Therapies for the Treatment of Cardiovascular Disease Associated with Type 2 Diabetes and Dyslipidemia

 , and
, and
Abstract
:1. Introduction
2. Therapies Based on the Incretin System
2.1. Biology of the Incretins
2.2. Degradation of the Incretins by Dipeptidyl Peptidase 4 (DPP4)
2.3. DPP4 Inhibitors as a Therapeutic Target for T2DM
2.4. Effects of DPP4 Inhibitors in CVD
2.4.1. Mechanisms of Gliptins in Experimental Atherosclerosis
2.4.2. Clinical Studies on DPP4 Inhibitors in T2DM with CVD
2.5. GLP1 Analogues as Therapeutic Strategies
2.5.1. Development of Drugs Based on GLP1 and Rational Design
2.5.2. Studies in Preclinical Models of T2DM, Atherosclerosis, and CVD
2.5.3. Clinical Studies on GLP1-Based Strategies in T2DM with CVD
2.6. GIP1 Emergent Therapies
2.6.1. Development of Drugs Based on GIP and Rational Design
2.6.2. Investigations of GIP Therapies in Preclinical Models
2.6.3. Studies in Humans with T2DM and CVD
3. Therapies to Inhibit Sodium-Glucose Co-Transporter 2
3.1. Structure of Sodium-Glucose Co-Transporters and Mechanism of Action
3.2. Development of SGLT Inhibitors
3.3. Effect of Gliflozins in Preclinical Models of CVD
3.4. Clinical Studies of Gliflozins in HF and CVD
4. Lipid-Lowering Therapies Based on the Proprotein Convertase Subtilisin Kexin 9 Inhibition
4.1. Biology of PCSK9 in Lipid Metabolism and Vascular Homeostasis
4.2. Rational for PCSK9 Inhibition as a Potential Therapy
4.2.1. PCSK9 Monoclonal Antibodies
4.2.2. Small Interfering RNA (siRNA)
4.3. Other Developing Approaches to PCSK9 Inhibition
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Naghavi, M.; Abajobir, A.A.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abera, S.F.; Aboyans, V.; Adetokunboh, O.; Ärnlöv, J.; Afshin, A.; et al. Global, regional, and national age-sex specifc mortality for 264 causes of death, 1980–2016: A systematic analysis for the global burden of disease study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef] [Green Version]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1: Evidence from genetic, epidemiologic, and clinical studies. A consensus statement fromthe European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American college of cardiology/American heart association task force on practice guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornfeldt, K.E. Uncomplicating the macrovascular complications of diabetes: The 2014 edwin bierman award lecture. Diabetes 2015, 64, 2689–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeva-Andany, M.M.; Martínez-Rodríguez, J.; González-Lucán, M.; Fernández-Fernández, C.; Castro-Quintela, E. Insulin resistance is a cardiovascular risk factor in humans. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 1449–1455. [Google Scholar] [CrossRef]
- Kubota, T.; Kubota, N.; Kumagai, H.; Yamaguchi, S.; Kozono, H.; Takahashi, T.; Inoue, M.; Itoh, S.; Takamoto, I.; Sasako, T.; et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011, 13, 294–307. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Hervás, S.; Vinué, Á.; Núñez, L.; Andrés-Blasco, I.; Piqueras, L.; TomásReal, J.; Ascaso, J.F.; Burks, D.J.; Sanz, M.J.; González-Navarro, H. Insulin resistance aggravates atherosclerosis by reducing vascular smoothmuscle cell survival and increasing CX3CL1/CX3CR1 axis. Cardiovasc. Res. 2014, 103, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, R.A.; Duinisveld, A.J.F.; Schaapherder, A.F.; Mulder-Stapel, A.; Hamming, J.F.; Kuiper, J.; de Boer, O.J.; van der Wal, A.C.; Kolodgie, F.D.; Virmani, R.; et al. A change in inflammatory footprint precedes plaque instability: A systematic evaluation of cellular aspects of the adaptive immune response in human atherosclerosis. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Lytvyn, Y.; Bjornstad, P.; Pun, N.; Cherney, D.Z.I. New and old agents in the management of diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2016, 25, 232–239. [Google Scholar] [CrossRef] [Green Version]
- White, W.B.; Baker, W.L. Cardiovascular effects of incretin-based therapies. Annu. Rev. Med. 2016, 67, 245–260. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Drucker, D.J. Cardiovascular actions of incretin-based therapies. Circ. Res. 2014, 114, 1788–1803. [Google Scholar] [CrossRef] [Green Version]
- Lambeir, A.M.; Durinx, C.; Scharpé, S.; De Meester, I. Dipeptidyl-peptidase IV from bench to bedside: An update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294. [Google Scholar] [CrossRef]
- Lamers, D.; Famulla, S.; Wronkowitz, N.; Hartwig, S.; Lehr, S.; Ouwens, D.M.; Eckardt, K.; Kaufman, J.M.; Ryden, M.; Müller, S.; et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes 2011, 60, 1917–1925. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Maiseyeu, A.; Davis, S.N.; Rajagopalan, S. DPP4 in cardiometabolic disease: Recent insights from the laboratory and clinical trials of DPP4 inhibition. Circ. Res. 2015, 116, 1491–1504. [Google Scholar] [CrossRef] [Green Version]
- Fisman, E.Z.; Tenenbaum, A. Antidiabetic treatment with gliptins: Focus on cardiovascular effects and outcomes. Cardiovasc. Diabetol. 2015, 14, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Röhrborn, D.; Wronkowitz, N.; Eckel, J. DPP4 in diabetes. Front. Immunol. 2015, 6, 386. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Rao, X.; Deiuliis, J.; Braunstein, Z.; Narula, V.; Hazey, J.; Mikami, D.; Needleman, B.; Satoskar, A.R.; Rajagopalan, S. A potential role for dendritic cell/macrophage-expressing DPP4 in obesity-induced visceral inflammation. Diabetes 2013, 62, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Ikushima, H.; Munakata, Y.; Iwata, S.; Ohnuma, K.; Kobayashi, S.; Dang, N.H.; Morimoto, C. Soluble CD26/dipeptidyl peptidase IV enhances transendothelial migration via its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cell. Immunol. 2002, 215, 106–110. [Google Scholar] [CrossRef]
- Ghorpade, D.S.; Ozcan, L.; Zheng, Z.; Nicoloro, S.M.; Shen, Y.; Chen, E.; Blüher, M.; Czech, M.P.; Tabas, I. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance HHS Public Access. Nature 2018, 555, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Blüher, M.; Klöting, N.; Schlich, R.; Willems, M.; Ruppe, F.; Knoefel, W.T.; Dietrich, A.; Fielding, B.A.; Arner, P.; et al. Adipose dipeptidyl peptidase-4 and obesity: Correlation with insulin resistance and depot-specific release from adipose tissue in vivo and in vitro. Diabetes Care 2013, 36, 4083–4090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wronkowitz, N.; Görgens, S.W.; Romacho, T.; Villalobos, L.A.; Sánchez-Ferrer, C.F.; Peiró, C.; Sell, H.; Eckel, J. Soluble DPP4 induces inflammation and proliferation of human smooth muscle cells via protease-activated receptor 2. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, N.; Teresa Julián, M.; Puig-Domingo, M.; Vives-Pi, M. Incretin hormones as immunomodulators of atherosclerosis. Front. Endocrinol. 2012, 3, 112. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Tavallaie, M.S.; Sun, R.; Wang, J.; Cai, Q.; Shen, J.; Lei, S.; Fu, L.; Jiang, F. Drug discovery approaches targeting the incretin pathway. Bioorg. Chem. 2020, 99. [Google Scholar] [CrossRef]
- Standl, E.; Schnell, O.; McGuire, D.K.; Ceriello, A.; Rydén, L. Integration of recent evidence into management of patients with atherosclerotic cardiovascular disease and type 2 diabetes. Lancet Diabetes Endocrinol. 2017, 5, 391–402. [Google Scholar] [CrossRef]
- McGuire, D.K.; van de Werf, F.; Armstrong, P.W.; Standl, E.; Koglin, J.; Green, J.B.; Bethel, M.A.; Cornel, J.H.; Lopes, R.D.; Halvorsen, S.; et al. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: Secondary analysis of a randomized clinical trial. JAMA Cardiol. 2016, 1, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Zannad, F.; Cannon, C.P.; Cushman, W.C.; Bakris, G.L.; Menon, V.; Perez, A.T.; Fleck, P.R.; Mehta, C.R.; Kupfer, S.; Wilson, C.; et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: A multicentre, randomised, double-blind trial. Lancet 2015, 385, 2067–2076. [Google Scholar] [CrossRef]
- McGuire, D.K.; Alexander, J.H.; Johansen, O.E.; Perkovic, V.; Rosenstock, J.; Cooper, M.E.; Wanner, C.; Kahn, S.E.; Toto, R.D.; Zinman, B.; et al. Linagliptin effects on heart failure and related outcomes in individuals with type 2 diabetes mellitus at high cardiovascular and renal risk in CARMELINA. Circulation 2019, 139, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Mcinnes, G.; Evans, M.; del Prato, S.; Stumvoll, M.; Schweizer, A.; Lukashevich, V.; Shao, Q.; Kothny, W. Cardiovascular and heart failure safety profile of vildagliptin: A meta-analysis of 17,000 patients. Diabetes Obes. Metab. 2015, 17, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Remm, F.; Franz, W.M.; Brenner, C. Gliptins and their target dipeptidyl peptidase 4: Implications for the treatment of vascular disease. Eur. Hear. J. Cardiovasc. Pharm. 2016, 2, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ervinna, N.; Mita, T.; Yasunari, E.; Azuma, K.; Tanaka, R.; Fujimura, S.; Sukmawati, D.; Nomiyama, T.; Kanazawa, A.; Kawamori, R.; et al. Anagliptin, a DPP-4 inhibitor, suppresses proliferation of vascular smooth muscles and monocyte inflammatory reaction and attenuates atherosclerosis in male apo e-deficient mice. Endocrinology 2013, 154, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Nishida, S.; Matsumura, T.; Senokuchi, T.; Murakami-Nishida, S.; Ishii, N.; Morita, Y.; Yagi, Y.; Motoshima, H.; Kondo, T.; Araki, E. Inhibition of inflammation-mediated DPP-4 expression by linagliptin increases M2 macrophages in atherosclerotic lesions. Biochem. Biophys. Res. Commun. 2020, 524, 8–15. [Google Scholar] [CrossRef]
- Kahles, F.; Liberman, A.; Halim, C.; Rau, M.; Möllmann, J.; Mertens, R.W.; Rückbeil, M.; Diepolder, I.; Walla, B.; Diebold, S.; et al. The incretin hormone GIP is upregulated in patients with atherosclerosis and stabilizes plaques in ApoE−/− mice by blocking monocyte/macrophage activation. Mol. Metab. 2018, 14, 150–157. [Google Scholar] [CrossRef]
- Nagashima, M.; Watanabe, T.; Terasaki, M.; Tomoyasu, M.; Nohtomi, K.; Kim-Kaneyama, J.; Miyazaki, A.; Hirano, T. Native incretins prevent the development of atherosclerotic lesions in apolipoprotein e knockout mice. Diabetologia 2011, 54, 2649–2659. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, Y.; Sato, K.; Watanabe, T.; Nohtomi, K.; Terasaki, M.; Nagashima, M.; Hirano, T. A glucagon-like peptide-1 analog liraglutide suppresses macrophage foam cell formation and atherosclerosis. Peptides 2014, 54, 19–26. [Google Scholar] [CrossRef]
- Bruen, R.; Curley, S.; Kajani, S.; Crean, D.; O’Reilly, M.E.; Lucitt, M.B.; Godson, C.G.; McGillicuddy, F.C.; Belton, O. Liraglutide dictates macrophage phenotype in apolipoprotein E null mice during early atherosclerosis. Cardiovasc. Diabetol. 2017, 16. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Lei, Y.; Inoue, A.; Piao, L.; Hu, L.; Jiang, H.; Sasaki, T.; Wu, H.; Xu, W.; Yu, C.; et al. Exenatide mitigated diet-induced vascular aging and atherosclerotic plaque growth in ApoE-deficient mice under chronic stress. Atherosclerosis 2017, 264, 1–10. [Google Scholar] [CrossRef]
- Arakawa, M.; Mita, T.; Azuma, K.; Ebato, C.; Goto, H.; Nomiyama, T.; Fujitani, Y.; Hirose, T.; Kawamori, R.; Watada, H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010, 59, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Parlevliet, E.T.; Geerling, J.J.; van der Tuin, S.J.L.; Zhang, H.; Bieghs, V.; Jawad, A.H.M.; Shiri-Sverdlov, R.; Bot, I.; de Jager, S.C.A.; et al. Exendin-4 decreases liver inflammation and atherosclerosis development simultaneously by reducing macrophage infiltration. Br. J. Pharm. 2014, 171, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Vinué, Á.; Navarro, J.; Herrero-Cervera, A.; García-Cubas, M.; Andrés-Blasco, I.; Martínez-Hervás, S.; Real, J.T.; Ascaso, J.F.; González-Navarro, H. The GLP-1 analogue lixisenatide decreases atherosclerosis in insulin-resistant mice by modulating macrophage phenotype. Diabetologia 2017, 60, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Rakipovski, G.; Rolin, B.; Nøhr, J.; Klewe, I.; Frederiksen, K.S.; Augustin, R.; Hecksher-Sørensen, J.; Ingvorsen, C.; Polex-Wolf, J.; Knudsen, L.B. The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE/− and LDLr/Mice by a mechanism that includes inflammatory pathways. JACC Basic Transl. Sci. 2018, 3, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Shah, Z.; Kampfrath, T.; Deiuliis, J.A.; Zhong, J.; Pineda, C.; Ying, Z.; Xu, X.; Lu, B.; Moffatt-Bruce, S.; Durairaj, R.; et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation 2011, 124, 2338–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmstädter, J.; Frenis, K.; Filippou, K.; Grill, A.; Dib, M.; Kalinovic, S.; Pawelke, F.; Kus, K.; Kröller-Schön, S.; Oelze, M.; et al. Endothelial GLP-1 (Glucagon-Like Peptide-1) receptor mediates cardiovascular protection by liraglutide in mice with experimental arterial hypertension. Arter. Thromb. Vasc. Biol. 2020, 40, 145–158. [Google Scholar] [CrossRef]
- Noyan-Ashraf, M.H.; Shikatani, E.A.; Schuiki, I.; Mukovozov, I.; Wu, J.; Li, R.K.; Volchuk, A.; Robinson, L.A.; Billia, F.; Drucker, D.J.; et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation 2013, 127, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Noyan-Ashraf, M.H.; Abdul Momen, M.; Ban, K.; Sadi, A.M.; Zhou, Y.Q.; Riazi, A.M.; Baggio, L.L.; Henkelman, R.M.; Husain, M.; Drucker, D.J. GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 2009, 58, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Wohlfart, P.; Linz, W.; Hübschle, T.; Linz, D.; Huber, J.; Hess, S.; Crowther, D.; Werner, U.; Ruetten, H. Cardioprotective effects of lixisenatide in rat myocardial ischemia-reperfusion injury studies. J. Transl. Med. 2013, 11, 84. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Mori, Y. Anti-atherogenic and anti-inflammatory properties of glucagon-like peptide-1, glucose-dependent insulinotropic polypepide, and dipeptidyl peptidase-4 inhibitors in experimental animals. J. Diabetes Investig. 2016, 7, 80–86. [Google Scholar] [CrossRef]
- Li, J.; Liu, X.; Fang, Q.; Ding, M.; Li, C. Liraglutide attenuates atherosclerosis via inhibiting ER-induced macrophage derived microvesicles production in T2DM rats. Diabetol. Metab. Syndr. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Lee, G.Y.; Park, H.S.; Lee, D.H.; Jung, O.T.; Min, K.K.; Kim, Y.B.; Jun, H.S.; Chul, J.H.; Park, K.S. Attenuation of carotid neointimal formation after direct delivery of a recombinant adenovirus expressing glucagon-like peptide-1 in diabetic rats. Cardiovasc. Res. 2017, 113, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Yamashita, S.; Takahashi, M.; Hashimoto, H.; Mori, Y.; Goto, M. Anagliptin, a dipeptidyl peptidase-4 inhibitor, decreases macrophage infiltration and suppresses atherosclerosis in aortic and coronary arteries in cholesterol-fed rabbits. Metabolism 2016, 65, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Sudo, M.; Li, Y.; Hiro, T.; Takayama, T.; Mitsumata, M.; Shiomi, M.; Sugitani, M.; Matsumoto, T.; Hao, H.; Hirayama, A. Inhibition of plaque progression and promotion of plaque stability by glucagon-like peptide-1 receptor agonist: Serial In vivo findings from iMap-IVUS in Watanabe heritable hyperlipidemic rabbits. Atherosclerosis 2017, 265, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Oyama, J.I.; Higashi, Y.; Node, K. Do incretins improve endothelial function? Cardiovasc. Diabetol. 2014, 13, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.-Y.; Shih, C.-M.; Tsao, N.-W.; Lin, Y.-W.; Huang, P.-H.; Wu, S.-C.; Lee, A.-W.; Kao, Y.-T.; Chang, N.-C.; Nakagami, H.; et al. Dipeptidyl peptidase-4 inhibitor improves neovascularization by increasing circulating endothelial progenitor cells. Br. J. Pharm. 2012, 167, 1506–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akoumianakis, I.; Badi, I.; Douglas, G.; Chuaiphichai, S.; Herdman, L.; Akawi, N.; Margaritis, M.; Antonopoulos, A.S.; Oikonomou, E.K.; Psarros, C.; et al. Insulin-induced vascular redox dysregulation in human atherosclerosis is ameliorated by dipeptidyl peptidase 4 inhibition. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Nagamine, A.; Hasegawa, H.; Hashimoto, N.; Yamada-Inagawa, T.; Hirose, M.; Kobara, Y.; Tadokoro, H.; Kobayashi, Y.; Takano, H. The effects of DPP-4 inhibitor on hypoxia-induced apoptosis in human umbilical vein endothelial cells. J. Pharm. Sci. 2017, 133, 42–48. [Google Scholar] [CrossRef]
- Chihara, A.; Tanaka, A.; Morimoto, T.; Sakuma, M.; Shimabukuro, M.; Nomiyama, T.; Arasaki, O.; Ueda, S.; Node, K. Differences in lipid metabolism between anagliptin and sitagliptin in patients with type 2 diabetes on statin therapy: A secondary analysis of the REASON trial. Cardiovasc. Diabetol. 2019, 18, 158. [Google Scholar] [CrossRef] [Green Version]
- Green, J.B.; Bethel, M.A.; Armstrong, P.W.; Buse, J.B.; Engel, S.S.; Garg, J.; Josse, R.; Kaufman, K.D.; Koglin, J.; Korn, S.; et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2015, 373, 232–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenstock, J.; Kahn, S.E.; Johansen, O.E.; Zinman, B.; Espeland, M.A.; Woerle, H.J.; Pfarr, E.; Keller, A.; Mattheus, M.; Baanstra, D.; et al. Effect of linagliptin vs. glimepiride on major adverse cardiovascular outcomes in patients with type 2 diabetes: The Carolina randomized clinical trial. JAMA J. Am. Med. Assoc. 2019, 322, 1155–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkovic, V.; Toto, R.; Cooper, M.E.; Mann, J.F.E.; Rosenstock, J.; McGuire, D.K.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Zinman, B.; et al. Effects of linagliptin on cardiovascular and kidney outcomes in people with normal and reduced kidney function: Secondary analysis of the carmelina randomized trial. Diabetes Care 2020, 43, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Claggett, B.; Diaz, R.; Dickstein, K.; Gerstein, H.C.; Køber, L.V.; Lawson, F.C.; Ping, L.; Wei, X.; Lewis, E.F.; et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N. Engl. J. Med. 2015, 373, 2247–2257. [Google Scholar] [CrossRef]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef]
- Angelyn Bethel, M.; Mentz, R.J.; Merrill, P.; Buse, J.B.; Chan, J.C.; Goodman, S.G.; Iqbal, N.; Jakuboniene, N.; Katona, B.; Lokhnygina, Y.; et al. Microvascular and cardiovascular outcomes according to renal function in patients treated with once-weekly exenatide: Insights from the EXSCEL trial. Diabetes Care 2020, 43. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Frandsen, K.B.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Bhatt, D.L.; Bain, S.C.; Buse, J.B.; Mann, J.F.E.; Marso, S.P.; Nauck, M.A.; Poulter, N.R.; Pratley, R.E.; Zinman, B.; et al. Effect of liraglutide on cardiovascular events in patients with type 2 diabetes mellitus and polyvascular disease results of the LEADER trial. Circulation 2018, 137, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Poulter, N.R.; Bhatt, D.L.; Bain, S.C.; Buse, J.B.; Leiter, L.A.; Nauck, M.A.; Pratley, R.E.; Zinman, B.; Ørsted, D.D.; et al. Effects of liraglutide on cardiovascular outcomes in patients with type 2 diabetes mellitus with or without history of myocardial infarction or stroke: Post hoc analysis from the leader trial. Circulation 2018, 138, 2884–2894. [Google Scholar] [CrossRef]
- Nauck, M.A.; Tornøe, K.; Rasmussen, S.; Treppendahl, M.B.; Marso, S.P. Cardiovascular outcomes in patients who experienced a myocardial infarction while treated with liraglutide versus placebo in the LEADER trial. Diabetes Vasc. Dis. Res. 2018, 15, 465–468. [Google Scholar] [CrossRef]
- Mann, J.F.E.; Ørsted, D.D.; Brown-Frandsen, K.; Marso, S.P.; Poulter, N.R.; Rasmussen, S.; Tornøe, K.; Zinman, B.; Buse, J.B. Liraglutide and renal outcomes in type 2 diabetes. N. Engl. J. Med. 2017, 377, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jodar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husain, M.; Birkenfeld, A.L.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.G.; Franco, D.R.; Jeppesen, O.K.; Lingvay, I.; Mosenzon, O.; Pedersen, S.D.; et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D’Agostino, R.B.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): A double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Dagenais, G.R.; Rydén, L.; Leiter, L.A.; Lakshmanan, M.; Dyal, L.; Probstfield, J.L.; Atisso, C.M.; Shaw, J.E.; Conget, I.; Cushman, W.C.; et al. Total cardiovascular or fatal events in people with type 2 diabetes and cardiovascular risk factors treated with dulaglutide in the REWIND trail: A post hoc analysis. Cardiovasc. Diabetol. 2020, 19, 199. [Google Scholar] [CrossRef]
- Frias, J.P.; Bastyr, E.J.; Vignati, L.; Tschöp, M.H.; Schmitt, C.; Owen, K.; Christensen, R.H.; DiMarchi, R.D. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. 2017, 26, 343–352. [Google Scholar] [CrossRef]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Coskun, T.; Sloop, K.W.; Loghin, C.; Alsina-Fernandez, J.; Urva, S.; Bokvist, K.B.; Cui, X.; Briere, D.A.; Cabrera, O.; Roell, W.C.; et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol. Metab. 2018, 18, 3–14. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-Like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [Green Version]
- Baggio, L.L.; Drucker, D.J. Harnessing the Therapeutic Potential of Glucagon-Like Peptide-1: A Critical Review. Treat Endocrinol. 2002, 1, 117–125. [Google Scholar] [CrossRef]
- Aroda, V.R. A Review of GLP-1 Receptor Agonists: Evolution and Advancement, Through the Lens of Randomised Controlled Trials. Diabetes Obes. Metab. 2018, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front. Endocrinol. 2019, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Eng, J.; Kleinman, W.A.; Singh, L.; Singh, G.; Raufman, J.P. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem. 1992, 267, 7402–7405. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 Receptor Agonists in the Treatment of type 2 Diabetes—State-of-the-Art. Mol. Metab. 2020, 101–102. [Google Scholar] [CrossRef]
- Deacon, C.F.; Knudsen, L.B.; Madsen, K.; Wiberg, F.C.; Jacobsen, O.; Holst, J.J. Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity. Diabetologia 1998, 41, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, J.G.; Hamilton, A.; Ramracheya, R.; Tarasov, A.I.; Brereton, M.; Haythorne, E.; Chibalina, M.V.; Spégel, P.; Mulder, H.; Zhang, Q.; et al. Dysregulation of glucagon secretion by hyperglycemia-induced sodium-dependent reduction of ATP production. Cell Metab. 2019, 29, 430–442. [Google Scholar] [CrossRef] [Green Version]
- Gaspari, T.; Welungoda, I.; Widdop, R.E.; Simpson, R.W.; Dear, A.E. The GLP-1 receptor agonist liraglutide inhibits progression of vascular disease via effects on atherogenesis, plaque stability and endothelial function in an ApoE/mouse model. Diabetes Vasc. Dis. Res. 2013, 10, 353–360. [Google Scholar] [CrossRef]
- Shiraishi, D.; Fujiwara, Y.; Komohara, Y.; Mizuta, H.; Takeya, M. Glucagon-like peptide-1 (GLP-1) induces M2 polarization of human macrophages via STAT3 activation. Biochem. Biophys. Res. Commun. 2012, 425, 304–308. [Google Scholar] [CrossRef]
- Ding, L.; Zhang, J. Glucagon-like peptide-1 activates endothelial nitric oxide synthase in human umbilical vein endothelial cells. Acta Pharm. Sin. 2012, 33, 75–81. [Google Scholar] [CrossRef]
- Dai, Y.; Mehta, J.L.; Chen, M. Glucagon-like peptide-1 receptor agonist liraglutide inhibits endothelin-1 in endothelial cell by repressing nuclear factor-kappa B activation. Cardiovasc. Drugs Ther. 2013, 27, 371–380. [Google Scholar] [CrossRef]
- Krasner, N.M.; Ido, Y.; Ruderman, N.B.; Cacicedo, J.M. Glucagon-Like Peptide-1 (GLP-1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogdu, Ö.; Eriksson, L.; Nyström, T.; Sjöholm, Å.; Zhang, Q. Exendin-4 restores glucolipotoxicity-induced gene expression in human coronary artery endothelial cells. Biochem. Biophys. Res. Commun. 2012, 419, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Matsui, T.; Hirano, T.; Yamagishi, S.I. Gip as a potential therapeutic target for atherosclerotic cardiovascular disease–a systematic review. Int. J. Mol. Sci. 2020, 21, 1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finan, B.; Ma, T.; Ottaway, N.; Müller, T.D.; Habegger, K.M.; Heppner, K.M.; Kirchner, H.; Holland, J.; Hembree, J.; Raver, C.; et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Sachs, S.; Niu, L.; Geyer, P.; Jall, S.; Kleinert, M.; Feuchtinger, A.; Stemmer, K.; Brielmeier, M.; Finan, B.; DiMarchi, R.D.; et al. Plasma proteome profiles treatment efficacy of incretin dual agonism in diet-induced obese female and male mice. Diabetes Obes. Metab. 2020. [Google Scholar] [CrossRef]
- Khoo, B.; Tan, T.M.M. Combination gut hormones: Prospects and questions for the future of obesity and diabetes therapy. J. Endocrinol. 2020, 246, R65–R74. [Google Scholar] [CrossRef]
- Yamagishi, S.I.; Fukami, K.; Matsui, T. Crosstalk between advanced glycation end products (AGEs)-receptor RAGE axis and dipeptidyl peptidase-4-incretin system in diabetic vascular complications. Cardiovasc. Diabetol. 2015, 14, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Nogi, Y.; Nagashima, M.; Terasaki, M.; Nohtomi, K.; Watanabe, T.; Hirano, T. Glucose-dependent insulinotropic polypeptide prevents the progression of macrophage-driven atherosclerosis in diabetic apolipoprotein E-null mice. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Watanabe, A.; Choe, S.; Chaptal, V.; Rosenberg, J.M.; Wright, E.M.; Grabe, M.; Abramson, J. The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature 2010, 468, 988–991. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.M.; Loo, D.D.F.L.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakris, G.L.; Fonseca, V.A.; Sharma, K.; Wright, E.M. Renal sodium-glucose transport: Role in diabetes mellitus and potential clinical implications. Kidney Int. 2009, 75, 1272–1277. [Google Scholar] [CrossRef] [Green Version]
- Rieg, T.; Vallon, V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia 2018, 61, 2079–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossetti, L.; Smith, D.; Shulman, G.I.; Papachristou, D.; DeFronzo, R.A. Correction of hyperglycemia with phlorizin normalizes tissues sensitivity to insulin in diabetic rats. J. Clin. Investig. 1987, 79, 1510–1515. [Google Scholar] [CrossRef]
- Oku, A.; Ueta, K.; Arakawa, K.; Ishihara, T.; Nawano, M.; Kuronuma, Y.; Matsumoto, M.; Saito, A.; Tsujihara, K.; Anai, M.; et al. T-1095, an inhibitor of renal Na+-glucose cotransporters, may provide a novel approach to treating diabetes. Diabetes 1999, 48, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.; Rajeev, S.P.; Cuthbertson, D.J.; Wilding, J.P.H. A review of the mechanism of action, metabolic profile and hemodynamic effects of sodium-glucose co-transporter-2 inhibitors. Diabetes Obes. Metab. 2019, 21, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, C.; Kerr-Conte, J.; Gmyr, V.; Queniat, G.; Moerman, E.; Thévenet, J.; Beaucamps, C.; Delalleau, N.; Popescu, I.; Malaisse, W.J.; et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat. Med. 2015, 21, 512–517. [Google Scholar] [CrossRef]
- Merovci, A.; Solis-Herrera, C.; Daniele, G.; Eldor, R.; Vanessa Fiorentino, T.; Tripathy, D.; Xiong, J.; Perez, Z.; Norton, L.; Abdul-Ghani, M.A.; et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J. Clin. Investig. 2014, 124, 509–514. [Google Scholar] [CrossRef]
- Tahara, A.; Takasu, T.; Yokono, M.; Imamura, M.; Kurosaki, E. Characterization and comparison of SGLT2 inhibitors: Part 3. Effects on diabetic complications in type 2 diabetic mice. Eur. J. Pharm. 2017, 809, 163–171. [Google Scholar] [CrossRef]
- Aroor, A.R.; Das, N.A.; Carpenter, A.J.; Habibi, J.; Jia, G.; Ramirez-Perez, F.I.; Martinez-Lemus, L.; Manrique-Acevedo, C.M.; Hayden, M.R.; Duta, C.; et al. Glycemic control by the SGLT2 inhibitor empagliflozin decreases aortic stiffness, renal resistivity index and kidney injury. Cardiovasc. Diabetol. 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Battson, M.L.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Weir, T.L.; Gentile, C.L. SGLT2 inhibition via dapagliflozin improves generalized vascular dysfunction and alters the gut microbiota in type 2 diabetic mice. Cardiovasc. Diabetol. 2018, 17, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terasaki, M.; Hiromura, M.; Mori, Y.; Kohashi, K.; Nagashima, M.; Kushima, H.; Watanabe, T.; Hirano, T. Amelioration of hyperglycemia with a sodium-glucose cotransporter 2 inhibitor prevents macrophage-driven atherosclerosis through macrophage foam cell formation suppression in type 1 and type 2 diabetic mice. PLoS ONE 2015, 10, e143396. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.; Spizzo, I.; Liu, H.; Hu, Y.; Simpson, R.W.; Widdop, R.E.; Dear, A.E. Dapagliflozin attenuates human vascular endothelial cell activation and induces vasorelaxation: A potential mechanism for inhibition of atherogenesis. Diabetes Vasc. Dis. Res. 2018, 15, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.H.; Oh, T.J.; Lee, G.; Maeng, H.J.; Lee, D.H.; Kim, K.M.; Choi, S.H.; Jang, H.C.; Lee, H.S.; Park, K.S.; et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE −/− mice fed a western diet. Diabetologia 2017, 60, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Nasiri-Ansari, N.; Dimitriadis, G.K.; Agrogiannis, G.; Perrea, D.; Kostakis, I.D.; Kaltsas, G.; Papavassiliou, A.G.; Randeva, H.S.; Kassi, E.; Nasiri-Ansari, Ν.; et al. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc. Diabetol. 2018, 17, 106. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Bajaj, M.; Yang, H.C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC Inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc. Drugs 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Nassif, M.E.; Windsor, S.; Tang, F.; Khariton, Y.; Husain, M.; Inzucchi, S.; McGuire, D.; Pitt, B.; Scirica, B.; Austin, B.; et al. Dapagliflozin effects on biomarkers, symptoms, and functional status in patients with heart failure with reduced ejection fraction. Circulation 2019, 140, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular outcomes with ertugliflozin in type 2 diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in patients with diabetes and chronic kidney disease. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Szarek, M.; Steg, P.G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; Metra, M.; et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Marx, N. Cardiovascular protection in type 2 diabetes: Insights from recent outcome trials. Diabetes Obes. Metab. 2019, 21, 3–14. [Google Scholar] [CrossRef]
- Seidah, N.G.; Awan, Z.; Chrétien, M.; Mbikay, M. PCSK9: A key modulator of cardiovascular health. Circ. Res. 2014, 114, 1022–1036. [Google Scholar] [CrossRef]
- Abifadel, M.; Varret, M.; Rabès, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Stoekenbroek, R.M.; Lambert, G.; Cariou, B.; Hovingh, G.K. Inhibiting PCSK9—Biology beyond LDL control. Nat. Rev. Endocrinol. 2018, 15, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A.; Pirillo, A.; Catapano, A.L.; Danilo Norata, G. Novel strategies to target proprotein convertase subtilisin kexin 9: Beyond monoclonal antibodies. Cardiovasc. Pathol. 2019, 115, 510–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Bélanger Jasmin, S.; Stifani, S.; Basak, A.; Prat, A.; Chrétien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9 from basic science discoveries to clinical trials. Circ. Res. 2018, 122, 1420–1438. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wang, X.; Liu, S.; Shahanawaz, J.; Theus, S.; Fan, Y.; Deng, X.; Zhou, S.; Mehta, J.L. PCSK9 expression in the ischemic heart and its relationship to infarct size, cardiac function, and development of autophagy. Cardiovasc. Res. 2018, 114, 1738–1751. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef]
- Lagace, T.A. PCSK9 and LDLR degradation: Regulatory mechanisms in circulation and in cells. Curr. Opin. Lipidol. 2014, 25, 387–393. [Google Scholar] [CrossRef]
- Lan, H.; Pang, L.; Smith, M.M.; Levitan, D.; Ding, W.; Liu, L.; Shan, L.; Shah, V.V.; Laverty, M.; Arreaza, G.; et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) affects gene expression pathways beyond cholesterol metabolism in liver cells. J. Cell. Physiol. 2010, 224, 273–281. [Google Scholar] [CrossRef]
- Tang, Z.H.; Li, T.H.; Peng, J.; Zheng, J.; Li, T.T.; Liu, L.S.; Jiang, Z.S.; Zheng, X.L. PCSK9: A novel inflammation modulator in atherosclerosis? J. Cell. Physiol. 2019, 234, 2345–2355. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Shahanawaz, J.; Reis, R.J.S.; Varughese, K.I.; Sawamura, T.; Mehta, J.L. Cross-Talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc. Res. 2015, 107, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Pothineni, N.V.K.; Goel, A.; Lüscher, T.F.; Mehta, J.L. PCSK9 and inflammation: Role of shear stress, pro-inflammatory cytokines, and LOX-1. Cardiovasc. Res. 2020, 116, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Giunzioni, I.; Tavori, H.; Covarrubias, R.; Major, A.S.; Ding, L.; Zhang, Y.; Devay, R.M.; Hong, L.; Fan, D.; Predazzi, I.M.; et al. Local effects of human PCSK9 on the atherosclerotic lesion. J. Pathol. 2016, 238, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.H.; Peng, J.; Ren, Z.; Yang, J.; Li, T.T.; Li, T.H.; Wang, Z.; Wei, D.H.; Liu, L.S.; Zheng, X.L.; et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis 2017, 262, 113–122. [Google Scholar] [CrossRef]
- Guo, Y.; Yan, B.; Gui, Y.; Tang, Z.; Tai, S.; Zhou, S.; Zheng, X.L. Physiology and role of PCSK9 in vascular disease: Potential impact of localized PCSK9 in vascular wall. J. Cell. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Urban, D.; Pöss, J.; Böhm, M.; Laufs, U. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J. Am. Coll. Cardiol. 2013, 62, 1401–1408. [Google Scholar] [CrossRef] [Green Version]
- Rashid, S.; Curtis, D.E.; Garuti, R.; Anderson, N.H.; Bashmakov, Y.; Ho, Y.K.; Hammer, R.E.; Moon, Y.A.; Horton, J.D. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc. Natl. Acad. Sci. USA 2005, 102, 5374–5379. [Google Scholar] [CrossRef] [Green Version]
- Denis, M.; Marcinkiewicz, J.; Zaid, A.; Gauthier, D.; Poirier, S.; Lazure, C.; Seidah, N.G.; Prat, A. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation 2012, 125, 894–901. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Luo, S.; Zhu, Z.; Xu, J. Small Molecules as Inhibitors of PCSK9: Current Status and Future Challenges. Eur. J. Med. Chem. 2019, 162, 212–233. [Google Scholar] [CrossRef]
- Warden, B.A.; Fazio, S.; Shapiro, M.D. The PCSK9 Revolution: Current Status, Controversies and Future Directions: The PCSK9 Revolution. Trends Cardiovasc. Med. 2020, 30, 179–185. [Google Scholar] [CrossRef]
- Nishikido, T.; Ray, K.K. Non-antibody approaches to proprotein convertase subtilisin kexin 9 inhibition: SiRNA, antisense oligonucleotides, adnectins, vaccination, and new attempts at small-molecule inhibitors based on new discoveries. Front. Cardiovasc. Med. 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Kühnast, S.; van der Hoorn, J.W.A.; Pieterman, E.J.; van den Hoek, A.M.; Sasiela, W.J.; Gusarova, V.; Peyman, A.; Schäfer, H.L.; Schwahn, U.; Jukema, J.W.; et al. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J. Lipid Res. 2014, 55, 2103–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.C.Y.; Piper, D.E.; Cao, Q.; Liu, D.; King, C.; Wang, W.; Tang, J.; Liu, Q.; Higbee, J.; Xia, Z.; et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. USA 2009, 106, 9820–9825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaparro-Riggers, J.; Liang, H.; deVay, R.M.; Bai, L.; Sutton, J.E.; Chen, W.; Geng, T.; Lindquist, K.; Casas, M.G.; Boustany, L.M.; et al. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J. Biol. Chem. 2012, 287, 11090–11097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank-Kamenetsky, M.; Grefhorst, A.; Anderson, N.N.; Racie, T.S.; Bramlage, B.; Akinc, A.; Butler, D.; Charisse, K.; Dorkin, R.; Fan, Y.; et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA 2008, 105, 11915–11920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; el Shahawy, M.; et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Brégeault, M.F.; Dalby, A.J.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; et al. Effect of alirocumab on mortality after acute coronary syndromes: An analysis of the ODYSSEY OUTCOMES randomized clinical trial. Circulation 2019, 140, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.; Frank-Kamenetsky, M.; Shulga-Morskaya, S.; Liebow, A.; Bettencourt, B.R.; Sutherland, J.E.; Hutabarat, R.M.; Clausen, V.A.; Karsten, V.; Cehelsky, J.; et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: A randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 2014, 383, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.T.; Turner, T.; Visseren, F.L.J.; et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, V.; Khantalin, I.; Ramin-Mangata, S.; Chémello, K.; Nativel, B.; Lambert, G. PCSK9: From biology to clinical applications. Pathology 2019, 51, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, B.; Hu, M.; Zhang, Y.; Chan, P.; Liu, Z.M. Alirocumab for the treatment of hypercholesterolemia. Expert Opin. Biol. 2017, 17, 633–643. [Google Scholar] [CrossRef]
- Kasichayanula, S.; Grover, A.; Emery, M.G.; Gibbs, M.A.; Somaratne, R.; Wasserman, S.M.; Gibbs, J.P. Clinical pharmacokinetics and pharmacodynamics of evolocumab, a PCSK9 inhibitor. Clin. Pharm. 2018, 57, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Soffer, G.; Pavlyha, M.; Ngai, C.; Thomas, T.; Holleran, S.; Ramakrishnan, R.; Karmally, W.; Nandakumar, R.; Fontanez, N.; Obunike, J.; et al. Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation 2017, 135, 352–362. [Google Scholar] [CrossRef]
- Ogura, M. PCSK9 inhibition in the management of familial hypercholesterolemia. J. Cardiol. 2018, 71, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Stoekenbroek, R.M.; Kallend, D.; Wijngaard, P.L.; Kastelein, J.J. Inclisiran for the treatment of cardiovascular disease: The ORION clinical development program. Future Cardiol. 2018, 14, 433–442. [Google Scholar] [CrossRef]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef] [Green Version]
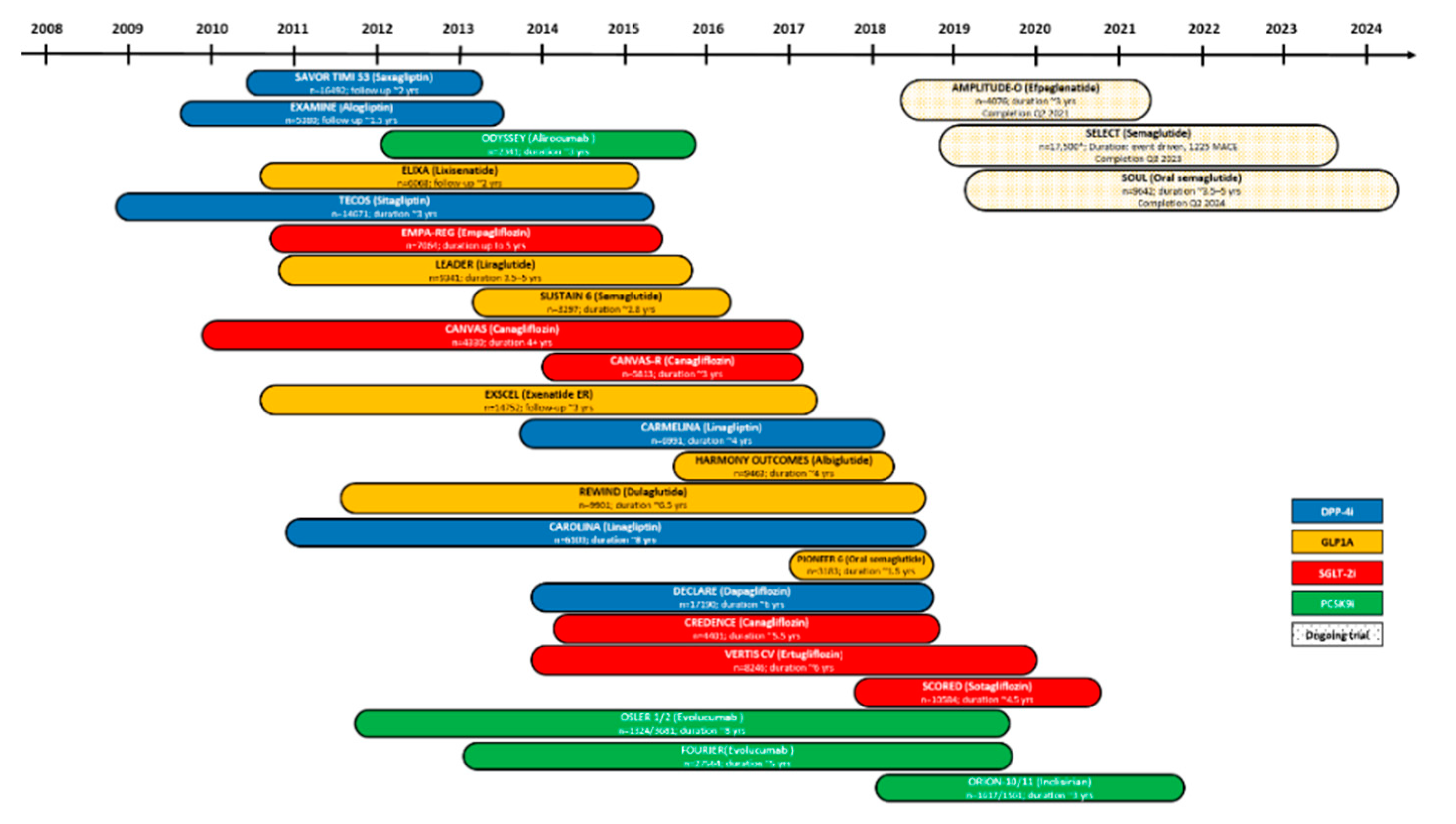
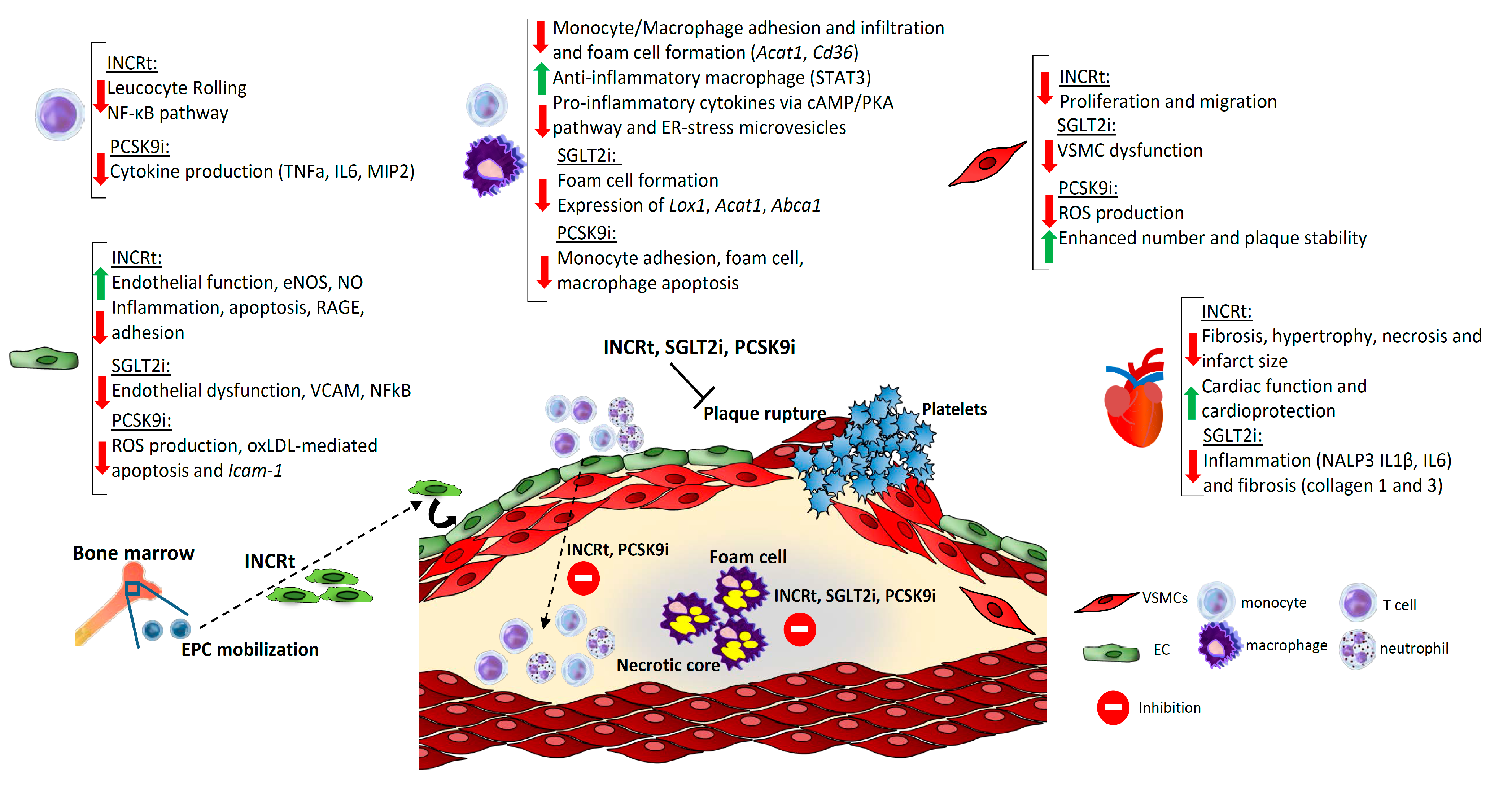

{kind=link}
{kind=link}
| Animal Model | Incretin Therapy | Mechanism of Action | Reported Effects | References |
|---|---|---|---|---|
| Apoe-/- mice | DPP4i Anagliptin | Suppressed VSMCs proliferation and macrophages inflammatory responses | Restrained atherosclerosis | [34] |
| DPP4i Linagliptin+HFD | Anti-inflammatory phenotype of macrophages | Improved atherosclerosis | [35] | |
| GIP active forms | Decreased VSMCs proliferation, monocyte infiltration, foam cell formation and related genes (Cd36, Acat1), and NF-kB-mediated inflammation in macrophages | Stabilization of atherosclerotic plaque | [36] | |
| GIP and GLP1 | Suppressed foam cell formation | Reduced atheroma plaque | [37] | |
| Liraglutide | Suppressed Acat1 expression and foam cell formation | Decreased atherosclerosis | [38] | |
| Anti-inflammatory macrophage polarization | Reduced atherosclerosis | [39] | ||
| Exenatide+CS | Reduced oxidative stress and inflammation | Reduced plaque | [40] | |
| Exendin-4 | Reduced monocyte adhesion and pro-inflammatory cytokines via cAMP/PKA pathway | Decreased lesion size | [41] | |
| APOE*3-Leiden.CETP mice | Exendin-4 | Decreased monocyte recruitment and adhesion and foam cell formation | Reduced atherosclerotic lesions | [42] |
| Apoe-/-Irs2+/- mice | Liraglutide Lixisenatide | STAT3-mediated macrophage polarization to an anti-inflammatory phenotype | Decreased atherosclerosis, necrotic core | [43] |
| Apoe-/- and Ldlr-/- mice | Liraglutide Semaglutide+WD | Changes in inflammatory markers | Reduced lesion size | [44] |
| Lldr-/- mice | DPP4i | Decreased pro-inflammatory genes expression and macrophage content | Decreased plaque size | [45] |
| Arterial hypertension Angiotensin II-mouse model | Liraglutide | Reduced leukocyte rolling and neutrophils infiltration | — | [46] |
| C57Bl6 mice | Liraglutide+45% HFD | Reduced eNOS expression and ER-stress response | Reduced cardiac fibrosis, hypertrophy, and necrosis | [47] |
| Myocardial injury mouse model | Liraglutide | Enhanced GSK3β, PPARβ-δ, Nrf-2, and HO-1 genes | Reduced mortality, infarct size, and rupture | [48] |
| Ischemia-reperfusion injury rats | Lixisenatide | --- | Reduced infarct-size, improved cardiac function | [49] |
| Restenosis mouse/rat model | Lixisenatide Exendin-4 | Reduced VSMC proliferation | Neointimal hyperplasia | [50] |
| Diabetic rats | Liraglutide | Decreased macrophage ER-stress-induced secretion of microvesicles | Diminished atherosclerosis and intima thickening | [51] |
| GLP1+adenovirus-mediated delivery | Reduced VSMC and monocyte migration and inflammation | Reduced intima thickening | [52] | |
| Rabbits | DPP4i Anagliptin+CD | Reduced macrophage infiltration | Restrained atherosclerosis | [53] |
| WHHL rabbits | Lixisenatide | Reduced macrophage, calcium deposition, necrosis | Prevention of plaque growth and instability | [54] |
| Incretin-Therapy | Clinical Trial | Patients | Reported Effects | References |
|---|---|---|---|---|
| DPP4i saxagliptin | SAVOR-TIMI 53 [NCT01107866] | T2DM patients with CV risk | -Unaffected CV risk -Increased HF hospitalization rate | [29] |
| DPP4i alogliptin | EXAMINE [NCT00968708] | T2DM patients with ACS | -Unaffected CV death and hospital admission for HF | [30] |
| DPP4i sitagliptin | TECOS [NCT00790205] | T2DM patients with established CVD | -Unaffected MACE or hospitalization for HF risk | [60] |
| DPP4i Linagliptin | CAROLINA [NCT01243424] | T2DM patients and CV risk | -Unaffected CV risk | [61] |
| CARMELINA [NCT01897532] | T2DM patients and CV risk and kidney disease | -Unaffected HF incidence -No influence of kidney disease -Reduced albuminuria | [31,62] | |
| Lixisenatide (exendin-4 based) | ELIXA [NCT01147250] | T2DM patients with a recent ACS | -No effects on MACE, hospitalization for HF -Decreased SBP and heart rate | [63] |
| Exenatide (exendin-4 synthetic) | EXSCEL [NCT01144338] | T2DM patients with or without CVD | -Unaffected incidence of MACE, retinopathy or renal outcomes -Modest reduction in SBP but increased heart rate | [64] |
| -Modest reduction in CV risk | [65] | |||
| Liraglutide (human GLP1A) | LEADER [NCT01179048] | T2DM patients at high CV risk | -Reduced rates of MACE and death -Reduced SBP and microvascular renal and retinal complications -Enhanced heart rate | [66] |
| -Same benefits for polyvascular and single vascular disease | [67] | |||
| -Reduced CV outcomes (MI/stroke) and CVD | [68] | |||
| -Unaffected HF hospitalization and death risk after MI | [69] | |||
| -Decreased rates of diabetic kidney disease | [70] | |||
| Semaglutide (human GLP1A) | SUSTAIN-6 [NCT01720446] | T2DM patients at high CV risk | -Reduced rates of CV death and non-fatal MI/stroke -Decreased SBP but enhanced mean pulse rate | [71] |
| PIONEER 6 [NCT02692716] | T2DM patients with high CV risk | -Unaffected CV risk -Decreased SBP and LDL-C -Gastrointestinal adverse events | [72] | |
| Albiglutide (modified human GLP1) | Harmony Outcomes [NCT02465515] | T2DM and CVD patients | -Decreased SBP but augmented heart rate -improved glomerular filtration rate -Reduced risk of MACE -Unaffected CV death | [73] |
| Dulaglutide (modified human GLP1) | REWIND [NCT01394952] | T2DM patients at high CV risk | -Unaffected all-cause mortality rate -Decreased SBP, pulse pressure and arterial pressure but enhanced heart rate -Reduced risk of CV outcomes, total CV, or fatal event burden | [74,75] |
| Dulaglutide (modified human GLP-1) and tirzepatide (LY3298176) | SURPASS-CVOT [NCT04255433] | T2DM patients with atherosclerotic CVD | Estimated completion date: October 2024 | https://clinicaltrials.gov/NCT04255433 |
| Efpeglenatide | AMPLITUDE-O [NCT03496298] | T2DM patients with high CDV risk | Estimated completion date: April 2021 | https://clinicaltrials.gov/NCT03496298 |
| NNC0090-2746 (RG7697) [NCT02205528] | Phase 2 trial | T2DM patients | -Improved glycemia control -Diminished body weight, cholesterol, and leptin. | [76] |
| Tirzepatide (LY3298176) [NCT03131687] | Phase 2 trial | T2DM patients | -Improved glycemic and body weight control -Enhanced pulse rate -Acceptable safety and tolerability profile | [77,78] |
| Drug | Animal Model | Effect on Lipids | Effect on Atherosclerosis | References |
|---|---|---|---|---|
| Dapagliflozin | KK/Ay mice | Decreased T-Chol, TG, and NEFAs (ipragliflozin and dapagliflozin) | -Decreased ED (CAMs and E-selectin) and plasmatic inflammatory parameters | [110] |
| Ipragliflozin | ||||
| Canagliflozin | ||||
| Luseogliflozin | ||||
| Empagliflozin | ||||
| Tofogliflozin | ||||
| Empagliflozin | db/db mice | No change in TG and T-Chol | -Reduced aortic and endothelial cell stiffness | [111] |
| Empagliflozin | ZDF rats | No change in TG, T-Chol, HDL, and LDL | -Reduced oxidative stress and inflammation -ED partially prevented | [112] |
| Dapagliflozin | db/db mice | --- | -Lower arterial stiffness -Improved ED and VSMC dysfunction | [113] |
| Ipragliflozin Dapagliflozin | STZ Apoe-/- mice db/db mice | No changes in TG, HDL and T-chol | -Dapagliflozin decreased macrophage infiltration, atherosclerotic lesions and plaque size -Ipragliflozin decreased foam cell formation | [114] |
| Dapagliflozin | Apoe-/- mice | --- | -Attenuated ED and VCAMs expression -Induced vasorelaxation | [115] |
| Empagliflozin | Apoe-/- mice | Decreased TG and increased HDL | -Decreased atherosclerotic plaque and inflammation | [116] |
| Canagliflozin | Apoe-/- mice | Decreased TG, T-chol and LDL | -Increased plaque stability -Reduced atherosclerosis and inflammatory parameters | [117] |
| Dapagliflozin | ob/ob mice | Decreased of TG | -Reduced expression of inflammatory parameters | [118] |
| Drug | Trial | Structural basis | -Effect on vascular and blood parameters, and MACE | References |
| Empagliflozin | EMPA-REG OUTCOME | C-glycosyl compound | -Decreased CV death (HR = 0.86), SBP and DBP -Increased HDL-C and LDL-C | [119] |
| Canagliflozin | CANVAS/CANVAS-R | -No effect (HR = 0.86) -Increased HDL-C and LDL-C | [120] | |
| Canagliflozin | CREDENCE | -Decreased nonfatal stroke/MI and CV death (HR = 0.80) | [121] | |
| Dapagliflozin | DECLARE-TIMI 58 | -No effect (HR = 0.93) -Decreased SBP and DBP | [122] | |
| Dapagliflozin | DEFINE-HF | -No decrease in HF (HR = 0.84) | [123] | |
| Ertugliflozin | VERTIS-CV | -No effect (HR = 0.97) -Decreased SBP | [124] | |
| Sotagliflozin | SCORED | -Decreased CV death (HR = 0.84) | [125] | |
| Sotagliflozin | SOLOIST-WHF | -Decreased CV death (HR = 0.72) | [126] |
| Inhibition Strategy | Animal Models | Effects on Lipids | Effects on Atherosclerosis | References |
|---|---|---|---|---|
| PCSK9 mAb | Cynomolgus monkeys | Decreased LDL-C (80%), T-Chol (48%) | --- | [153] |
| PCSK9 mAb | C57BL/6 mice Cynomolgus monkeys | Decreased LDL-C (40%) | --- | [154] |
| PCSK9 mAb: alirocumab | APOE*3Leiden.CETP mice | Decreased non-HDL-C and TGs | -Decreased inflammation and atherosclerotic lesion -Increased plaque stability | [152] |
| siRNA PCSK9 | Cynomolgus monkeys C57BL/6 mice Sprague–Dawley rats | Decrease of LDL-C and T-Chol (60%) | --- | [155] |
| Inhibition Strategy | Trial Name | Type of Patients | Reported Effects | References |
| PCSK9 mAb: evolocumab | FOURIER/OSLER [NCT01764633; NCT01439880] Phase III | Patients with atherosclerotic CVD | -Increased LDLR and HDL-C -Decreased LDL-C (61%), Total-C (36.1%), TG (12.6%), and Lp(a) (25.5%). -Increased Apolipoprotein A1 and HDL-C -Reduced CV (12–19%) | [156,157] |
| PCAK9 mAb: alirocumab | ODYSSEY [NCT01507831] Phase III | ACS patients | -Increased hepatic LDLR and HDL-C (4%) -Decreased LDL-C (58%), Total-C (37.8%), TG (15.6%), and Lp(a) (29.3%) and MACE -Increased ApoA1 and HDL-C -Reduced CV events (48%) | [158,159] |
| PCSK9 mAb: alirocumab, evolocumab | Meta-analysis | Alirocumab or evolocumab-treated patients | -Decreased nonfatal CV events and mortality. -Improved atherogenic events | [150,160] |
| PCSK9 siRNA: ALN-PCS | Phase I dose-escalation study [NCT01437059] | Healthy adult volunteers | -Reduced LDL-C (40%) | [161] |
| PCSK9 siRNA: ALN-60212 (Inclisirian) | ORION 1 Phase II: [NCT02597127] Phase III 10, 11: [NCT03399370; NCT03400800] | Patients at CVD risk with elevated LDL-C and some receiving statins | -Reduced LDL-C (52.6%) relative to base-line, as well as apoB and non-HDL-C. | [162,163] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar-Ballester, M.; Hurtado-Genovés, G.; Taberner-Cortés, A.; Herrero-Cervera, A.; Martínez-Hervás, S.; González-Navarro, H. Therapies for the Treatment of Cardiovascular Disease Associated with Type 2 Diabetes and Dyslipidemia. Int. J. Mol. Sci. 2021, 22, 660. https://doi.org/10.3390/ijms22020660
Aguilar-Ballester M, Hurtado-Genovés G, Taberner-Cortés A, Herrero-Cervera A, Martínez-Hervás S, González-Navarro H. Therapies for the Treatment of Cardiovascular Disease Associated with Type 2 Diabetes and Dyslipidemia. International Journal of Molecular Sciences. 2021; 22(2):660. https://doi.org/10.3390/ijms22020660
Chicago/Turabian StyleAguilar-Ballester, María, Gema Hurtado-Genovés, Alida Taberner-Cortés, Andrea Herrero-Cervera, Sergio Martínez-Hervás, and Herminia González-Navarro. 2021. "Therapies for the Treatment of Cardiovascular Disease Associated with Type 2 Diabetes and Dyslipidemia" International Journal of Molecular Sciences 22, no. 2: 660. https://doi.org/10.3390/ijms22020660