Hear 2017, 103, A134–A135. [Google Scholar] [CrossRef][Green Version]
- Olson, N.; van der Vliet, A. Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric. Oxide 2011, 25, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Singel, D.J.; Stamler, J.S. Chemical physiology of blood flow regulation by red blood cells: The role of nitric oxide and S-nitrosohemoglobin. Annu. Rev. Physiol. 2005, 67, 99–145. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Steenbergen, C.; Murphy, E. S-Nitrosylation: NO-Related Redox Signaling to Protect Against Oxidative Stress. Antioxidants Redox Signal. 2006, 8, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Qiu, H. Post-Translational S-Nitrosylation of Proteins in Regulating Cardiac Oxidative Stress. Antioxidants 2020, 9, 1051. [Google Scholar] [CrossRef] [PubMed]
- Dulce, R.A.; Mayo, V.; Rangel, E.B.; Balkan, W.; Hare, J.M. Interaction between neuronal nitric oxide synthase signaling and temperature influences sarcoplasmic reticulum calcium leak: Role of nitroso-redox balance. Circ. Res. 2014, 116, 46–55. [Google Scholar] [CrossRef]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91. [Google Scholar] [CrossRef]
- Zhu, W.Z.; Zheng, M.; Koch, W.J.; Lefkowitz, R.J.; Kobilka, B.K.; Xiao, R.P. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 1607–1612. [Google Scholar] [CrossRef]
- Ferro, A.; Queen, L.R.; Priest, R.M.; Xu, B.; Ritter, J.M.; Poston, L.; Ward, J.P. Activation of nitric oxide synthase by beta 2-adrenoceptors in human umbilical vein endothelium in vitro. Br. J. Pharmacol. 1999, 126, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Dessy, C.; Balligand, J.L. Beta3-adrenergic receptors in cardiac and vascular tissues emerging concepts and therapeutic perspectives. Adv. Pharmacol. 2010, 59, 135–163. [Google Scholar] [PubMed]
- Shenoy, S.K. Regulation of Receptor Fate by Ubiquitination of Activated beta 2-Adrenergic Receptor and beta-Arrestin. Science 2001, 294, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.T.; Chen, W.; Shenoy, S.; Cong, M.; Exum, S.T.; Lefkowitz, R.J. Phosphorylation of beta-arrestin2 regulates its function in internalization of beta(2)-adrenergic receptors. Biochemistry 2002, 41, 10692–10699. [Google Scholar] [CrossRef] [PubMed]
- Aronstam, R.S.; Martin, D.C.; Dennison, R.L.; Cooley, H.G. S-nitrosylation of m2 muscarinic receptor thiols disrupts receptor-G-protein coupling. Ann. N. Y. Acad. Sci. 1995, 757, 215–217. [Google Scholar] [CrossRef]
- Miyamoto, A.; Laufs, U.; Pardo, C.; Liao, J.K. Modulation of Bradykinin Receptor Ligand Binding Affinity and Its Coupled G-proteins by Nitric Oxide. J. Biol. Chem. 1997, 272, 19601–19608. [Google Scholar] [CrossRef]
- Nozik-Grayck, E.; Whalen, E.J.; Stamler, J.S.; McMahon, T.J.; Chitano, P.; Piantadosi, C.A. S-nitrosoglutathione inhibits α1-adrenergic receptor-mediated vasoconstriction and ligand binding in pulmonary artery. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L136–L143. [Google Scholar] [CrossRef]
- Leclerc, P.C.; Lanctot, P.M.; Auger-Messier, M.; Escher, E.; LeDuc, R.; Guillemette, G. S-nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. Br. J. Pharmacol. 2006, 148, 306–313. [Google Scholar] [CrossRef]
- Adam, L.; Bouvier, M.; Jones, T.L. Nitric oxide modulates beta(2)-adrenergic receptor palmitoylation and signaling. J. Biol. Chem. 1999, 274, 26337–26343. [Google Scholar] [CrossRef]
- Reiter, E.; Lefkowitz, R.J. GRKs and beta-arrestins: Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 2006, 17, 159–165. [Google Scholar] [CrossRef]
- Traynham, J.C.; Hullmann, J.; Koch, W.J. Canonical and non-canonical actions of GRK5 in the heart. J. Mol. Cell Cardiol 2016, 92, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Koch, W.J.; Rockman, H.A.; Samama, P.; Hamilton, R.A.; Bond, R.A.; Milano, C.A.; Lefkowitz, R.J. Cardiac function in mice overexpressing the beta-adrenergic receptor kinase or a beta ARK inhibitor. Science 1995, 268, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Rockman, H.A.; Choi, D.J.; Akhter, S.A.; Jaber, M.; Giros, B.; Lefkowitz, R.J.; Caron, M.G.; Koch, W.J. Control of myocardial contractile function by the level of beta-adrenergic receptor kinase 1 in gene-targeted mice. J. Biol. Chem. 1998, 273, 18180–18184. [Google Scholar] [CrossRef] [PubMed]
- Lafarga, V.; Aymerich, I.; Tapia, O.; Mayor, J.F.; Penela, P. A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J. 2011, 31, 856–869. [Google Scholar] [CrossRef]
- Pitcher, J.A.; Hall, R.A.; Daaka, Y.; Zhang, J.; Ferguson, S.S.G.; Hester, S.; Miller, S.; Caron, M.G.; Lefkowitz, R.J.; Barak, L.S. The G Protein-coupled Receptor Kinase 2 Is a Microtubule-associated Protein Kinase That Phosphorylates Tubulin. J. Biol. Chem. 1998, 273, 12316–12324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, M.; Sato, P.Y.; Chuprun, J.K.; Peroutka, R.J.; Otis, N.J.; Ibetti, J.; Pan, S.; Sheu, S.-S.; Gao, E.; Koch, W.J.; et al. Prodeath signaling of G protein-coupled receptor kinase 2 in cardiac myocytes after ischemic stress occurs via extracellular signal-regulated kinase-dependent heat shock protein 90-mediated mitochondrial targeting. Circ. Res. 2013, 112, 1121–1134. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Chuprun, J.K.; Rengo, G.; Gao, E.; Wei, Z.; Peroutka, R.J.; Gold, J.I.; Gumpert, A.; Chen, M.; Otis, N.J.; et al. G Protein–Coupled Receptor Kinase 2 Activity Impairs Cardiac Glucose Uptake and Promotes Insulin Resistance After Myocardial Ischemia. Circulation 2011, 123, 1953–1962. [Google Scholar] [CrossRef]
- Hullmann, J.E.; Grisanti, L.A.; Makarewich, C.A.; Gao, E.; Gold, J.I.; Chuprun, J.K.; Tilley, D.G.; Houser, S.R.; Koch, W.J. GRK5-Mediated Exacerbation of Pathological Cardiac Hypertrophy Involves Facilitation of Nuclear NFAT Activity. Circ. Res. 2014, 115, 976–985. [Google Scholar] [CrossRef]
- Chuang, T.T.; LeVine, H., 3rd; De Blasi, A. Phosphorylation and activation of beta-adrenergic receptor kinase by protein kinase C. J. Biol. Chem. 1995, 270, 18660–18665. [Google Scholar] [CrossRef]
- Cong, M.; Perry, S.J.; Lin, F.-T.; Fraser, I.D.; Hu, L.A.; Chen, W.; Pitcher, J.A.; Scott, J.D.; Lefkowitz, R.J. Regulation of Membrane Targeting of the G Protein-coupled Receptor Kinase 2 by Protein Kinase A and Its Anchoring Protein AKAP79. J. Biol. Chem. 2001, 276, 15192–15199. [Google Scholar] [CrossRef]
- Penela, P.; Elorza, A.; Sarnago, S.; Mayor, F. Beta-arrestin- and c-Src-dependent degradation of G-protein-coupled receptor kinase 2. EMBO J. 2001, 20, 5129–5138. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.N.; Benovic, J.L. Regulation of the G Protein-coupled Receptor Kinase GRK5 by Protein Kinase C. J. Biol. Chem. 1997, 272, 3806–3812. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V.; Lisanti, M.P.; Benovic, J.L. Regulation of G protein-coupled receptor kinases by caveolin. J. Biol. Chem. 1999, 274, 8858–8864. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.N.; Satpaev, D.K.; Slepak, V.Z.; Benovic, J.L. Regulation of G Protein-coupled Receptor Kinases by Calmodulin and Localization of the Calmodulin Binding Domain. J. Biol. Chem. 1997, 272, 18273–18280. [Google Scholar] [CrossRef] [PubMed]
- Que, L.G.; Liu, L.; Yan, Y.; Whitehead, G.S.; Gavett, S.H.; Schwartz, D.A.; Stamler, J.S. Protection from Experimental Asthma by an Endogenous Bronchodilator. Science 2005, 308, 1618–1621. [Google Scholar] [CrossRef] [PubMed]
- Whalen, E.J.; Johnson, A.K.; Lewis, S.J. Beta-adrenoceptor dysfunction after inhibition of NO synthesis. Hypertens 2000, 36, 376–382. [Google Scholar] [CrossRef]
- Wu, W.; Sung, C.C.; Yu, P.; Li, J.; Chung, K.K. S-Nitrosylation of G protein-coupled receptor kinase 6 and Casein kinase 2 alpha modulates their kinase activity toward alpha-synuclein phosphorylation in an animal model of Parkinson’s disease. PLoS ONE 2020, 15, e0232019. [Google Scholar]
- Lieu, M.; Traynham, C.J.; De Lucia, C.; Pfleger, J.; Piedepalumbo, M.; Roy, R.; Petovic, J.; Landesberg, G.; Forrester, S.J.; Hoffman, M.; et al. Loss of Dynamic Regulation of G Protein-Coupled Receptor Kinase 2 by Nitric Oxide Leads to Cardiovascular Dysfunction with Aging. Am. J. Physiol. Circ. Physiol. 2020, 318, H1162–H1175. [Google Scholar] [CrossRef]
- Wang, G.; Moniri, N.H.; Ozawa, K.; Stamler, J.S.; Daaka, Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc. Natl. Acad. Sci. USA 2006, 103, 1295–1300. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nat. Cell Biol. 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Drake, M.T.; Shenoy, S.K.; Lefkowitz, R.J. Trafficking of G protein-coupled receptors. Circ. Res. 2006, 99, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Gupta, P.; Shukla, A.K. GPCR Signaling: β-arrestins Kiss and Remember. Curr. Biol. 2016, 26, R285–R288. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- Witherow, D.S.; Garrison, T.R.; Miller, W.E.; Lefkowitz, R.J. beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc. Natl. Acad. Sci. USA 2004, 101, 8603–8607. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.J.; Baillie, G.S.; Kohout, T.A.; McPhee, I.; Magiera, M.M.; Ang, K.L.; Miller, W.E.; McLean, A.J.; Conti, M.; Houslay, M.D.; et al. Targeting of Cyclic AMP Degradation to beta 2-Adrenergic Receptors by beta -Arrestins. Science 2002, 298, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.D.; Perry, S.J.; Regier, D.S.; Prescott, S.M.; Topham, M.K.; Lefkowitz, R.J. Targeting of diacylglycerol degradation to M1 muscarinic receptors by beta-arrestins. Science 2007, 315, 663–666. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Sotnikova, T.D.; Marion, S.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 2005, 122, 261–273. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Benovic, J.L. Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J. Biol. Chem. 1993, 268, 11628–11638. [Google Scholar] [CrossRef]
- Lin, F.T.; Miller, W.E.; Luttrell, L.M.; Lefkowitz, R.J. Feedback regulation of beta-arrestin1 function by extracellular signal-regulated kinases. J. Biol. Chem. 1999, 274, 15971–15974. [Google Scholar] [CrossRef]
- Kuhr, F.K.; Zhang, Y.; Brovkovych, V.; Skidgel, R.A. Beta-arrestin 2 is required for B1 receptor-dependent post-translational activation of inducible nitric oxide synthase. FASEB J. 2010, 24, 2475–2483. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayki-Mutlu, G.; Koch, W.J. Nitric Oxide and S-Nitrosylation in Cardiac Regulation: G Protein-Coupled Receptor Kinase-2 and β-Arrestins as Targets. Int. J. Mol. Sci. 2021, 22, 521. https://doi.org/10.3390/ijms22020521
Kayki-Mutlu G, Koch WJ. Nitric Oxide and S-Nitrosylation in Cardiac Regulation: G Protein-Coupled Receptor Kinase-2 and β-Arrestins as Targets. International Journal of Molecular Sciences. 2021; 22(2):521. https://doi.org/10.3390/ijms22020521
Chicago/Turabian StyleKayki-Mutlu, Gizem, and Walter J. Koch. 2021. "Nitric Oxide and S-Nitrosylation in Cardiac Regulation: G Protein-Coupled Receptor Kinase-2 and β-Arrestins as Targets" International Journal of Molecular Sciences 22, no. 2: 521. https://doi.org/10.3390/ijms22020521
APA StyleKayki-Mutlu, G., & Koch, W. J. (2021). Nitric Oxide and S-Nitrosylation in Cardiac Regulation: G Protein-Coupled Receptor Kinase-2 and β-Arrestins as Targets. International Journal of Molecular Sciences, 22(2), 521. https://doi.org/10.3390/ijms22020521