
Disruption of Chromatin Dynamics by Hypotonic Stress Suppresses HR and Shifts DSB Processing to Error-Prone SSA

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
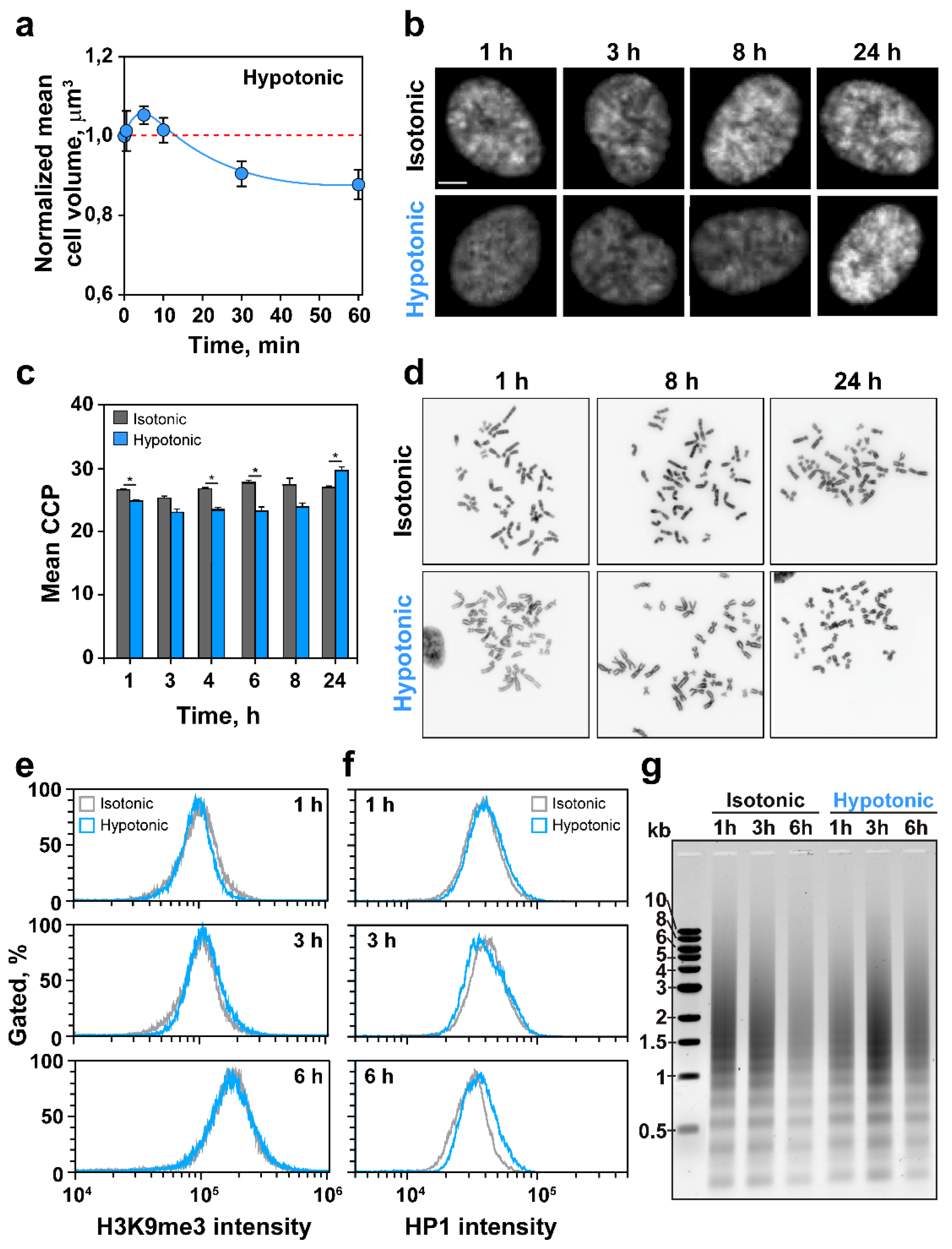
2.1. HypoS Causes Chromatin Decondensation
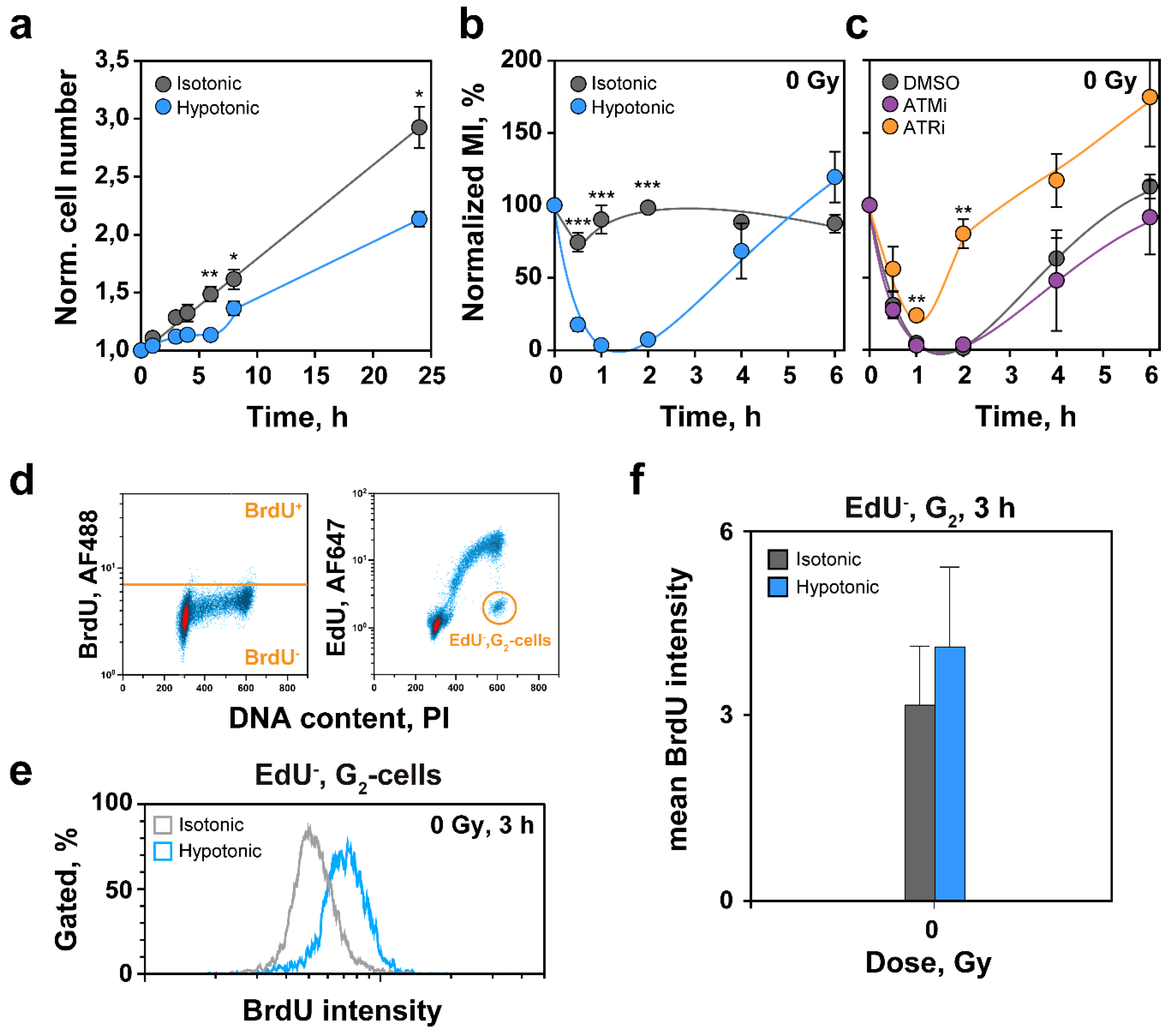
2.2. Effects of HypoS on Cell Proliferation and Cell Cycle Progression
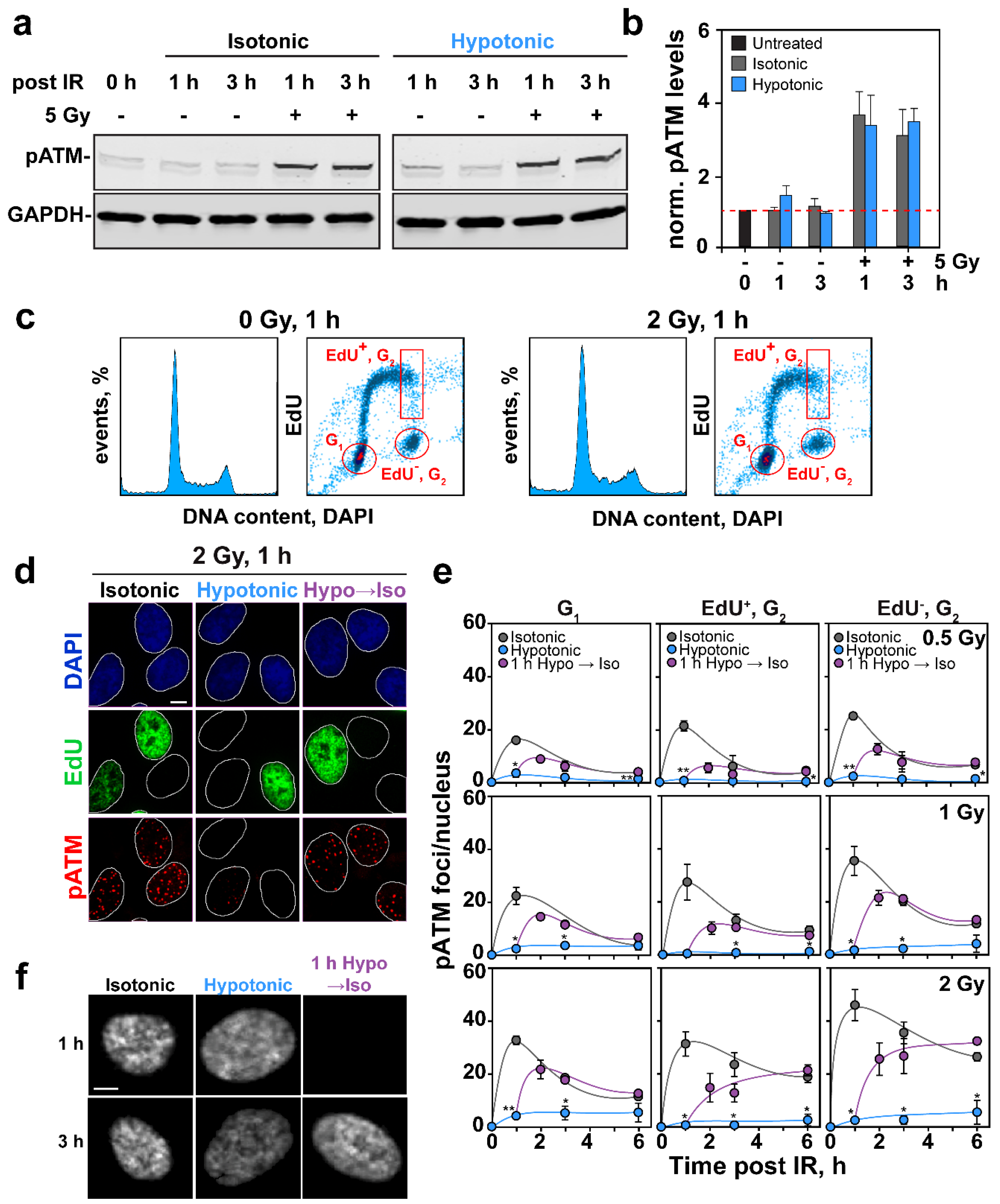
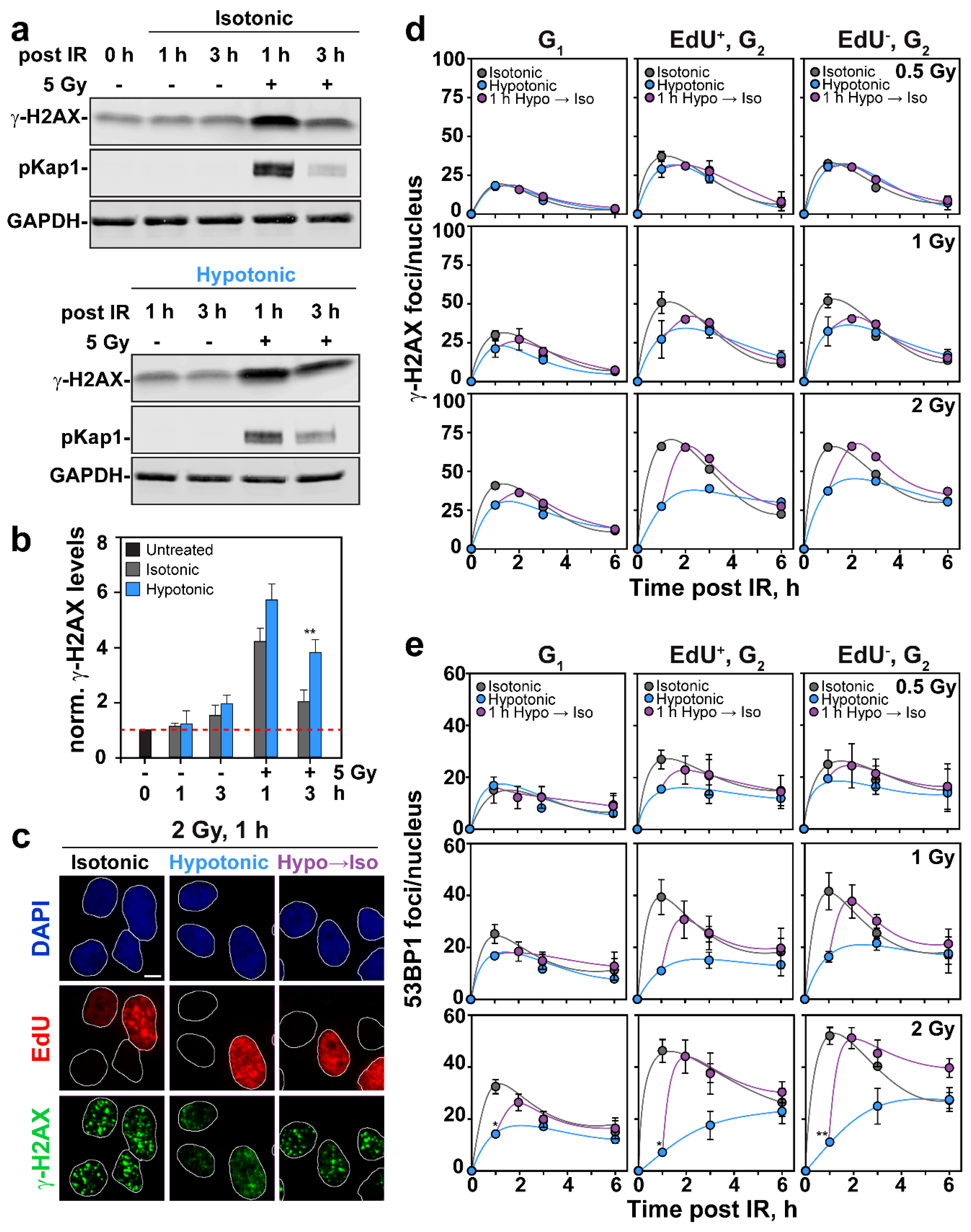
2.3. Effects of HypoS on DDR
2.4. Effects of HypoS on c-NHEJ and alt-EJ
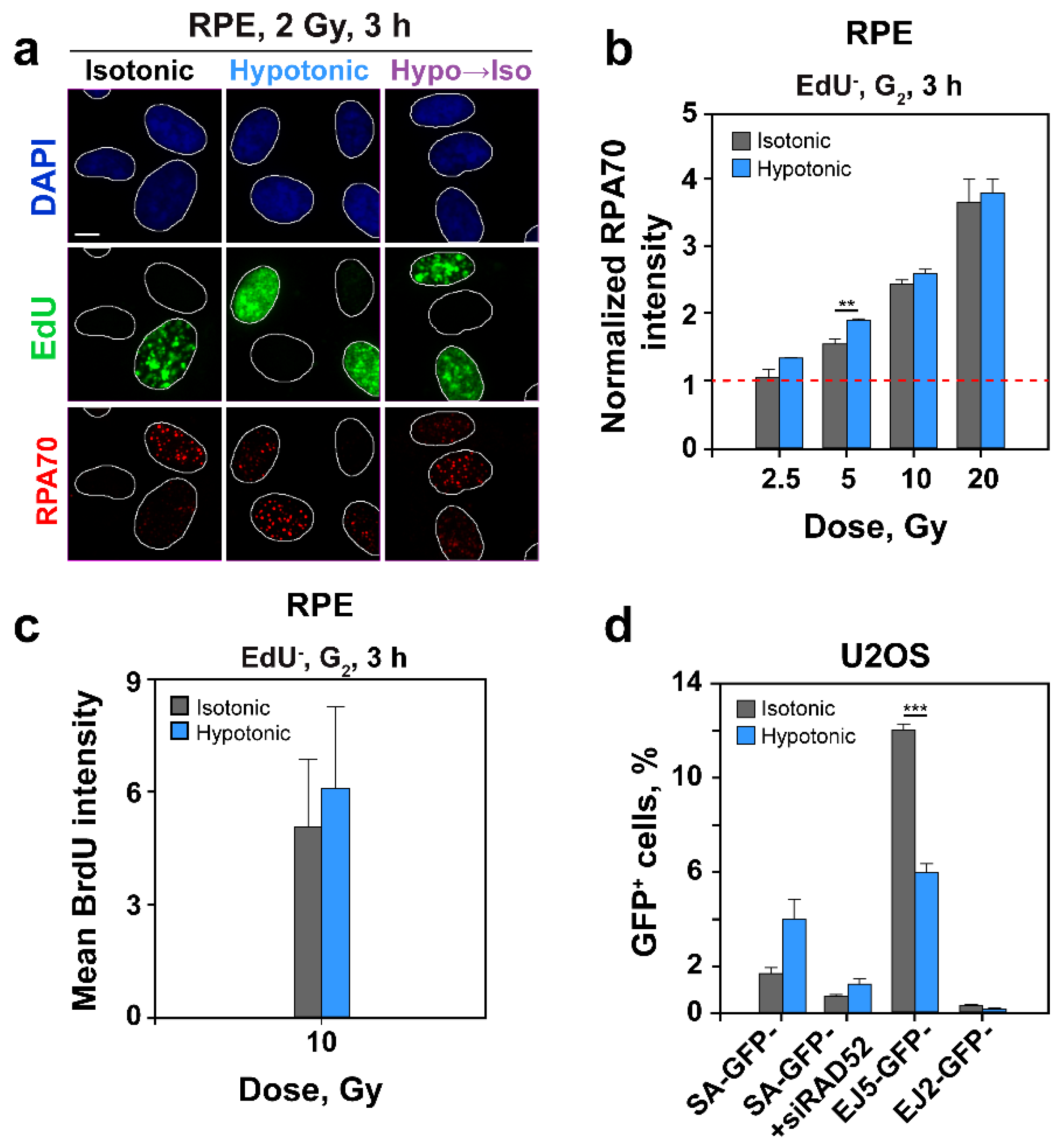
2.5. HypoS Profoundly Suppresses HR
2.6. HypoS Activates SSA to Compensate for HR Inhibition
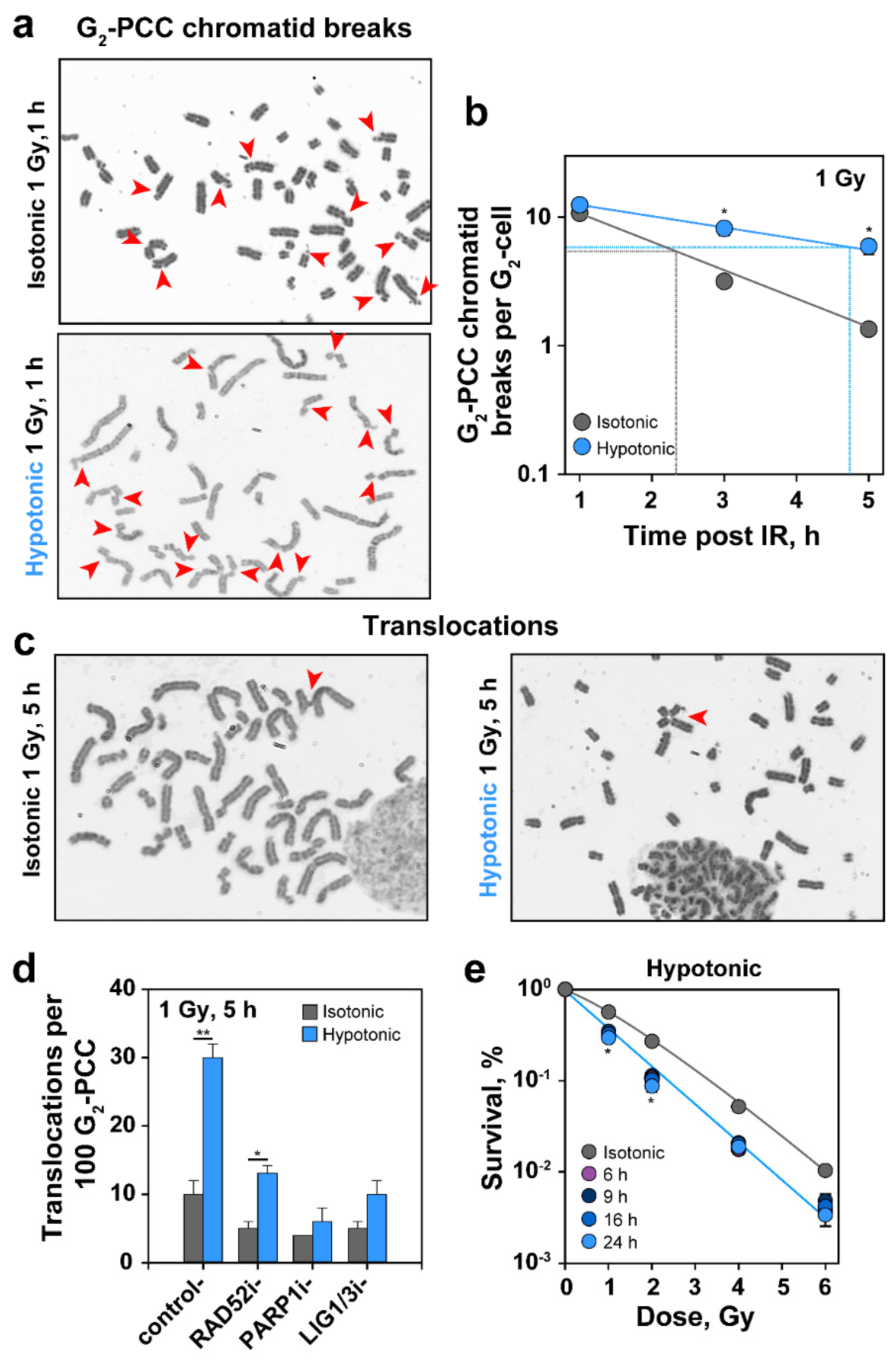
2.7. HypoS Suppresses PCC Break Repair in G2, Increases Formation of Translocations and Radiosensitizes Cells to Killing
3. Discussion
3.1. General Physiological Effects of Hypos
3.2. Effects of HypoS on Cell Cycle Progression and Cell Proliferation
3.3. Effects of HypoS on Chromatin
3.4. Effects of HypoS on DSB Signaling
3.5. HypoS Differentially Affects DSB Processing by Different Repair Pathways
3.6. Effects of HypoS on Chromatid Break Repair, Formation of Translocations and Cell Survival
4. Materials and Methods
4.1. Cell Culture and Cell Volume Measurements
4.2. Radiation Exposure
4.3. Inhibitors
4.4. Micrococcal Nuclease (MNase) Assay
4.5. Pulsed-Field Gel Electrophoresis (PFGE)
4.6. Indirect Immunofluorescence and Image Analysis
4.7. Flow Cytometry (FC) Analysis
4.8. Protein Extraction, SDS-PAGE and Western Blot Analysis
4.9. RNA Interference
4.10. Analysis of HR, c-NHEJ, alt-EJ and SSA Function Using GFP Reporter Cell Lines
4.11. Cytogenetics
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.W.; Elledge, S.J. The DNA Damage Response: Ten Years After. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef]
- Janssen, A.; Breuer, G.A.; Brinkman, E.K.; van der Meulen, A.I.; Borden, S.V.; van Steensel, B.; Bindra, R.S.; LaRocque, J.R.; Karpen, G.H. A single double-strand break system reveals repair dynamics and mechanisms in heterochromatin and euchromatin. Genes Dev. 2016, 30, 1645–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaurasia, P.; Sen, R.; Pandita, T.K.; Bhaumik, S.R. Preferential Repair of DNA Double-strand Break at the Active Gene in Vivo. J. Biol. Chem. 2012, 287, 36414–36422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordelet, H.; Dubrana, K. Keep moving and stay in a good shape to find your homologous recombination partner. Curr. Genet. 2019, 65, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Oberdoerffer, P. Chromatin dynamics in DNA double-strand break repair. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2012, 1819, 811–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeenk, G.; van Attikum, H. The Chromatin Response to DNA Breaks: Leaving a Mark on Genome Integrity. Annu. Rev. Biochem. 2013, 82, 55–80. [Google Scholar] [CrossRef] [PubMed]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-Strand Breaks in Heterochromatin Move Outside of a Dynamic HP1a Domain to Complete Recombinational Repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.-O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Richly, H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. Int. J. Mol. Sci. 2017, 18, 1715. [Google Scholar] [CrossRef] [Green Version]
- Price, B.D.; D’Andrea, A.D. Chromatin Remodeling at DNA Double-Strand Breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Sun, Y.; Jiang, X.; Ayrapetov, M.K.; Moskwa, P.; Yang, S.; Weinstock, D.M.; Price, B.D. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J. Cell Biol. 2010, 191, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Luijsterburg, M.S.; Dinant, C.; Lans, H.; Stap, J.; Wiernasz, E.; Lagerwerf, S.; Warmerdam, D.O.; Lindh, M.; Brink, M.C.; Dobrucki, J.W.; et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 2009, 185, 577–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG Island. PLoS Genet. 2008, 4, e1000155. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-Dependent Chromatin Changes Silence Transcription In cis to DNA Double-Strand Breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soutoglou, E.; Dorn, J.F.; Sengupta, K.; Jasin, M.; Nussenzweig, A.; Ried, T.; Danuser, G.; Misteli, T. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007, 9, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, M.; Derheimer, F.A.; Tait-Mulder, J.; Kastan, M.B. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2013, 110, 16874–16879. [Google Scholar] [CrossRef] [Green Version]
- Dabin, J.; Fortuny, A.; Polo, S.E. Epigenome Maintenance in Response to DNA Damage. Mol. Cell 2016, 62, 712–727. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Lukásová, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to g-radiation. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [Green Version]
- Dewey, W.C.; Noel, J.S.; Dettor, C.M. Changes in radiosensitivity and dispersion of chromatin during the cell cycle of synchronous Chinese hamster cells. Radiat. Res. 1972, 52, 373–394. [Google Scholar] [CrossRef]
- Iliakis, G. Radiation induced potentially lethal damage: DNA lesions susceptible to fixation. (Review Article). Int. J. Radiat. Biol. 1988, 53, 541–584. [Google Scholar] [CrossRef]
- Iliakis, G.; Bryant, P.E.; Ngo, F.Q.H. Independent forms of potentially lethal damage fixed in plateau-phase Chinese hamster cells by post-irradiation treatment in hypertonic salt solution or araA. Radiat. Res. 1985, 104, 329–345. [Google Scholar] [CrossRef]
- Iliakis, G. Evidence for the induction of two types of potentially lethal damage after exposure of plateau phase Chinese hamster V79 cells to γ-rays. Radiat. Environ. Biophys. 1985, 24, 185–202. [Google Scholar] [CrossRef]
- Iliakis, G.; Okayasu, R.; Varlotto, J.; Shernoff, C.; Wang, Y. Hypertonic treatment during premature chromosome condensation allows visualization of interphase chromosome breaks repaired with fast kinetics in irradiated CHO cells. Radiat. Res. 1993, 135, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef]
- Dmitrieva, N.I.; Burg, M.B. Hypertonic stress response. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 2005, 569, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.B.; Kwon, E.D.; Kultz, D. Regulation of gene expression by hypertonicity. Annu. Rev. Physiol. 1997, 59, 437–455. [Google Scholar] [CrossRef]
- Raaphorst, G.P.; Dewey, W.C. Fixation of potentially lethal radiation damage by post-irradiation exposure of Chinese hamster cells to 0-5M or 1-5M NaCl solutions. Int. J. Radiat. Biol. 1979, 36, 303–315. [Google Scholar]
- Tsai, H.J.; Nelliat, A.R.; Choudhury, M.I.; Kucharavy, A.; Bradford, W.D.; Cook, M.E.; Kim, J.; Mair, D.B.; Sun, S.X.; Schatz, M.C.; et al. Hypo-osmotic-like stress underlies general cellular defects of aneuploidy. Nature 2019, 570, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Irianto, J.; Lee, D.A.; Knight, M.M. Quantification of chromatin condensation level by image processing. Med. Eng. Phys. 2014, 36, 412–417. [Google Scholar] [CrossRef]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G(2)-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM epistatically suppress DNA end resection and hyperactivation of ATR-dependent G(2)-checkpoint in S-phase irradiated cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. γ-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong suppression of gene conversion with increasing DNA double-strand break load delimited by 53BP1 and RAD52. Nucleic Acids Res. 2020, 48, 1905–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscariello, M.; Iliakis, G. Effects of chromatin decondensation on alternative NHEJ. DNA Repair 2013, 12, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.; Stark, J.M. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand-breaks. Methods Mol. Biol. 2012, 920, 379–391. [Google Scholar]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [Green Version]
- Tutt, A.; Bertwistle, D.; Valentine, J.; Gabriel, A.; Swift, S.; Ross, G.; Griffin, C.; Thacker, J.; Ashworth, A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001, 20, 4704–4716. [Google Scholar] [CrossRef]
- Do, A.T.; Brooks, J.T.; Le Neveu, M.K.; LaRocque, J.R. Double-Strand Break Repair Assays Determine Pathway Choice and Structure of Gene Conversion Events in Drosophila melanogaster. G3 Genes Genomes Genet. 2014, 4, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Pantelias, G.E.; Iliakis, G. Chromosome breaks generated by low doses of ionizing radiation in G(2)-phase are processed exclusively by gene conversion. DNA Repair 2020, 89, 102828. [Google Scholar] [CrossRef]
- Hubert, A.; Cauliez, B.; Chedeville, A.; Husson, A.; Lavoinne, A. Osmotic stress, a proinflammatory signal in Caco-2 cells. Biochimie 2004, 86, 533–541. [Google Scholar] [CrossRef]
- Nako, Y.; Shiozaki, A.; Ichikawa, D.; Komatsu, S.; Konishi, H.; Iitaka, D.; Ishii, H.; Ikoma, H.; Kubota, T.; Fujiwara, H.; et al. Enhancement of the cytocidal effects of hypotonic solution using a chloride channel blocker in pancreatic cancer cells. Pancreatology 2012, 12, 440–448. [Google Scholar] [CrossRef]
- Kudou, M.; Shiozaki, A.; Kosuga, T.; Ichikawa, D.; Konishi, H.; Morimura, R.; Komatsu, S.; Ikoma, H.; Fujiwara, H.; Okamoto, K.; et al. Inhibition of Regulatory Volume Decrease Enhances the Cytocidal Effect of Hypotonic Shock in Hepatocellular Carcinoma. J. Cancer 2016, 7, 1524–1533. [Google Scholar] [CrossRef] [Green Version]
- Lima, A.F.; May, G.; Díaz-Colunga, J.; Pedreiro, S.; Paiva, A.; Ferreira, L.; Enver, T.; Iborra, F.J.; Pires das Neves, R. Osmotic modulation of chromatin impacts on efficiency and kinetics of cell fate modulation. Sci. Rep. 2018, 8, 7210. [Google Scholar] [CrossRef] [Green Version]
- Erickson, G.R.; Alexopoulos, L.G.; Guilak, F. Hyper-osmotic stress induces volume change and calcium transients in chondrocytes by transmembrane, phospholipid, and G-protein pathways. J. Biomech. 2001, 34, 1527–1535. [Google Scholar] [CrossRef]
- Nandigama, R.; Padmasekar, M.; Wartenberg, M.; Sauer, H. Feed forward cycle of hypotonic stress-induced ATP release, purinergic receptor activation, and growth stimulation of prostate cancer cells. J. Biol. Chem. 2006, 281, 5686–5693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Rhee, P.L.; Rhee, J.C.; Kim, Y.I.; So, I.; Kim, K.W.; Park, M.K.; Uhm, D.Y.; Kang, T.M. Hypotonic swelling increases L-type calcium current in smooth muscle cells of the human stomach. Exp. Physiol. 2000, 85, 497–504. [Google Scholar] [CrossRef]
- Huang, C.; Li, Y.; Wang, H. The observation of high hypotonicity manipulating cell division. Heliyon 2019, 5, e02095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, G.Z.; Rott, L.S.; Lowe, A.W.; Omary, M.B. Hyposmotic stress induces cell growth arrest via proteasome activation and cyclin/cyclin-dependent kinase degradation. J. Biol. Chem. 2002, 277, 19295–19303. [Google Scholar] [CrossRef] [Green Version]
- Jäckle, T.; Hasel, C.; Melzner, I.; Brüderlein, S.; Jehle, P.M.; Möller, P. Sustained hyposmotic stress induces cell death: Apoptosis by defeat. Am. J. Physiol.-Cell Physiol. 2001, 281, C1716–C1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dettor, C.M.; Dewey, W.C.; Winans, L.F.; Noel, J.S. Enhancement of X-ray damage in synchronous Chinese hamster cells by hypertonic treatments. Radiat. Res. 1972, 52, 352–372. [Google Scholar] [CrossRef] [PubMed]
- Raaphorst, G.P.; Dewey, W.C. Alterations in the radiosensitivity of CHO cells by anisotonic treatments: Correlations between cell lethality and chromosomal aberrations. Radiat. Res. 1979, 79, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Brooker, A.S.; Berkowitz, K.M. The roles of cohesins in mitosis, meiosis, and human health and disease. Methods Mol. Biol. 2014, 1170, 229–266. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T. Chromosome shaping by two condensins. Cell Cycle 2004, 3, 26–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowell, I.G.; Sunter, N.J.; Singh, P.B.; Austin, C.A.; Durkacz, B.W.; Tilby, M.J. gH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS ONE 2007, 2, e1057. [Google Scholar] [CrossRef]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-resolution profiling of gH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019, 38, e101379. [Google Scholar] [CrossRef]
- Yasuda, S.; Tsuchiya, H.; Kaiho, A.; Guo, Q.; Ikeuchi, K.; Endo, A.; Arai, N.; Ohtake, F.; Murata, S.; Inada, T.; et al. Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 2020, 578, 296–300. [Google Scholar] [CrossRef]
- Khurana, S.; Kruhlak, M.J.; Kim, J.; Tran, A.D.; Liu, J.; Nyswaner, K.; Shi, L.; Jailwala, P.; Sung, M.-H.; Hakim, O.; et al. A Macrohistone Variant Links Dynamic Chromatin Compaction to BRCA1-Dependent Genome Maintenance. Cell Rep. 2014, 8, 1049–1062. [Google Scholar] [CrossRef] [Green Version]
- Adam, S.; Dabin, J.; Polo, S.E. Chromatin plasticity in response to DNA damage: The shape of things to come. DNA Repair 2015, 32, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Elia, M.C.; Bradley, M.O. Influence of chromatin structure on the induction of DNA double strand breaks by ionizing radiation. Cancer Res. 1992, 52, 1580–1586. [Google Scholar]
- Warters, R.L.; Lyons, B.W. Variation in radiation-induced formation of DNA double-strand breaks as a function of chromatin structure. Radiat. Res. 1992, 130, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin compaction protects genomic DNA from radiation damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef] [Green Version]
- Lobrich, M.; Jeggo, P. A Process of Resection-Dependent Nonhomologous End Joining Involving the Goddess Artemis. Trends Biochem. Sci. 2017, 42, 690–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoda, E.; Hochegger, H.; Saberi, A.; Taniguchi, Y.; Takeda, S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair 2006, 5, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yu, H. The Smc complexes in DNA damage response. Cell Biosci. 2012, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, M.C.; Laulier, C.; Gunn, A.; Cheng, A.; Robbiani, D.F.; Nussenzweig, A.; Stark, J.M. Ring Finger Nuclear Factor RNF168 Is Important for Defects in Homologous Recombination Caused by Loss of the Breast Cancer Susceptibility Factor BRCA1. J. Biol. Chem. 2012, 287, 40618–40628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, L.; Dong, C.; Li, J.; Chen, J.; Yuan, J.; Huang, J.; Chan, K.M.; Yu, C.H.; Huen, M.S.Y. RNF169 limits 53BP1 deposition at DSBs to stimulate single-strand annealing repair. Proc. Natl. Acad. Sci. USA 2018, 115, E8286–E8295. [Google Scholar] [CrossRef] [Green Version]
- Elliott, B.; Richardson, C.; Jasin, M. Chromosomal Translocation Mechanisms at Intronic Alu Elements in Mammalian Cells. Mol. Cell 2005, 17, 885–894. [Google Scholar] [CrossRef]
- Manthey, G.M.; Bailis, A.M. Rad51 Inhibits Translocation Formation by Non-Conservative Homologous Recombination in Saccharomyces cerevisiae. PLoS ONE 2010, 5, e11889. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, X. Microhomology-mediated end joining: New players join the team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Magin, S.; Meher, P.K.; Iliakis, G. Nucleoside Analogs Radiosensitize G0 Cells by Activating DNA End Resection and Alternative End-Joining. Radiat. Res. 2021, 195, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Magin, S.; Papaioannou, M.; Saha, J.; Staudt, C.; Iliakis, G.E. Inhibition of homologous recombination and promotion of mutagenic repair of DNA double-strand breaks underpins arabinoside-nucleoside analog-radiosensitization. Mol. Cancer Ther. 2015, 14, 1424–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, A.; Mladenov, E.; Iliakis, G. Proficiency in homologous recombination repair is prerequisite for activation of G(2)-checkpoint at low radiation doses. DNA Repair 2021, 101, 103076. [Google Scholar] [CrossRef]
- Murmann-Konda, T.; Soni, A.; Stuschke, M.; Iliakis, G. Analysis of chromatid-break-repair detects a homologous recombination to non-homologous end-joining switch with increasing load of DNA double-strand breaks. Mutat. Res. 2021, 867, 503372. [Google Scholar] [CrossRef]
- Roukos, V.; Voss, T.C.; Schmidt, C.K.; Lee, S.; Wangsa, D.; Misteli, T. Spatial Dynamics of Chromosome Translocations in Living Cells. Science 2013, 341, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Loucas, B.D.; Cornforth, M.N. The LET Dependence of Unrepaired Chromosome Damage in Human Cells: A Break Too Far? Radiat. Res. 2013, 179, 393–405. [Google Scholar] [CrossRef]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Okayasu, R.; Varlotto, J.; Iliakis, G. Hypertonic treatment does not affect the radiation yield of interphase chromosome breaks in DNA double-strand break repair deficient xrs-5 cells. Radiat. Res. 1993, 135, 171–177. [Google Scholar] [CrossRef]
- Okayasu, R.; Iliakis, G. Ionizing radiation induces two forms of interphase chromosome breaks in CHO cells that rejoin with different kinetics and show different sensitivity to treatment in hypertonic medium or b-araA. Radiat. Res. 1993, 136, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.A.; Tolmach, L.J. Repair of potentially lethal damage in x-irradiated hela cells. Radiat. Res. 1966, 29, 413–432. [Google Scholar] [CrossRef]
- Moscariello, M.; Wieloch, R.; Kurosawa, A.; Li, F.; Adachi, N.; Mladenov, E.; Iliakis, G. Role for Artemis nuclease in the repair of radiation-induced DNA double strand breaks by alternative end joining. DNA Repair 2015, 31, 29–40. [Google Scholar] [CrossRef]
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem. Biol. 2015, 22, 1491–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhong, S.; Zhu, X.; Dziegielewska, B.; Ellenberger, T.; Wilson, G.M.; MacKerell, A.D., Jr.; Tomkinson, A.E. Rational Design of Human DNA Ligase Inhibitors that Target Cellular DNA Replication and Repair. Cancer Res. 2008, 68, 3169–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Wang, M.; Wu, W.; Singh, S.K.; Mussfeldt, T.; Iliakis, G. Repair of radiation induced DNA double strand breaks by backup NHEJ is enhanced in G2. DNA Repair 2008, 7, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Metzger, L.; Iliakis, G. Kinetics of DNA double strand breaks throughout the cell cycle as assayed by pulsed field gel electrophoresis in CHO cells. Int. J. Radiat. Biol. 1991, 59, 1325–1339. [Google Scholar] [CrossRef]
- Kenny, M.K.; Schlegel, U.; Furneaux, H.; Hurwitz, J. The role of human single-stranded DNA binding protein and its individual subunits in simian virus 40 DNA replication. J. Biol. Chem. 1990, 265, 7693–7700. [Google Scholar] [CrossRef]
- Soni, A.; Siemann, M.; Grabos, M.; Murmann, T.; Pantelias, G.E.; Iliakis, G. Requirement for Parp-1 and DNA ligases 1 or 3 but not of Xrcc1 in chromosomal translocation formation by backup end joining. Nucleic Acids Res. 2014, 42, 6380–6392. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krieger, L.M.; Mladenov, E.; Soni, A.; Demond, M.; Stuschke, M.; Iliakis, G. Disruption of Chromatin Dynamics by Hypotonic Stress Suppresses HR and Shifts DSB Processing to Error-Prone SSA. Int. J. Mol. Sci. 2021, 22, 10957. https://doi.org/10.3390/ijms222010957
Krieger LM, Mladenov E, Soni A, Demond M, Stuschke M, Iliakis G. Disruption of Chromatin Dynamics by Hypotonic Stress Suppresses HR and Shifts DSB Processing to Error-Prone SSA. International Journal of Molecular Sciences. 2021; 22(20):10957. https://doi.org/10.3390/ijms222010957
Chicago/Turabian StyleKrieger, Lisa Marie, Emil Mladenov, Aashish Soni, Marilen Demond, Martin Stuschke, and George Iliakis. 2021. "Disruption of Chromatin Dynamics by Hypotonic Stress Suppresses HR and Shifts DSB Processing to Error-Prone SSA" International Journal of Molecular Sciences 22, no. 20: 10957. https://doi.org/10.3390/ijms222010957
APA StyleKrieger, L. M., Mladenov, E., Soni, A., Demond, M., Stuschke, M., & Iliakis, G. (2021). Disruption of Chromatin Dynamics by Hypotonic Stress Suppresses HR and Shifts DSB Processing to Error-Prone SSA. International Journal of Molecular Sciences, 22(20), 10957. https://doi.org/10.3390/ijms222010957