Role of CD4+ T Cells in the Control of Viral Infections: Recent Advances and Open Questions


Abstract

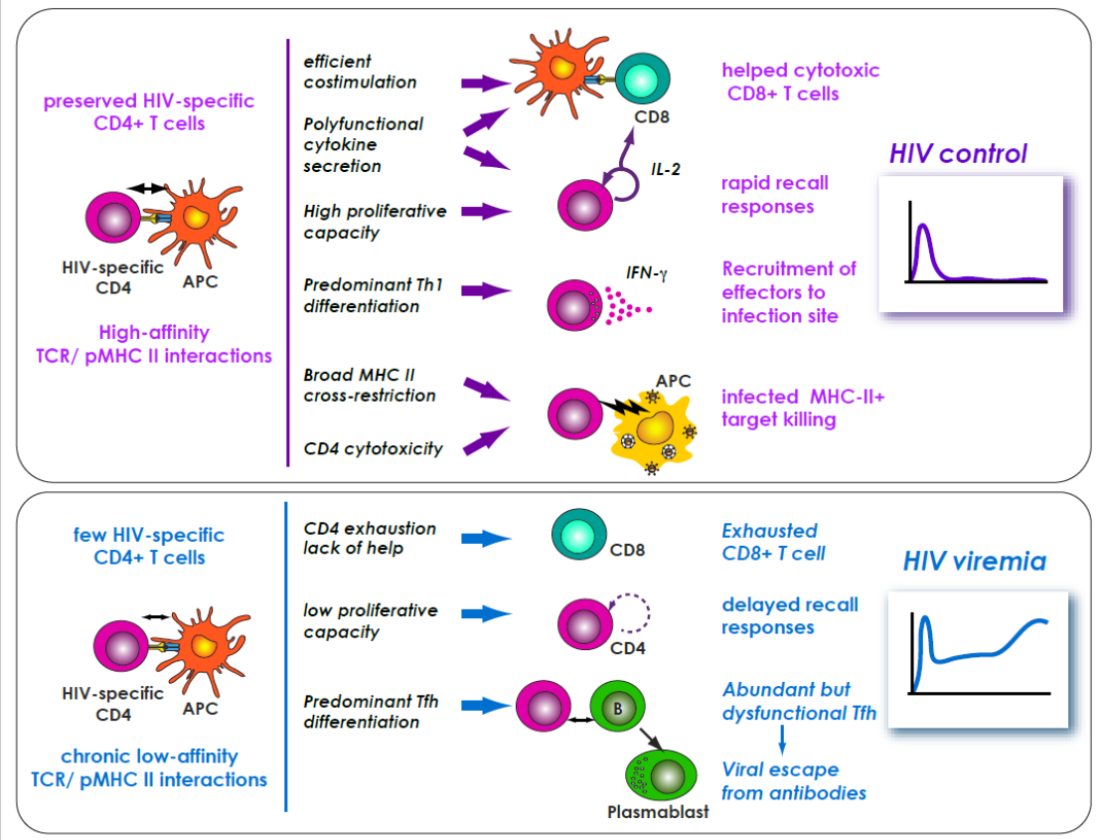{kind=link}
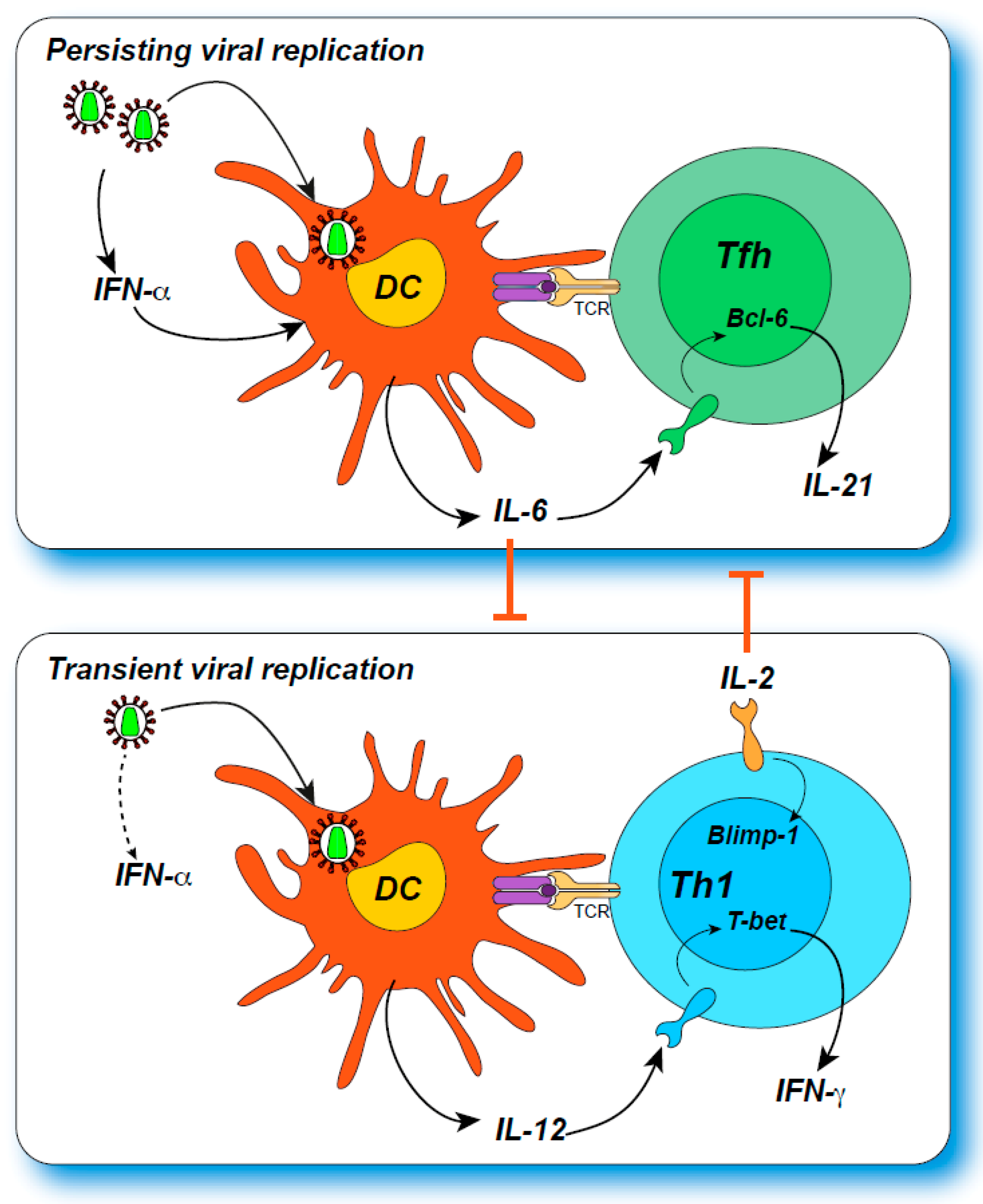{kind=link}
1. Introduction
2. Rapid Kinetics of CD4+ T Cell Responses in Viral Infections
3. The Multiple Components of CD4+ T Cell Help Provided to CD8+ T Cells
4. Defective CD4+ T Cell Help in Chronic Viral Infections Leading to CD8+ T Cell Exhaustion
4.1. The “Exhaustion” T Cell Differentiation Program
4.2. Immunotherapy Aimed at “Helpless” Pre-Exhausted Cells
5. Direct Effector Functions of Antiviral CD4+ T Cells
5.1. Antiviral Effect of T Helper 1 (Th1) Cells
5.2. Antiviral Effect of Cytotoxic CD4+ T Cells
6. Immunoregulation in Chronic Viral Infections
6.1. Upregulation of Inhibitory Receptors
6.2. Interleukin-10 (IL-10)-Dependent Suppression by Tr1 Cells
6.3. Regulatory T Cell (Treg)-Dependent Immunoregulation
6.4. Perturbed Treg/Th17 Balance in Chronic HIV Infection
7. Shifting Th1/T Follicular Helper (Tfh) Balance in Chronic Viral Infections
7.1. Multiple Parameters Control Tfh Differentiation
7.2. Preferential Expansion of Tfh Cells in Chronic Viral Infections
7.3. Pertubed Tfh Function in Progressive HIV Infection
8. Influence of TCR Affinity on T Cell Function and Dynamics
9. The Role of TCR Signal Strength in Viral Control
9.1. Role of TCR Affinity in Antiviral CD8+ T Cell Potency
9.2. Role of TCR Affinity in CD4+ T Cell Helper and Antiviral Functions
9.3. High-Affinity Public TCR Clonotypes Sustain an Efficient CD4+ T Cell Response in Controlled HIV Infection
10. Influence of TCR Affinity on T Helper Differentiation in Viral Infections
10.1. Regulation of the Th1/Th2 Balance
10.2. Regulation of the Th1/Tfh Balance
11. TCR Clonotypic Analyses Shed New Light on the Dynamics of Antiviral Responses
12. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDS | Acuired Immunodeficiency Syndrome |
| AP-1 | Activator Protein-1 |
| APC | Antigen Presenting Cell |
| APOBEC3G | Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G |
| Ascl-2 | Achaete-scute complex homolog 2 |
| BATF | Basic leucine zipper ATF-like transcription factor |
| Bcl-6 | B-Cell Lymphoma-6 |
| bNAb | Broadly Neutralizing Antibody |
| CCL3 | Chemokine C-C Ligand-3 |
| CD | Cluster of Differentiation |
| CMV | Cytomegalovirus |
| COVID-19 | Coronavirus Disease -year 2019 |
| CRTAM | Cytotoxic and Regulatory T Cell Molecule |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated protein 4 |
| CXCR5 | C-X-C chemokine receptor type 5 |
| DC | Dendritic Cells |
| EBI-2 | Epstein-Barr virus-Induced Gene 2 |
| GC | Germinal Center |
| HAV | Hepatitis A Virus |
| HCV | Hepatitis C Virus |
| HIV | Human Immunodeficiency Virus |
| IAV | Influenza A Virus |
| ICOS | Inducible T cell Costimulator |
| IDO | Indoleamine 2,3-dioxygenase |
| IFITM3 | Interferon-induced transmembrane protein 3 |
| IFN | Interferon |
| IL | Interleukin |
| IRF4 | Interferon Regulatory Factor 4 |
| ISG | Interferon-Stimulated Genes |
| JC polyomavirus | John Cunningham polyomavirus |
| LAG-3 | Lymphocyte Activation gene-3 |
| LCMV | Lymphocytic Choriomeningitis Virus |
| MAF | Musculoaponeurotic Fibrosarcoma oncogene |
| MHC | Major Histocompatibility Complex |
| MX2 | MX Dynamin Like GTPase 2 |
| NFAT2 | Nuclear Factor of Activated T cells-2 |
| NK | Natural Killer |
| PD-1 | Programmed cell death-1 |
| pMHC | Peptide-MHC complex |
| RSV | Respiratory Syncytial Virus |
| Runx3 | Runt-related Transcription Factor-3 |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome-related Coronavirus 2 |
| SIV | Simian Immunodeficiency Virus |
| TCM | T Central Memory |
| TCF-1 | T cell factor-1 |
| TCR | T Cell Receptor |
| TF | Transcription Factor |
| Tfh | T follicular helper |
| Th | T helper |
| Treg | Regulatory T cell |
| TIGIT | T cell Immunoreceptor with Ig and ITIM Domains |
| TNF-α | Tumor Necrosis factor-alpha |
| TRM | T resident Memory |
| TGF-β | Transforming Growth Factor-beta |
| VSV | Vesicular Stomatitis Virus |
| VZV | Varicella Zoster Virus |
References
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef]
- Huang, J.; Brameshuber, M.; Zeng, X.; Xie, J.; Li, Q.J.; Chien, Y.H.; Valitutti, S.; Davis, M.M. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity 2013, 39, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F. Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Jost, S.; Tomezsko, P.J.; Rands, K.; Toth, I.; Lichterfeld, M.; Gandhi, R.T.; Altfeld, M. CD4+ T-cell help enhances NK cell function following therapeutic HIV-1 vaccination. J. Virol. 2014, 88, 8349–8354. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4(+) T cells in immunity to viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Serroukh, Y.; Gu-Trantien, C.; Hooshiar Kashani, B.; Defrance, M.; Vu Manh, T.P.; Azouz, A.; Detavernier, A.; Hoyois, A.; Das, J.; Bizet, M.; et al. The transcription factors Runx3 and ThPOK cross-regulate acquisition of cytotoxic function by human Th1 lymphocytes. Elife 2018, 7, e30496. [Google Scholar] [CrossRef]
- Juno, J.A.; van Bockel, D.; Kent, S.J.; Kelleher, A.D.; Zaunders, J.J.; Munier, C.M. Cytotoxic CD4 T Cells-Friend or Foe during Viral Infection? Front. Immunol. 2017, 8, 19. [Google Scholar] [CrossRef]
- Yarchoan, R.; Uldrick, T.S. HIV-Associated Cancers and Related Diseases. N. Engl. J. Med. 2018, 378, 1029–1041. [Google Scholar] [CrossRef]
- Minervina, A.A.; Pogorelyy, M.V.; Komech, E.A.; Karnaukhov, V.K.; Bacher, P.; Rosati, E.; Franke, A.; Chudakov, D.M.; Mamedov, I.Z.; Lebedev, Y.B.; et al. Primary and secondary anti-viral response captured by the dynamics and phenotype of individual T cell clones. Elife 2020, 9, e53704. [Google Scholar] [CrossRef]
- Zhou, Y.; Callendret, B.; Xu, D.; Brasky, K.M.; Feng, Z.; Hensley, L.L.; Guedj, J.; Perelson, A.S.; Lemon, S.M.; Lanford, R.E.; et al. Dominance of the CD4+ T helper cell response during acute resolving hepatitis A virus infection. J. Exp. Med. 2012, 209, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Schulze Zur Wiesch, J.; Ciuffreda, D.; Lewis-Ximenez, L.; Kasprowicz, V.; Nolan, B.E.; Streeck, H.; Aneja, J.; Reyor, L.L.; Allen, T.M.; Lohse, A.W.; et al. Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J. Exp. Med. 2012, 209, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.C.; Sung, P.S.; Park, S.H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Sacha, J.B.; Giraldo-Vela, J.P.; Buechler, M.B.; Martins, M.A.; Maness, N.J.; Chung, C.; Wallace, L.T.; Leon, E.J.; Friedrich, T.C.; Wilson, N.A.; et al. Gag- and Nef-specific CD4+ T cells recognize and inhibit SIV replication in infected macrophages early after infection. Proc. Natl. Acad. Sci. USA 2009, 106, 9791–9796. [Google Scholar] [CrossRef]
- Castellino, F.; Huang, A.Y.; Altan-Bonnet, G.; Stoll, S.; Scheinecker, C.; Germain, R.N. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 2006, 440, 890–895. [Google Scholar] [CrossRef]
- Oh, S.; Perera, L.P.; Terabe, M.; Ni, L.; Waldmann, T.A.; Berzofsky, J.A. IL-15 as a mediator of CD4+ help for CD8+ T cell longevity and avoidance of TRAIL-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 5201–5206. [Google Scholar] [CrossRef]
- Wolkers, M.C.; Gerlach, C.; Arens, R.; Janssen, E.M.; Fitzgerald, P.; Schumacher, T.N.; Medema, J.P.; Green, D.R.; Schoenberger, S.P. Nab2 regulates secondary CD8+ T-cell responses through control of TRAIL expression. Blood 2012, 119, 798–804. [Google Scholar] [CrossRef][Green Version]
- Ahrends, T.; Spanjaard, A.; Pilzecker, B.; Babala, N.; Bovens, A.; Xiao, Y.; Jacobs, H.; Borst, J. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity 2017, 47, 848–861.e5. [Google Scholar] [CrossRef]
- Cullen, J.G.; McQuilten, H.A.; Quinn, K.M.; Olshansky, M.; Russ, B.E.; Morey, A.; Wei, S.; Prier, J.E.; La Gruta, N.L.; Doherty, P.C.; et al. CD4(+) T help promotes influenza virus-specific CD8(+) T cell memory by limiting metabolic dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 4481–4488. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Lu, B.; Gerard, C.; Iwasaki, A. CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature 2009, 462, 510–513. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Zhang, N.; Marshall, H.D.; Staron, M.M.; Guan, T.; Hu, Y.; Cauley, L.S.; Craft, J.; Kaech, S.M. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity 2014, 41, 633–645. [Google Scholar] [CrossRef]
- Wiesel, M.; Oxenius, A. From crucial to negligible: Functional CD8(+) T-cell responses and their dependence on CD4(+) T-cell help. Eur. J. Immunol. 2012, 42, 1080–1088. [Google Scholar] [CrossRef]
- Durlanik, S.; Loyal, L.; Stark, R.; Sercan Alp, Ö.; Hartung, A.; Radbruch, A.; von Herrath, M.; Matzmohr, N.; Frentsch, M.; Thiel, A. CD40L expression by CD4(+) but not CD8(+) T cells regulates antiviral immune responses in acute LCMV infection in mice. Eur. J. Immunol. 2016, 46, 2566–2573. [Google Scholar] [CrossRef]
- Wiesel, M.; Kratky, W.; Oxenius, A. Type I IFN substitutes for T cell help during viral infections. J. Immunol. 2011, 186, 754–763. [Google Scholar] [CrossRef]
- Sun, J.C.; Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef]
- Flinsenberg, T.W.; Spel, L.; Jansen, M.; Koning, D.; de Haar, C.; Plantinga, M.; Scholman, R.; van Loenen, M.M.; Nierkens, S.; Boon, L.; et al. Cognate CD4 T-cell licensing of dendritic cells heralds anti-cytomegalovirus CD8 T-cell immunity after human allogeneic umbilical cord blood transplantation. J. Virol. 2015, 89, 1058–1069. [Google Scholar] [CrossRef]
- Lim, E.Y.; Jackson, S.E.; Wills, M.R. The CD4+ T Cell Response to Human Cytomegalovirus in Healthy and Immunocompromised People. Front. Cell. Infect. Microbiol. 2020, 10, 202. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Bengsch, B.; Ohtani, T.; Khan, O.; Setty, M.; Manne, S.; O’Brien, S.; Gherardini, P.F.; Herati, R.S.; Huang, A.C.; Chang, K.M.; et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity 2018, 48, 1029–1045.e5. [Google Scholar] [CrossRef]
- Saez-Cirion, A.; Manel, N. Immune Responses to Retroviruses. Annu. Rev. Immunol. 2018, 36, 193–220. [Google Scholar] [CrossRef]
- Walker, B.D.; Yu, X.G. Unravelling the mechanisms of durable control of HIV-1. Nat. Rev. Immunol. 2013, 13, 487–498. [Google Scholar] [CrossRef]
- Chakrabarti, L.A. The Different Modes of Resistance to AIDS: Lessons from HIV/SIV Controllers and SIV Natural Hosts. In Natural Hosts of SIV: Implications in AIDS; Ansari, A.A., Silvestri, G., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2014; pp. 287–352. [Google Scholar]
- Younes, S.A.; Yassine-Diab, B.; Dumont, A.R.; Boulassel, M.R.; Grossman, Z.; Routy, J.P.; Sekaly, R.P. HIV-1 viremia prevents the establishment of interleukin 2-producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J. Exp. Med. 2003, 198, 1909–1922. [Google Scholar] [CrossRef]
- Guihot, A.; Tubiana, R.; Breton, G.; Marcelin, A.G.; Samri, A.; Assoumou, L.; Goncalves, E.; Bricaire, F.; Costagliola, D.; Calvez, V.; et al. Immune and virological benefits of 10 years of permanent viral control with antiretroviral therapy. AIDS 2010, 24, 614–617. [Google Scholar]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Scott, A.C.; Dundar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; Gonzalez-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; Lopez-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef]
- Yao, C.; Sun, H.W.; Lacey, N.E.; Ji, Y.; Moseman, E.A.; Shih, H.Y.; Heuston, E.F.; Kirby, M.; Anderson, S.; Cheng, J.; et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat. Immunol. 2019, 20, 890–901. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- He, R.; Hou, S.; Liu, C.; Zhang, A.; Bai, Q.; Han, M.; Yang, Y.; Wei, G.; Shen, T.; Yang, X.; et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature 2016, 537, 412–428. [Google Scholar] [CrossRef]
- Liu, S.Y.; Sanchez, D.J.; Aliyari, R.; Lu, S.; Cheng, G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. USA 2012, 109, 4239–4244. [Google Scholar] [CrossRef]
- Kane, M.; Zang, T.M.; Rihn, S.J.; Zhang, F.; Kueck, T.; Alim, M.; Schoggins, J.; Rice, C.M.; Wilson, S.J.; Bieniasz, P.D. Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell Host Microbe 2016, 20, 392–405. [Google Scholar] [CrossRef]
- Vetter, M.L.; D’Aquila, R.T. Cytoplasmic APOBEC3G restricts incoming Vif-positive human immunodeficiency virus type 1 and increases two-long terminal repeat circle formation in activated T-helper-subtype cells. J. Virol. 2009, 83, 8646–8654. [Google Scholar] [CrossRef]
- Vingert, B.; Benati, D.; Lambotte, O.; de Truchis, P.; Slama, L.; Jeannin, P.; Galperin, M.; Perez-Patrigeon, S.; Boufassa, F.; Kwok, W.W.; et al. HIV controllers maintain a population of highly efficient Th1 effector cells in contrast to patients treated in the long term. J. Virol. 2012, 86, 10661–10674. [Google Scholar] [CrossRef]
- Morou, A.; Brunet-Ratnasingham, E.; Dube, M.; Charlebois, R.; Mercier, E.; Darko, S.; Brassard, N.; Nganou-Makamdop, K.; Arumugam, S.; Gendron-Lepage, G.; et al. Altered differentiation is central to HIV-specific CD4(+) T cell dysfunction in progressive disease. Nat. Immunol. 2019, 20, 1059–1070. [Google Scholar] [CrossRef]
- Bustamante, J.; Boisson-Dupuis, S.; Abel, L.; Casanova, J.L. Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin. Immunol. 2014, 26, 454–470. [Google Scholar] [CrossRef]
- Takeuchi, A.; Badr Mel, S.; Miyauchi, K.; Ishihara, C.; Onishi, R.; Guo, Z.; Sasaki, Y.; Ike, H.; Takumi, A.; Tsuji, N.M.; et al. CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J. Exp. Med. 2016, 213, 123–138. [Google Scholar] [CrossRef]
- McKinstry, K.K.; Strutt, T.M.; Kuang, Y.; Brown, D.M.; Sell, S.; Dutton, R.W.; Swain, S.L. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J. Clin. Investig. 2012, 122, 2847–2856. [Google Scholar] [CrossRef]
- Munier, C.M.L.; van Bockel, D.; Bailey, M.; Ip, S.; Xu, Y.; Alcantara, S.; Liu, S.M.; Denyer, G.; Kaplan, W.; group, P.S.; et al. The primary immune response to Vaccinia virus vaccination includes cells with a distinct cytotoxic effector CD4 T-cell phenotype. Vaccine 2016, 34, 5251–5261. [Google Scholar] [CrossRef]
- Casazza, J.P.; Betts, M.R.; Price, D.A.; Precopio, M.L.; Ruff, L.E.; Brenchley, J.M.; Hill, B.J.; Roederer, M.; Douek, D.C.; Koup, R.A. Acquisition of direct antiviral effector functions by CMV-specific CD4+ T lymphocytes with cellular maturation. J. Exp. Med. 2006, 203, 2865–2877. [Google Scholar] [CrossRef]
- Pachnio, A.; Ciaurriz, M.; Begum, J.; Lal, N.; Zuo, J.; Beggs, A.; Moss, P. Cytomegalovirus Infection Leads to Development of High Frequencies of Cytotoxic Virus-Specific CD4+ T Cells Targeted to Vascular Endothelium. PLoS Pathog. 2016, 12, e1005832. [Google Scholar] [CrossRef]
- Huygens, A.; Lecomte, S.; Tackoen, M.; Olislagers, V.; Delmarcelle, Y.; Burny, W.; Van Rysselberge, M.; Liesnard, C.; Larsen, M.; Appay, V.; et al. Functional Exhaustion Limits CD4+ and CD8+ T-Cell Responses to Congenital Cytomegalovirus Infection. J. Infect. Dis. 2015, 212, 484–494. [Google Scholar] [CrossRef]
- Appay, V.; Zaunders, J.J.; Papagno, L.; Sutton, J.; Jaramillo, A.; Waters, A.; Easterbrook, P.; Grey, P.; Smith, D.; McMichael, A.J.; et al. Characterization of CD4(+) CTLs ex vivo. J. Immunol. 2002, 168, 5954–5958. [Google Scholar] [CrossRef]
- Zaunders, J.J.; Dyer, W.B.; Wang, B.; Munier, M.L.; Miranda-Saksena, M.; Newton, R.; Moore, J.; Mackay, C.R.; Cooper, D.A.; Saksena, N.K.; et al. Identification of circulating antigen-specific CD4+ T lymphocytes with a CCR5+, cytotoxic phenotype in an HIV-1 long-term nonprogressor and in CMV infection. Blood 2004, 103, 2238–2247. [Google Scholar] [CrossRef]
- Zaunders, J.J.; Munier, M.L.; Kaufmann, D.E.; Ip, S.; Grey, P.; Smith, D.; Ramacciotti, T.; Quan, D.; Finlayson, R.; Kaldor, J.; et al. Early proliferation of CCR5(+) CD38(+++) antigen-specific CD4(+) Th1 effector cells during primary HIV-1 infection. Blood 2005, 106, 1660–1667. [Google Scholar] [CrossRef]
- Soghoian, D.Z.; Jessen, H.; Flanders, M.; Sierra-Davidson, K.; Cutler, S.; Pertel, T.; Ranasinghe, S.; Lindqvist, M.; Davis, I.; Lane, K.; et al. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci. Transl. Med. 2012, 4, 123ra25. [Google Scholar] [CrossRef]
- Phetsouphanh, C.; Aldridge, D.; Marchi, E.; Munier, C.M.L.; Meyerowitz, J.; Murray, L.; Van Vuuren, C.; Goedhals, D.; Fidler, S.; Kelleher, A.; et al. Maintenance of Functional CD57+ Cytolytic CD4+ T Cells in HIV+ Elite Controllers. Front. Immunol. 2019, 10, 1844. [Google Scholar] [CrossRef]
- Sauce, D.; Larsen, M.; Fastenackels, S.; Pauchard, M.; Ait-Mohand, H.; Schneider, L.; Guihot, A.; Boufassa, F.; Zaunders, J.; Iguertsira, M.; et al. HIV disease progression despite suppression of viral replication is associated with exhaustion of lymphopoiesis. Blood 2011, 117, 5142–5151. [Google Scholar] [CrossRef]
- Kaufmann, D.E.; Kavanagh, D.G.; Pereyra, F.; Zaunders, J.J.; Mackey, E.W.; Miura, T.; Palmer, S.; Brockman, M.; Rathod, A.; Piechocka-Trocha, A.; et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 2007, 8, 1246–1254. [Google Scholar] [CrossRef]
- Chevalier, M.F.; Didier, C.; Petitjean, G.; Karmochkine, M.; Girard, P.M.; Barré-Sinoussi, F.; Scott-Algara, D.; Weiss, L. Phenotype alterations in regulatory T-cell subsets in primary HIV infection and identification of Tr1-like cells as the main interleukin 10-producing CD4+ T cells. J. Infect. Dis. 2015, 211, 769–779. [Google Scholar] [CrossRef]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E.; et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef] [PubMed]
- Parish, I.A.; Marshall, H.D.; Staron, M.M.; Lang, P.A.; Brustle, A.; Chen, J.H.; Cui, W.; Tsui, Y.C.; Perry, C.; Laidlaw, B.J.; et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J. Clin. Investig. 2014, 124, 3455–3468. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Gregori, S.; Bacchetta, R.; Battaglia, M.; Gagliani, N. The Biology of T Regulatory Type 1 Cells and Their Therapeutic Application in Immune-Mediated Diseases. Immunity 2018, 49, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.J.; Duffy, M.; Brady, M.T.; McKiernan, S.; Hall, W.; Hegarty, J.; Curry, M.; Mills, K.H. CD4 T helper type 1 and regulatory T cells induced against the same epitopes on the core protein in hepatitis C virus-infected persons. J. Infect. Dis. 2002, 185, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Hasenkrug, K.J.; Chougnet, C.A.; Dittmer, U. Regulatory T cells in retroviral infections. PLoS Pathog. 2018, 14, e1006776. [Google Scholar] [CrossRef]
- Brincks, E.L.; Roberts, A.D.; Cookenham, T.; Sell, S.; Kohlmeier, J.E.; Blackman, M.A.; Woodland, D.L. Antigen-specific memory regulatory CD4+Foxp3+ T cells control memory responses to influenza virus infection. J. Immunol. 2013, 190, 3438–3446. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, J.; Gu, Q.; Huang, M.; Zhang, W.; Guo, J.; Zhou, X. Reciprocal Expression of IL-35 and IL-10 Defines Two Distinct Effector Treg Subsets that Are Required for Maintenance of Immune Tolerance. Cell Rep. 2017, 21, 1853–1869. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, Q.; Li, C.; Deng, H.; Si, C.; Xiong, H. Interleukin-35 in immune-related diseases: Protection or destruction. Immunology 2019, 157, 13–20. [Google Scholar] [CrossRef]
- Shao, X.; Ma, J.; Jia, S.; Yang, L.; Wang, W.; Jin, Z. Interleukin-35 Suppresses Antiviral Immune Response in Chronic Hepatitis B Virus Infection. Front. Cell. Infect. Microbiol. 2017, 7, 472. [Google Scholar] [CrossRef]
- Zelinskyy, G.; Dietze, K.; Sparwasser, T.; Dittmer, U. Regulatory T cells suppress antiviral immune responses and increase viral loads during acute infection with a lymphotropic retrovirus. PLoS Pathog. 2009, 5, e1000406. [Google Scholar] [CrossRef]
- Baban, B.; Chandler, P.R.; Sharma, M.D.; Pihkala, J.; Koni, P.A.; Munn, D.H.; Mellor, A.L. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J. Immunol. 2009, 183, 2475–2483. [Google Scholar] [CrossRef]
- Shacklett, B.L. Mucosal Immunity in HIV/SIV Infection: T Cells, B Cells and Beyond. Curr. Immunol. Rev. 2019, 15, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Gosselin, A.; Wacleche, V.S.; El-Far, M.; Said, E.A.; Kared, H.; Grandvaux, N.; Boulassel, M.R.; Routy, J.P.; Ancuta, P. Memory CCR6+CD4+ T cells are preferential targets for productive HIV type 1 infection regardless of their expression of integrin beta7. J. Immunol. 2011, 186, 4618–4630. [Google Scholar] [CrossRef] [PubMed]
- Sandler, N.G.; Douek, D.C. Microbial translocation in HIV infection: Causes, consequences and treatment opportunities. Nat. Rev. Microbiol. 2012, 10, 655–666. [Google Scholar] [CrossRef]
- Loiseau, C.; Requena, M.; Nayrac, M.; Mavigner, M.; Cazabat, M.; Iscache, A.L.; Carrere, N.; Suc, B.; Alric, L.; Izopet, J.; et al. Increased CXCR3+ T Cells Impairs Recruitment of T-Helper Type 17 Cells via Interferon gamma and Interleukin 18 in the Small Intestine Mucosa During Treated HIV-1 Infection. J. Infect. Dis. 2019, 220, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Greczmiel, U.; Krautler, N.J.; Pedrioli, A.; Bartsch, I.; Agnellini, P.; Bedenikovic, G.; Harker, J.; Richter, K.; Oxenius, A. Sustained T follicular helper cell response is essential for control of chronic viral infection. Sci. Immunol. 2017, 2, eaam8686. [Google Scholar] [CrossRef]
- Xiao, N.; Eto, D.; Elly, C.; Peng, G.; Crotty, S.; Liu, Y.C. The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells. Nat. Immunol. 2014, 15, 657–666. [Google Scholar] [CrossRef]
- Bentebibel, S.E.; Lopez, S.; Obermoser, G.; Schmitt, N.; Mueller, C.; Harrod, C.; Flano, E.; Mejias, A.; Albrecht, R.A.; Blankenship, D.; et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci. Transl. Med. 2013, 5, 176ra32. [Google Scholar] [CrossRef]
- Koutsakos, M.; Wheatley, A.K.; Loh, L.; Clemens, E.B.; Sant, S.; Nussing, S.; Fox, A.; Chung, A.W.; Laurie, K.L.; Hurt, A.C.; et al. Circulating TFH cells, serological memory, and tissue compartmentalization shape human influenza-specific B cell immunity. Sci. Transl. Med. 2018, 10, eaan8405. [Google Scholar] [CrossRef]
- Shi, J.; Hou, S.; Fang, Q.; Liu, X.; Liu, X.; Qi, H. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity 2018, 49, 264–274.e4. [Google Scholar] [CrossRef] [PubMed]
- DiToro, D.; Winstead, C.J.; Pham, D.; Witte, S.; Andargachew, R.; Singer, J.R.; Wilson, C.G.; Zindl, C.L.; Luther, R.J.; Silberger, D.J.; et al. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science 2018, 361, eaao2933. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, N.; Liu, Y.; Bentebibel, S.E.; Ueno, H. Molecular Mechanisms Regulating T Helper 1 versus T Follicular Helper Cell Differentiation in Humans. Cell Rep. 2016, 16, 1082–1095. [Google Scholar] [CrossRef]
- Weinstein, J.S.; Laidlaw, B.J.; Lu, Y.; Wang, J.K.; Schulz, V.P.; Li, N.; Herman, E.I.; Kaech, S.M.; Gallagher, P.G.; Craft, J. STAT4 and T-bet control follicular helper T cell development in viral infections. J. Exp. Med. 2018, 215, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, E.; Yi, T.; Cyster, J.G. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature 2016, 533, 110–114. [Google Scholar] [CrossRef] [PubMed]
- De Giovanni, M.; Cutillo, V.; Giladi, A.; Sala, E.; Maganuco, C.G.; Medaglia, C.; Di Lucia, P.; Bono, E.; Cristofani, C.; Consolo, E.; et al. Spatiotemporal regulation of type I interferon expression determines the antiviral polarization of CD4(+) T cells. Nat. Immunol. 2020, 21, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Harker, J.A.; Lewis, G.M.; Mack, L.; Zuniga, E.I. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science 2011, 334, 825–829. [Google Scholar] [CrossRef]
- Fahey, L.M.; Wilson, E.B.; Elsaesser, H.; Fistonich, C.D.; McGavern, D.B.; Brooks, D.G. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J. Exp. Med. 2011, 208, 987–999. [Google Scholar] [CrossRef]
- Lindqvist, M.; van Lunzen, J.; Soghoian, D.Z.; Kuhl, B.D.; Ranasinghe, S.; Kranias, G.; Flanders, M.D.; Cutler, S.; Yudanin, N.; Muller, M.I.; et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J. Clin. Investig. 2012, 122, 3271–3280. [Google Scholar] [CrossRef]
- Osokine, I.; Snell, L.M.; Cunningham, C.R.; Yamada, D.H.; Wilson, E.B.; Elsaesser, H.J.; de la Torre, J.C.; Brooks, D. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc. Natl. Acad. Sci. USA 2014, 111, 7409–7414. [Google Scholar] [CrossRef]
- Xu, L.; Cao, Y.; Xie, Z.; Huang, Q.; Bai, Q.; Yang, X.; He, R.; Hao, Y.; Wang, H.; Zhao, T.; et al. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat. Immunol. 2015, 16, 991–999. [Google Scholar] [CrossRef]
- Rutishauser, R.L.; Deguit, C.D.T.; Hiatt, J.; Blaeschke, F.; Roth, T.L.; Wang, L.; Raymond, K.A.; Starke, C.E.; Mudd, J.C.; Chen, W.; et al. TCF-1 regulates HIV-specific CD8+ T cell expansion capacity. JCI Insight 2020, 136648. [Google Scholar] [CrossRef]
- Papillion, A.; Powell, M.D.; Chisolm, D.A.; Bachus, H.; Fuller, M.J.; Weinmann, A.S.; Villarino, A.; O’Shea, J.J.; Leon, B.; Oestreich, K.J.; et al. Inhibition of IL-2 responsiveness by IL-6 is required for the generation of GC-TFH cells. Sci. Immunol. 2019, 4, eaaw7636. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.D.; Ray, J.P.; Laidlaw, B.J.; Zhang, N.; Gawande, D.; Staron, M.M.; Craft, J.; Kaech, S.M. The transforming growth factor beta signaling pathway is critical for the formation of CD4 T follicular helper cells and isotype-switched antibody responses in the lung mucosa. Elife 2015, 4, e04851. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, N.; Kuo, H.H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 2020, 183, 143–157.e13. [Google Scholar] [CrossRef]
- Elsner, R.A.; Shlomchik, M.J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150. [Google Scholar] [CrossRef]
- Moir, S.; Fauci, A.S. B-cell responses to HIV infection. Immunol. Rev. 2017, 275, 33–48. [Google Scholar] [CrossRef]
- Perreau, M.; Savoye, A.L.; De Crignis, E.; Corpataux, J.M.; Cubas, R.; Haddad, E.K.; De Leval, L.; Graziosi, C.; Pantaleo, G. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J. Exp. Med. 2012, 210, 143–156. [Google Scholar] [CrossRef]
- Amet, T.; Son, Y.M.; Jiang, L.; Cheon, I.S.; Huang, S.; Gupta, S.K.; Dent, A.L.; Montaner, L.J.; Yu, Q.; Sun, J. BCL6 represses antiviral resistance in follicular T helper cells. J. Leukoc. Biol. 2017, 102, 527–536. [Google Scholar] [CrossRef]
- Cubas, R.A.; Mudd, J.C.; Savoye, A.L.; Perreau, M.; van Grevenynghe, J.; Metcalf, T.; Connick, E.; Meditz, A.; Freeman, G.J.; Abesada-Terk, G., Jr.; et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat. Med. 2013, 19, 494–499. [Google Scholar] [CrossRef]
- Buranapraditkun, S.; Pissani, F.; Teigler, J.E.; Schultz, B.T.; Alter, G.; Marovich, M.; Robb, M.L.; Eller, M.A.; Martin, J.; Deeks, S.; et al. Preservation of Peripheral T Follicular Helper Cell Function in HIV Controllers. J. Virol. 2017, 91, e00497-17. [Google Scholar] [CrossRef] [PubMed]
- Claireaux, M.; Galperin, M.; Benati, D.; Nouel, A.; Mukhopadhyay, M.; Klingler, J.; de Truchis, P.; Zucman, D.; Hendou, S.; Boufassa, F.; et al. A High Frequency of HIV-Specific Circulating Follicular Helper T Cells Is Associated with Preserved Memory B Cell Responses in HIV Controllers. mBio 2018, 9, e00317-18. [Google Scholar] [CrossRef] [PubMed]
- Buckner, C.M.; Kardava, L.; Zhang, X.; Gittens, K.; Justement, J.S.; Kovacs, C.; McDermott, A.B.; Li, Y.; Sajadi, M.M.; Chun, T.W.; et al. Maintenance of HIV-Specific Memory B-Cell Responses in Elite Controllers Despite Low Viral Burdens. J. Infect. Dis. 2016, 214, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Rouers, A.; Klingler, J.; Su, B.; Samri, A.; Laumond, G.; Even, S.; Avettand-Fenoel, V.; Richetta, C.; Paul, N.; Boufassa, F.; et al. HIV-Specific B Cell Frequency Correlates with Neutralization Breadth in Patients Naturally Controlling HIV-Infection. EBioMedicine 2017, 21, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D.; Wrin, T.; Little, S.J.; Petropoulos, C.J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 2003, 100, 4144–4149. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M. Broadly neutralizing antibodies for the treatment and prevention of HIV infection. Curr. Opin. HIV AIDS 2020, 15, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Malherbe, L.; Hausl, C.; Teyton, L.; McHeyzer-Williams, M.G. Clonal selection of helper T cells is determined by an affinity threshold with no further skewing of TCR binding properties. Immunity 2004, 21, 669–679. [Google Scholar] [CrossRef]
- Williams, M.A.; Ravkov, E.V.; Bevan, M.J. Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity 2008, 28, 533–545. [Google Scholar] [CrossRef]
- Gett, A.V.; Sallusto, F.; Lanzavecchia, A.; Geginat, J. T cell fitness determined by signal strength. Nat. Immunol. 2003, 4, 355–360. [Google Scholar] [CrossRef]
- Kim, C.; Wilson, T.; Fischer, K.F.; Williams, M.A. Sustained interactions between T cell receptors and antigens promote the differentiation of CD4(+) memory T cells. Immunity 2013, 39, 508–520. [Google Scholar] [CrossRef]
- Tubo, N.J.; Jenkins, M.K. TCR signal quantity and quality in CD4 T cell differentiation. Trends Immunol. 2014, 35, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zarnitsyna, V.I.; Liu, B.; Edwards, L.J.; Jiang, N.; Evavold, B.D.; Zhu, C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature 2010, 464, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Govern, C.C.; Paczosa, M.K.; Chakraborty, A.K.; Huseby, E.S. Fast on-rates allow short dwell time ligands to activate T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 8724–8729. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.A.; Boniface, J.J.; Davis, M.M. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity 1999, 10, 485–492. [Google Scholar] [CrossRef]
- McHeyzer-Williams, L.J.; Panus, J.F.; Mikszta, J.A.; McHeyzer-Williams, M.G. Evolution of antigen-specific T cell receptors in vivo: Preimmune and antigen-driven selection of preferred complementarity-determining region 3 (CDR3) motifs. J. Exp. Med. 1999, 189, 1823–1838. [Google Scholar] [CrossRef] [PubMed]
- Cukalac, T.; Chadderton, J.; Handel, A.; Doherty, P.C.; Turner, S.J.; Thomas, P.G.; La Gruta, N.L. Reproducible selection of high avidity CD8+ T-cell clones following secondary acute virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 1485–1490. [Google Scholar] [CrossRef]
- Baumgartner, C.K.; Yagita, H.; Malherbe, L.P. A TCR affinity threshold regulates memory CD4 T cell differentiation following vaccination. J. Immunol. 2012, 189, 2309–2317. [Google Scholar] [CrossRef]
- Dolton, G.; Lissina, A.; Skowera, A.; Ladell, K.; Tungatt, K.; Jones, E.; Kronenberg-Versteeg, D.; Akpovwa, H.; Pentier, J.M.; Holland, C.J.; et al. Comparison of peptide-major histocompatibility complex tetramers and dextramers for the identification of antigen-specific T cells. Clin. Exp. Immunol. 2014, 177, 47–63. [Google Scholar] [CrossRef]
- Martinez, R.J.; Andargachew, R.; Martinez, H.A.; Evavold, B.D. Low-affinity CD4+ T cells are major responders in the primary immune response. Nat. Commun. 2016, 7, 13848. [Google Scholar] [CrossRef]
- Tungatt, K.; Bianchi, V.; Crowther, M.D.; Powell, W.E.; Schauenburg, A.J.; Trimby, A.; Donia, M.; Miles, J.J.; Holland, C.J.; Cole, D.K.; et al. Antibody stabilization of peptide-MHC multimers reveals functional T cells bearing extremely low-affinity TCRs. J. Immunol. 2015, 194, 463–474. [Google Scholar] [CrossRef]
- Merkenschlager, J.; Ploquin, M.J.; Eksmond, U.; Andargachew, R.; Thorborn, G.; Filby, A.; Pepper, M.; Evavold, B.; Kassiotis, G. Stepwise B-cell-dependent expansion of T helper clonotypes diversifies the T-cell response. Nat. Commun. 2016, 7, 10281. [Google Scholar] [CrossRef] [PubMed]
- Persaud, S.P.; Parker, C.R.; Lo, W.L.; Weber, K.S.; Allen, P.M. Intrinsic CD4+ T cell sensitivity and response to a pathogen are set and sustained by avidity for thymic and peripheral complexes of self peptide and MHC. Nat. Immunol. 2014, 15, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Alexander-Miller, M.A. High-avidity CD8+ T cells: Optimal soldiers in the war against viruses and tumors. Immunol. Res. 2005, 31, 13–24. [Google Scholar] [CrossRef]
- Messaoudi, I.; Guevara Patino, J.A.; Dyall, R.; LeMaoult, J.; Nikolich-Zugich, J. Direct link between mhc polymorphism, T cell avidity, and diversity in immune defense. Science 2002, 298, 1797–1800. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Haddad, E.K.; Marceau, J.; Morabito, K.M.; Rao, S.S.; Filali-Mouhim, A.; Sekaly, R.P.; Graham, B.S. A Numerically Subdominant CD8 T Cell Response to Matrix Protein of Respiratory Syncytial Virus Controls Infection with Limited Immunopathology. PLoS Pathog. 2016, 12, e1005486. [Google Scholar] [CrossRef][Green Version]
- Schober, K.; Voit, F.; Grassmann, S.; Muller, T.R.; Eggert, J.; Jarosch, S.; Weissbrich, B.; Hoffmann, P.; Borkner, L.; Nio, E.; et al. Reverse TCR repertoire evolution toward dominant low-affinity clones during chronic CMV infection. Nat. Immunol. 2020, 21, 434–441. [Google Scholar] [CrossRef]
- Neveu, B.; Debeaupuis, E.; Echasserieau, K.; le Moullac-Vaidye, B.; Gassin, M.; Jegou, L.; Decalf, J.; Albert, M.; Ferry, N.; Gournay, J.; et al. Selection of high-avidity CD8 T cells correlates with control of hepatitis C virus infection. Hepatology 2008, 48, 713–722. [Google Scholar] [CrossRef]
- Lichterfeld, M.; Yu, X.G.; Mui, S.K.; Williams, K.L.; Trocha, A.; Brockman, M.A.; Allgaier, R.L.; Waring, M.T.; Koibuchi, T.; Johnston, M.N.; et al. Selective depletion of high-avidity human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T cells after early HIV-1 infection. J. Virol. 2007, 81, 4199–4214. [Google Scholar] [CrossRef]
- Almeida, J.R.; Price, D.A.; Papagno, L.; Arkoub, Z.A.; Sauce, D.; Bornstein, E.; Asher, T.E.; Samri, A.; Schnuriger, A.; Theodorou, I.; et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 2007, 204, 2473–2485. [Google Scholar] [CrossRef]
- Berger, C.T.; Frahm, N.; Price, D.A.; Mothe, B.; Ghebremichael, M.; Hartman, K.L.; Henry, L.M.; Brenchley, J.M.; Ruff, L.E.; Venturi, V.; et al. High-functional-avidity cytotoxic T lymphocyte responses to HLA-B-restricted Gag-derived epitopes associated with relative HIV control. J. Virol. 2011, 85, 9334–9345. [Google Scholar] [CrossRef]
- Lissina, A.; Chakrabarti, L.A.; Takiguchi, M.; Appay, V. TCR clonotypes: Molecular determinants of T-cell efficacy against HIV. Curr. Opin. Virol. 2016, 16, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Conrad, J.A.; Ramalingam, R.K.; Smith, R.M.; Barnett, L.; Lorey, S.L.; Wei, J.; Simons, B.C.; Sadagopal, S.; Meyer-Olson, D.; Kalams, S.A. Dominant clonotypes within HIV-specific T cell responses are programmed death-1high and CD127low and display reduced variant cross-reactivity. J. Immunol. 2011, 186, 6871–6885. [Google Scholar] [CrossRef] [PubMed]
- Angin, M.; Volant, S.; Passaes, C.; Lecuroux, C.; Monceaux, V.; Dillies, M.A.; Valle-Casuso, J.C.; Pancino, G.; Vaslin, B.; Le Grand, R.; et al. Metabolic plasticity of HIV-specific CD8(+) T cells is associated with enhanced antiviral potential and natural control of HIV-1 infection. Nat. Metab. 2019, 1, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, E.L.; Baalwa, J.; Conrod, K.E.; Wang, S.; Wei, X.; Wong, M.; Turner, J.; Pellegrino, P.; Williams, I.; Shaw, G.M.; et al. Escape is a more common mechanism than avidity reduction for evasion of CD8+ T cell responses in primary human immunodeficiency virus type 1 infection. Retrovirology 2011, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Ladell, K.; Hashimoto, M.; Iglesias, M.C.; Wilmann, P.G.; McLaren, J.E.; Gras, S.; Chikata, T.; Kuse, N.; Fastenackels, S.; Gostick, E.; et al. A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity 2013, 38, 425–436. [Google Scholar] [CrossRef]
- Chen, H.; Ndhlovu, Z.M.; Liu, D.; Porter, L.C.; Fang, J.W.; Darko, S.; Brockman, M.A.; Miura, T.; Brumme, Z.L.; Schneidewind, A.; et al. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat. Immunol. 2012, 13, 691–700. [Google Scholar] [CrossRef]
- Almeida, J.R.; Sauce, D.; Price, D.A.; Papagno, L.; Shin, S.Y.; Moris, A.; Larsen, M.; Pancino, G.; Douek, D.C.; Autran, B.; et al. Antigen sensitivity is a major determinant of CD8+ T-cell polyfunctionality and HIV-suppressive activity. Blood 2009, 113, 6351–6360. [Google Scholar] [CrossRef]
- Foley, M.H.; Forcier, T.; McAndrew, E.; Gonzalez, M.; Chen, H.; Juelg, B.; Walker, B.D.; Irvine, D.J. High avidity CD8+ T cells efficiently eliminate motile HIV-infected targets and execute a locally focused program of anti-viral function. PLoS ONE 2014, 9, e87873. [Google Scholar] [CrossRef]
- Lima, N.S.; Takata, H.; Huang, S.H.; Haregot, A.; Mitchell, J.; Blackmore, S.; Garland, A.; Sy, A.; Cartwright, P.; Routy, J.P.; et al. CTL Clonotypes with Higher TCR Affinity Have Better Ability to Reduce the HIV Latent Reservoir. J. Immunol. 2020, 205, 699–707. [Google Scholar] [CrossRef]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef]
- Su, L.F.; Kidd, B.A.; Han, A.; Kotzin, J.J.; Davis, M.M. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity 2013, 38, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Campion, S.L.; Brodie, T.M.; Fischer, W.; Korber, B.T.; Rossetti, A.; Goonetilleke, N.; McMichael, A.J.; Sallusto, F. Proteome-wide analysis of HIV-specific naive and memory CD4(+) T cells in unexposed blood donors. J. Exp. Med. 2014, 211, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Young, G.R.; Ploquin, M.J.; Eksmond, U.; Wadwa, M.; Stoye, J.P.; Kassiotis, G. Negative selection by an endogenous retrovirus promotes a higher-avidity CD4+ T cell response to retroviral infection. PLoS Pathog. 2012, 8, e1002709. [Google Scholar] [CrossRef] [PubMed]
- Thorborn, G.; Ploquin, M.J.; Eksmond, U.; Pike, R.; Bayer, W.; Dittmer, U.; Hasenkrug, K.J.; Pepper, M.; Kassiotis, G. Clonotypic composition of the CD4+ T cell response to a vectored retroviral antigen is determined by its speed. J. Immunol. 2014, 193, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, S.; Peppers, G.; Kim, S.H.; Graham, B.S. Clonotype-specific avidity influences the dynamics and hierarchy of virus-specific regulatory and effector CD4(+) T-cell responses. Eur. J. Immunol. 2014, 44, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, M.; Hatta, Y.; Kawaoka, Y.; Malherbe, L.P. Antigen signal strength during priming determines effector CD4 T cell function and antigen sensitivity during influenza virus challenge. J. Immunol. 2014, 193, 2812–2820. [Google Scholar] [CrossRef]
- Ren, H.M.; Kolawole, E.M.; Ren, M.; Jin, G.; Netherby-Winslow, C.S.; Wade, Q.; Shwetank; Rahman, Z.S.M.; Evavold, B.D.; Lukacher, A.E. IL-21 from high-affinity CD4 T cells drives differentiation of brain-resident CD8 T cells during persistent viral infection. Sci. Immunol. 2020, 5, eabb5590. [Google Scholar] [CrossRef]
- Atkinson, A.L.; Atwood, W.J. Fifty Years of JC Polyomavirus: A Brief Overview and Remaining Questions. Viruses 2020, 12, 969. [Google Scholar] [CrossRef]
- Seth, N.; Kaufmann, D.; Lahey, T.; Rosenberg, E.S.; Wucherpfennig, K.W. Expansion and contraction of HIV-specific CD4 T cells with short bursts of viremia, but physical loss of the majority of these cells with sustained viral replication. J. Immunol. 2005, 175, 6948–6958. [Google Scholar] [CrossRef]
- Lubong Sabado, R.; Kavanagh, D.G.; Kaufmann, D.E.; Fru, K.; Babcock, E.; Rosenberg, E.; Walker, B.; Lifson, J.; Bhardwaj, N.; Larsson, M. In vitro priming recapitulates in vivo HIV-1 specific T cell responses, revealing rapid loss of virus reactive CD4 T cells in acute HIV-1 infection. PLoS ONE 2009, 4, e4256. [Google Scholar] [CrossRef]
- Schieffer, M.; Jessen, H.K.; Oster, A.F.; Pissani, F.; Soghoian, D.Z.; Lu, R.; Jessen, A.B.; Zedlack, C.; Schultz, B.T.; Davis, I.; et al. Induction of Gag-specific CD4 T cell responses during acute HIV infection is associated with improved viral control. J. Virol. 2014, 88, 7357–7366. [Google Scholar] [CrossRef] [PubMed]
- Vingert, B.; Perez-Patrigeon, S.; Jeannin, P.; Lambotte, O.; Boufassa, F.; Lemaitre, F.; Kwok, W.W.; Theodorou, I.; Delfraissy, J.F.; Theze, J.; et al. HIV controller CD4+ T cells respond to minimal amounts of Gag antigen due to high TCR avidity. PLoS Pathog. 2010, 6, e1000780. [Google Scholar] [CrossRef] [PubMed]
- Benati, D.; Galperin, M.; Lambotte, O.; Gras, S.; Lim, A.; Mukhopadhyay, M.; Nouel, A.; Campbell, K.A.; Lemercier, B.; Claireaux, M.; et al. Public T cell receptors confer high-avidity CD4 responses to HIV controllers. J. Clin. Investig. 2016, 126, 2093–2108. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.; Farenc, C.; Mukhopadhyay, M.; Jayasinghe, D.; Decroos, A.; Benati, D.; Tan, L.L.; Ciacchi, L.; Reid, H.H.; Rossjohn, J.; et al. CD4(+) T cell-mediated HLA class II cross-restriction in HIV controllers. Sci. Immunol. 2018, 3, eaat0687. [Google Scholar] [CrossRef] [PubMed]
- Ferre, A.L.; Hunt, P.W.; McConnell, D.H.; Morris, M.M.; Garcia, J.C.; Pollard, R.B.; Yee, H.F., Jr.; Martin, J.N.; Deeks, S.G.; Shacklett, B.L. HIV controllers with HLA-DRB1*13 and HLA-DQB1*06 alleles have strong, polyfunctional mucosal CD4+ T-cell responses. J. Virol. 2010, 84, 11020–11029. [Google Scholar] [CrossRef]
- Ranasinghe, S.; Cutler, S.; Davis, I.; Lu, R.; Soghoian, D.Z.; Qi, Y.; Sidney, J.; Kranias, G.; Flanders, M.D.; Lindqvist, M.; et al. Association of HLA-DRB1-restricted CD4 T cell responses with HIV immune control. Nat. Med. 2013, 19, 930–933. [Google Scholar] [CrossRef]
- Yamane, H.; Paul, W.E. Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol. Rev. 2013, 252, 12–23. [Google Scholar] [CrossRef]
- Iwata, A.; Durai, V.; Tussiwand, R.; Briseno, C.G.; Wu, X.; Grajales-Reyes, G.E.; Egawa, T.; Murphy, T.L.; Murphy, K.M. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat. Immunol. 2017, 18, 563–572. [Google Scholar] [CrossRef]
- van Panhuys, N.; Klauschen, F.; Germain, R.N. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization In Vivo. Immunity 2014, 41, 63–74. [Google Scholar] [CrossRef]
- Brenna, E.; Davydov, A.N.; Ladell, K.; McLaren, J.E.; Bonaiuti, P.; Metsger, M.; Ramsden, J.D.; Gilbert, S.C.; Lambe, T.; Price, D.A.; et al. CD4(+) T Follicular Helper Cells in Human Tonsils and Blood Are Clonally Convergent but Divergent from Non-Tfh CD4(+) Cells. Cell Rep. 2020, 30, 137–152.e5. [Google Scholar] [CrossRef]
- Fazilleau, N.; McHeyzer-Williams, L.J.; Rosen, H.; McHeyzer-Williams, M.G. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 2009, 10, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Tubo, N.J.; Pagan, A.J.; Taylor, J.J.; Nelson, R.W.; Linehan, J.L.; Ertelt, J.M.; Huseby, E.S.; Way, S.S.; Jenkins, M.K. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 2013, 153, 785–796. [Google Scholar] [CrossRef]
- Cho, Y.L.; Flossdorf, M.; Kretschmer, L.; Hofer, T.; Busch, D.H.; Buchholz, V.R. TCR Signal Quality Modulates Fate Decisions of Single CD4(+) T Cells in a Probabilistic Manner. Cell Rep. 2017, 20, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Kotov, D.I.; Mitchell, J.S.; Pengo, T.; Ruedl, C.; Way, S.S.; Langlois, R.A.; Fife, B.T.; Jenkins, M.K. TCR Affinity Biases Th Cell Differentiation by Regulating CD25, Eef1e1, and Gbp2. J. Immunol. 2019, 202, 2535–2545. [Google Scholar] [CrossRef] [PubMed]
- Snook, J.P.; Kim, C.; Williams, M.A. TCR signal strength controls the differentiation of CD4(+) effector and memory T cells. Sci. Immunol. 2018, 3, eaas9103. [Google Scholar] [CrossRef]
- Krishnamoorthy, V.; Kannanganat, S.; Maienschein-Cline, M.; Cook, S.L.; Chen, J.; Bahroos, N.; Sievert, E.; Corse, E.; Chong, A.; Sciammas, R. The IRF4 Gene Regulatory Module Functions as a Read-Write Integrator to Dynamically Coordinate T Helper Cell Fate. Immunity 2017, 47, 481–497.e7. [Google Scholar] [CrossRef]
- Künzli, M.; Reuther, P.; Pinschewer, D.D.; King, C.G. Opposing effects of T cell receptor signal strength on CD4 T cells responding to acute versus chronic viral infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef]
- Richter, K.; Perriard, G.; Oxenius, A. Reversal of chronic to resolved infection by IL-10 blockade is LCMV strain dependent. Eur. J. Immunol. 2013, 43, 649–654. [Google Scholar] [CrossRef]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science 2012, 335, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.C.; Dash, P.; McCullers, J.A.; Doherty, P.C.; Thomas, P.G. T cell receptor alphabeta diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci. Transl. Med. 2012, 4, 128ra42. [Google Scholar] [CrossRef]
- Connors, M.; Kovacs, J.A.; Krevat, S.; Gea-Banacloche, J.C.; Sneller, M.C.; Flanigan, M.; Metcalf, J.A.; Walker, R.E.; Falloon, J.; Baseler, M.; et al. HIV infection induces changes in CD4+ T-cell phenotype and depletions within the CD4+ T-cell repertoire that are not immediately restored by antiviral or immune-based therapies. Nat. Med. 1997, 3, 533–540. [Google Scholar] [CrossRef]
- Gorochov, G.; Neumann, A.U.; Kereveur, A.; Parizot, C.; Li, T.; Katlama, C.; Karmochkine, M.; Raguin, G.; Autran, B.; Debre, P. Perturbation of CD4+ and CD8+ T-cell repertoires during progression to AIDS and regulation of the CD4+ repertoire during antiviral therapy. Nat. Med. 1998, 4, 215–221. [Google Scholar] [CrossRef]
- Gantner, P.; Pagliuzza, A.; Pardons, M.; Ramgopal, M.; Routy, J.P.; Fromentin, R.; Chomont, N. Single-cell TCR sequencing reveals phenotypically diverse clonally expanded cells harboring inducible HIV proviruses during ART. Nat. Commun. 2020, 11, 4089. [Google Scholar] [CrossRef] [PubMed]
- Hey-Nguyen, W.J.; Bailey, M.; Xu, Y.; Suzuki, K.; Van Bockel, D.; Finlayson, R.; Leigh Brown, A.; Carr, A.; Cooper, D.A.; Kelleher, A.D.; et al. HIV-1 DNA Is Maintained in Antigen-Specific CD4+ T Cell Subsets in Patients on Long-Term Antiretroviral Therapy Regardless of Recurrent Antigen Exposure. AIDS Res. Hum. Retroviruses 2019, 35, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, P.; Jackson, J.R.; Oliveira, T.Y.; Gaebler, C.; Ramos, V.; Caskey, M.; Jankovic, M.; Nussenzweig, M.C.; Cohn, L.B. Antigen-responsive CD4+ T cell clones contribute to the HIV-1 latent reservoir. J. Exp. Med. 2020, 217, 7. [Google Scholar] [CrossRef]
- Herati, R.S.; Muselman, A.; Vella, L.; Bengsch, B.; Parkhouse, K.; Del Alcazar, D.; Kotzin, J.; Doyle, S.A.; Tebas, P.; Hensley, S.E.; et al. Successive annual influenza vaccination induces a recurrent oligoclonotypic memory response in circulating T follicular helper cells. Sci. Immunol. 2017, 2, eaag2152. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, M.; Galperin, M.; Patgaonkar, M.; Vasan, S.; Ho, D.D.; Nouel, A.; Claireaux, M.; Benati, D.; Lambotte, O.; Huang, Y.; et al. DNA Vaccination by Electroporation Amplifies Broadly Cross-Restricted Public TCR Clonotypes Shared with HIV Controllers. J. Immunol. 2017, 199, 3437–3452. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Cavanagh, M.M.; Le Saux, S.; NamKoong, H.; Kim, C.; Turgano, E.; Liu, Y.; Wang, C.; Mackey, S.; Swan, G.E.; et al. Diversification of the antigen-specific T cell receptor repertoire after varicella zoster vaccination. Sci. Transl. Med. 2016, 8, 332ra46. [Google Scholar] [CrossRef]
- Han, A.; Glanville, J.; Hansmann, L.; Davis, M.M. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol. 2014, 32, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.; Fiore-Gartland, A.J.; Hertz, T.; Wang, G.C.; Sharma, S.; Souquette, A.; Crawford, J.C.; Clemens, E.B.; Nguyen, T.H.O.; Kedzierska, K.; et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 2017, 547, 89–93. [Google Scholar] [CrossRef] [PubMed]
- La Gruta, N.L.; Gras, S.; Daley, S.R.; Thomas, P.G.; Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 2018, 18, 467–478. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, W.S., 3rd; Smith, A.; Schoch, G.; Hansen, J.A.; Matsen, F.A.; Bradley, P. Human T cell receptor occurrence patterns encode immune history, genetic background, and receptor specificity. Elife 2018, 7, e38358. [Google Scholar] [CrossRef]
- Schultheiss, C.; Paschold, L.; Simnica, D.; Mohme, M.; Willscher, E.; von Wenserski, L.; Scholz, R.; Wieters, I.; Dahlke, C.; Tolosa, E.; et al. Next-Generation Sequencing of T and B Cell Receptor Repertoires from COVID-19 Patients Showed Signatures Associated with Severity of Disease. Immunity 2020, 53, 442–455.e4. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Egan, D.N.; Bar, M.; Schmitt, T.M.; McAfee, M.S.; Paulson, K.G.; Voillet, V.; Gottardo, R.; Ragnarsson, G.B.; Bleakley, M.; et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019, 25, 1064–1072. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Haabeth, O.A.; Tveita, A.A.; Fauskanger, M.; Schjesvold, F.; Lorvik, K.B.; Hofgaard, P.O.; Omholt, H.; Munthe, L.A.; Dembic, Z.; Corthay, A.; et al. How Do CD4(+) T Cells Detect and Eliminate Tumor Cells That Either Lack or Express MHC Class II Molecules? Front. Immunol. 2014, 5, 174. [Google Scholar] [CrossRef]
- Migliori, E.; Chang, M.; Muranski, P. Restoring antiviral immunity with adoptive transfer of ex-vivo generated T cells. Curr. Opin. Hematol. 2018, 25, 486–493. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kervevan, J.; Chakrabarti, L.A. Role of CD4+ T Cells in the Control of Viral Infections: Recent Advances and Open Questions. Int. J. Mol. Sci. 2021, 22, 523. https://doi.org/10.3390/ijms22020523
Kervevan J, Chakrabarti LA. Role of CD4+ T Cells in the Control of Viral Infections: Recent Advances and Open Questions. International Journal of Molecular Sciences. 2021; 22(2):523. https://doi.org/10.3390/ijms22020523
Chicago/Turabian StyleKervevan, Jérôme, and Lisa A. Chakrabarti. 2021. "Role of CD4+ T Cells in the Control of Viral Infections: Recent Advances and Open Questions" International Journal of Molecular Sciences 22, no. 2: 523. https://doi.org/10.3390/ijms22020523
APA StyleKervevan, J., & Chakrabarti, L. A. (2021). Role of CD4+ T Cells in the Control of Viral Infections: Recent Advances and Open Questions. International Journal of Molecular Sciences, 22(2), 523. https://doi.org/10.3390/ijms22020523