
Mastocytosis and Mast Cell Activation Disorders: Clearing the Air



Abstract
:1. Introduction
2. Brief Overview of Mast Cell Biology
3. Stem Cell Factor and Its Receptor
4. Materials and Methods
5. Classification
6. Mastocytosis
- >25% of infiltrating mast cells are atypical or spindle-shaped
- KIT mutation in bone marrow or peripheral blood or extracutaneous tissue
- Other rarer mutations may be observed
- These include TET2, SRSF2, ASXL1, RUNX1, CBL, JAK2
- Mast cells express clonal markers such as CD25 or CD2 by immunocytochemistry or flow cytometry (besides CD117 or tryptase)
- Serum tryptase is >20 ng/mL (unless there is an underlying myeloid neoplasm)
- Meets all criteria for systemic mastocytosis
- Additional 2 or more type B symptoms are present
- >30% of cells in bone marrow or extracutaneous tissue are mast cells and tryptase levels are >200 ng/mL
- Dysplasia or myeloproliferation of non-mast cell lineage cells but not meeting criteria for hematological neoplasm
- Organomegaly (hepatomegaly, splenomegaly, lymphadenopathy) in the absence of functional organ impairment
- Meets criteria for systemic mastocytosis
- Meets criteria for associated hematological neoplasm
- Meets criteria for systemic mastocytosis
- Additional 1 or > C type findings but no evidence for mast cell leukemia
- Bone marrow dysfunction due to mast cell infiltration with associated cytopenia (neutropenia, anemia, thrombocytopenia)
- Hepatomegaly with abnormal liver functions, portal hypertension or ascites
- Osteolytic lesions with associated pathological fractures not related to osteoporosis
- Palpable splenomegaly with hypersplenism and cytopenia
- Gastrointestinal infiltration by mast cells with malabsorption
- Meets criteria for systemic mastocytosis
- Bone marrow is densely infiltrated by atypical or immature mast cells
- Bone marrow aspirate shows mast cells >30%
- Mast cells may represent >10% of circulating white cells in blood except in aleukemic variant
- Does not meet criteria for mastocytosis
- Highly malignant atypical mast cells with destructive and metastatic potential
- Mast cell activation symptoms are rare (<20%)
- Minority of patients express the canonical D816V mutation
7. Clonal Disorders: Cutaneous Mastocytosis
8. Clonal Disorders: Systemic Mastocytosis and Advanced Systemic Mastocytosis
9. Nonclonal Disorder: Mast Cell Sarcoma
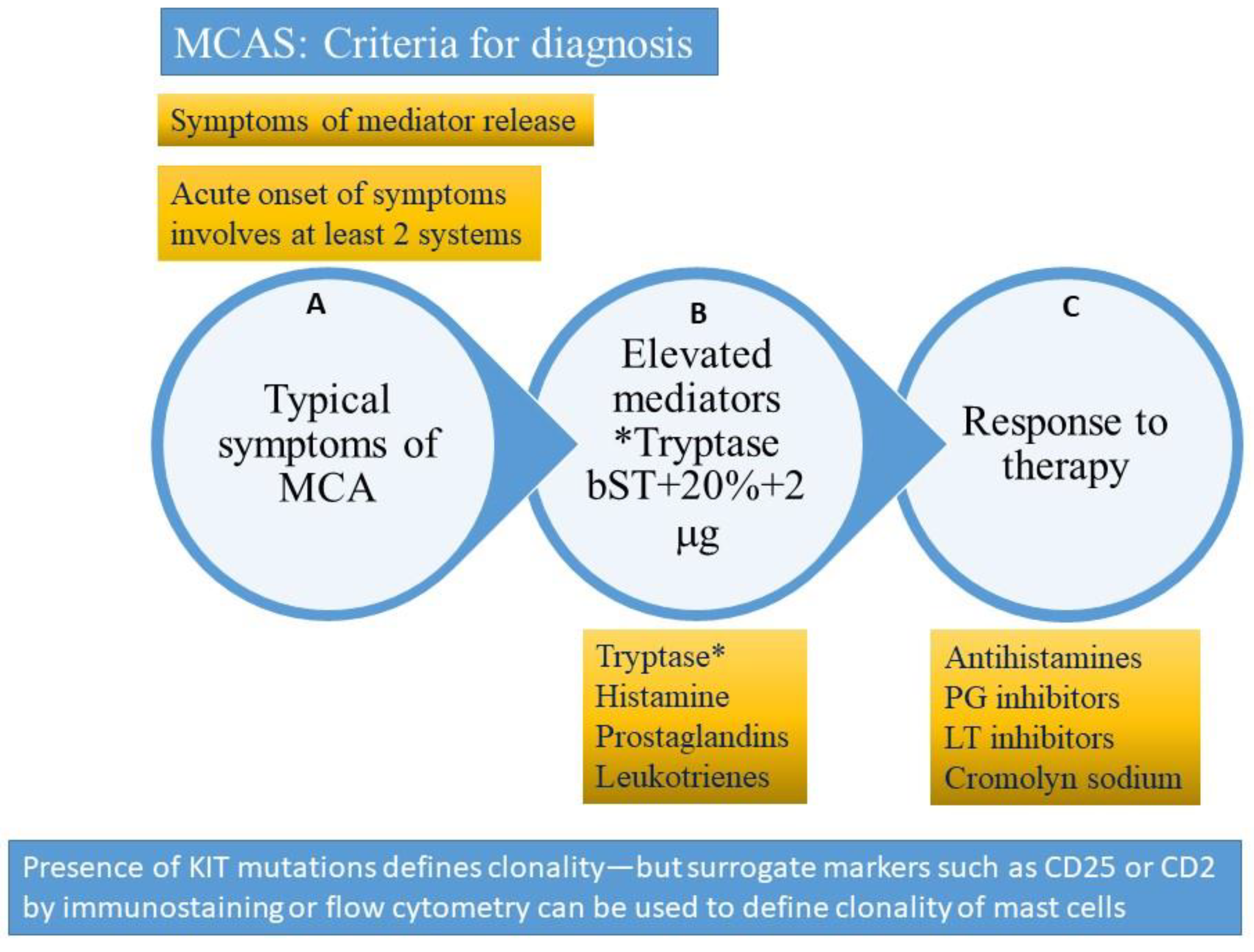
10. Mast Cell Activation Syndrome
- Acute and recurrent symptoms arising from mast cell activation and degranulation with at least 2 organ systems affected at presentation (skin, gastrointestinal, respiratory, neuropsychiatric or cardiovascular)
- Elevated mast cell-derived mediators (usually tryptase increase using formula 20% + 2 ng/mL over basal tryptase) during episode
- Improvement of symptoms after treatment with inhibitors of mast cell mediator release or function
- Does not meet criteria for systemic mastocytosis
- Meets criteria for MCAS as above
- Clonality is present
- KIT Asp816Val mutation in >80% cases (in tissue, peripheral blood or might require bone marrow evaluation) by next generation sequencing or allele-specific polymerase chain reaction
- Otherrarer mutations (TET2, SRSF2, ASXL1, RUNX1, CBL, JAK2) are observed more in systemic mastocytosis than MMAS
- Expressionof CD2 (lymphocyte function-associated antigen 2) and/or CD25 (alpha chain ofhigh affinity receptor for interleukin 2) markers on mast cells by immunostaining or by flow cytometry
- Does not meet criteria for systemic mastocytosis
- Meets criteria for MCAS
- Mast cell clonality is absent
- Underlying etiology for mast cell activation is usually present (allergy to medications or foods, autoimmunity, neoplasia or infection)
- Does not meet criteria for systemic mastocytosis
- Meets criteria for MCAS
- Mast cell clonality is absent
- Underlying trigger for mast cell activation is usually absent
11. Diagnosis of Mast Cell Activation Syndrome
12. Primary or Clonal Mast Cell Activation Syndrome (MMAS or CMCAS)
13. Secondary (Non-Clonal) Mast Cell Activating Syndrome
14. Combined MCAS
15. Idiopathic Anaphylaxis and Idiopathic Mast Cell Activation
16. Hymenoptera Reactions and Mast Cell Disorders
17. Bone Marrow Mastocytosis
18. Hereditary Alpha Tryptasemia
19. Combined Disorders
20. Management of Mast Cell Disorders
21. Unmet Needs and Areas of Uncertainty
22. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghably, J.; Saleh, H.; Vyas, H.; Peiris, E.; Misra, N.; Krishnaswamy, G. Paul Ehrlich’s mastzellen: A historical perspective of relevant developments in mast cell biology. Methods Mol. Biol. 2015, 1220, 3–10. [Google Scholar]
- Krishnaswamy, G.; Kelley, J.; Johnson, D.; Chi, D.S. The human mast cell: Functions in physiology and disease. Front. Biosci. 2001, 6, D1109–D1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnaswamy, G.; Ajitawi, O.; Chi, D.S. The human mast cell: An overview. Methods Mol. Biol. 2006, 315, 13–34. [Google Scholar] [PubMed]
- Elieh Ali, K.D.; WÃhrl, S.; Bielory, L. Mast Cell Biology at Molecular Level: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2020, 58, 342–365. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.M.T.; Rupprecht, C.P.; Haque, A.; Pattanaik, D.; Yusin, J.; Krishnaswamy, G. Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond. Int. J. Mol. Sci. 2021, 22, 7785. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, P.; Bonifacio, M.; Lombardo, C.; Zanotti, R. Hymenoptera Allergy and Mast Cell Activation Syndromes. Curr. Allergy Asthma Rep. 2016, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, P.; Bonifacio, M.; Zanotti, R. Mast Cell Disorders in Drug Hypersensitivity. Curr. Pharm. Des. 2016, 22, 6862–6869. [Google Scholar] [CrossRef]
- Butterfield, J.H.; Ravi, A.; Pongdee, T. Mast Cell Mediators of Significance in Clinical Practice in Mastocytosis. Immunol. Allergy Clin. N. Am. 2018, 38, 397–410. [Google Scholar] [CrossRef]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2015, 6, 620. [Google Scholar] [CrossRef] [Green Version]
- Gehlen, M.; Schmidt, N.; Pfeifer, M. Osteoporosis Caused by Systemic Mastocytosis: Prevalence in a Cohort of 8392 Patients with Osteoporosis. Calcif. Tissue Int. 2021. [Google Scholar] [CrossRef]
- da Silva, E.Z.; Jamur, M.C.; Oliver, C. Mast cell function: A new vision of an old cell. J. Histochem. Cytochem. 2014, 62, 698–738. [Google Scholar] [CrossRef]
- Prussin, C.; Metcalfe, D.D. 5. IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Immunol. 2006, 117, S450–S456. [Google Scholar] [CrossRef]
- Matito, A.; Escribese, M.M.; Longo, N. Clinical Approach to Mast Cell Activation Syndromes: A Practical Overview. J. Investig. Allergol Clin. Immunol. 2021. [Google Scholar] [CrossRef]
- Sabato, V.; Michel, M.; Blank, U.; Ebo, D.G.; Vitte, J. Mast cell activation syndrome: Is anaphylaxis part of the phenotype? A systematic review. Curr. Opin. Allergy Clin. Immunol. 2021, 21, 426–434. [Google Scholar] [CrossRef]
- Niedoszytko, M.; Valent, P.; Nedoszytko, B. Mastocytosis, MCAS, and Related Disorders-Diagnosis, Classification, and Therapy. Int. J. Mol. Sci. 2021, 22, 5024. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C. Doctor, I Think I Am Suffering from MCAS: Differential Diagnosis and Separating Facts from Fiction. J. Allergy Clin. Immunol. Pract. 2019, 7, 1109–1114. [Google Scholar] [CrossRef]
- Yu, Y.; Blokhuis, B.R.; Garssen, J.; Redegeld, F.A. Non-IgE mediated mast cell activation. Eur. J. Pharmacol. 2016, 778, 33–43. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Criscuolo, M.; GullÃ, C.; Petrosino, A.; Carlo, B.N.; Colosimo, C. Systemic mastocytosis revisited with an emphasis on skeletal manifestations. Radiol. Med. 2021, 126, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Gangireddy, M.; Ciofoaia, G.A. Systemic Mastocytosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Afrin, L.B.; Ackerley, M.B.; Bluestein, L.S.; Brewer, J.H.; Brook, J.B.; Buchanan, A.D.; Cuni, J.R.; Davey, W.P.; Dempsey, T.T.; Dorff, S.R.; et al. Diagnosis of mast cell activation syndrome: A global “consensus-2”. Diagnosis 2021, 8, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, L.B.; Pace, L.A.; Rezaie, A.; Afrin, L.B.; Molderings, G.J. Mast Cell Activation Syndrome: A Primer for the Gastroenterologist. Dig. Dis. Sci. 2021, 66, 965–982. [Google Scholar] [CrossRef]
- Schaffer, J.V. Pediatric Mastocytosis: Recognition and Management. Am. J. Clin. Dermatol. 2021, 22, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Sandru, F.; Petca, R.-C.; Costescu, M.; Dumitrașcu, M.; Popa, A.; Petca, A.; Miulescu, R.-G. Cutaneous Mastocytosis in Childhood-Update from the Literature. J. Clin. Med. 2021, 10, 1474. [Google Scholar] [CrossRef] [PubMed]
- Khokhar, D.; Akin, C. Mast Cell Activation: When the Whole Is Greater than the Sum of Its Parts. Med. Clin. N. Am. 2020, 104, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Bonadonna, P.; Hartmann, K.; Brockow, K.; Niedoszytko, M.; Nedoszytko, B.; Siebenhaar, F.; Sperr, W.R.; Oude Elberink, J.N.G.; et al. Proposed Diagnostic Algorithm for Patients with Suspected Mast Cell Activation Syndrome. J. Allergy Clin. Immunol. Pract. 2019, 7, 1125–1133. [Google Scholar] [CrossRef]
- Akin, C. Mast cell activation syndromes. J. Allergy Clin. Immunol. 2017, 140, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiler, C.R. Mast Cell Activation Syndrome: Tools for Diagnosis and Differential Diagnosis. J. Allergy Clin. Immunol. Pract. 2020, 8, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef]
- Li, Z. New Insights into the Pathogenesis of Systemic Mastocytosis. Int. J. Mol. Sci. 2021, 22, 4900. [Google Scholar] [CrossRef]
- Pardanani, A. Systemic mastocytosis in adults: 2021 Update on diagnosis, risk stratification and management. Am. J. Hematol. 2021, 96, 508–525. [Google Scholar] [CrossRef]
- Konnikova, L.; Robinson, T.O.; Owings, A.H.; Shirley, J.F.; Davis, E.; Tang, Y.; Wall, S.; Li, J.; Hasan, M.H.; Gharaibeh, R.Z.; et al. Small intestinal immunopathology and GI-associated antibody formation in hereditary alpha-tryptasemia. J. Allergy Clin. Immunol. 2021, 148, 813–821.e7. [Google Scholar] [CrossRef]
- Dahlin, J.S.; Maurer, M.; Metcalfe, D.D.; Pejler, G.; Sagi-Eisenberg, R.; Nilsson, G. The ingenious mast cell: Contemporary insights into mast cell behavior and function. Allergy 2021. [Google Scholar] [CrossRef]
- Krishnaswamy, G.; Lakshman, T.; Miller, A.; Srikanth, S.; Hall, K.; Huang, S.-K.; Suttles, J.; Smith, J.; Stout, R. Multifunctional cytokine expression by human mast cells: Regulation by T cell membrane contact and glucocorticoids. J. Interferon Cytokine Res. 1997, 17, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Shakoory, B.; Fitzgerald, S.M.; Lee, S.A.; Chi, D.S.; Krishnaswamy, G. The role of human mast cell-derived cytokines in eosinophil biology. J. Interferon Cytokine Res. 2004, 24, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Brazzelli, V.; Grassi, S.; Merante, S.; Grasso, V.; Ciccocioppo, R.; Bossi, G.; Borroni, G. Narrow-band UVB phototherapy and psoralen-ultraviolet A photochemotherapy in the treatment of cutaneous mastocytosis: A study in 20 patients. Photodermatol. Photoimmunol. Photomed. 2016, 32, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T. Gastrointestinal abnormalities and involvement in systemic mastocytosis. Hematol. Oncol. Clin. N. Am. 2000, 14, 579–623. [Google Scholar] [CrossRef]
- Nedoszytko, B.; Arock, M.; Lyons, J.; Bachelot, G.; Schwartz, L.; Reiter, A.; Jawhar, M.; Schwaab, J.; Lange, M.; Greiner, G.; et al. Clinical Impact of Inherited and Acquired Genetic Variants in Mastocytosis. Int. J. Mol. Sci. 2021, 22, 411. [Google Scholar] [CrossRef]
- Valent, P.; Bonadonna, P.; Hartmann, K.; Broesby-Olsen, S.; Brockow, K.; Butterfield, J.H.; Triggiani, M.; Lyons, J.J.; Elberink, J.N.O.; Arock, M.; et al. Why the 20% + 2 Tryptase Formula Is a Diagnostic Gold Standard for Severe Systemic Mast Cell Activation and Mast Cell Activation Syndrome. Int. Arch. Allergy Immunol. 2019, 180, 44–51. [Google Scholar] [CrossRef]
- Gulen, T.; Akin, C.; Bonadonna, P.; Siebenhaar, F.; Broesby-Olsen, S.; Brockow, K.; Niedoszytko, M.; Nedoszytko, B.; Oude Elberink, H.N.G.; Butterfield, J.H.; et al. Selecting the Right Criteria and Proper Classification to Diagnose Mast Cell Activation Syndromes: A Critical Review. J. Allergy Clin. Immunol. Pract. 2021. [Google Scholar] [CrossRef] [PubMed]
- Atiakshin, D.; Buchwalow, I.; Horny, P.; Tiemann, M. Protease profile of normal and neoplastic mast cells in the human bone marrow with special emphasis on systemic mastocytosis. Histochem. Cell Biol. 2021, 155, 561–580. [Google Scholar] [CrossRef]
- Dougherty, R.H.; Sidhu, S.S.; Raman, K.; Solon, M.; Solberg, O.D.; Caughey, G.H.; Woodruff, P.G.; Fahy, J.V. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J. Allergy Clin. Immunol. 2010, 125, 1046–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, A.M. Ultrastructural studies of human basophils and mast cells. J. Histochem. Cytochem. 2005, 53, 1043–1070. [Google Scholar] [CrossRef] [PubMed]
- Kinet, J.P. High Affinity IgE Receptor: From Physiology to Pathology. Annu. Rev. Immunol. 1999, 17, 931–972. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Kitaura, J. Tuning IgE: IgE-Associating Molecules and Their Effects on IgE-Dependent Mast Cell Reactions. Cells 2021, 10, 1697. [Google Scholar] [CrossRef]
- Paivandy, A.; Pejler, G. Novel Strategies to Target Mast Cells in Disease. J. Innate Immun. 2021, 13, 131–147. [Google Scholar] [CrossRef]
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast cell mediators: Their differential release and the secretory pathways involved. Front. Immunol. 2014, 5, 569. [Google Scholar] [CrossRef] [Green Version]
- Atiakshin, D.; Buchwalow, I.; Samoilova, V.; Tiemann, M. Tryptase as a polyfunctional component of mast cells. Histochem. Cell Biol. 2018, 149, 461–477. [Google Scholar] [CrossRef]
- Atiakshin, D.; Buchwalow, I.; Tiemann, M. Mast cell chymase: Morphofunctional characteristics. Histochem. Cell Biol. 2019, 152, 253–269. [Google Scholar] [CrossRef]
- Atiakshin, D.; Samoilova, V.; Buchwalow, I.; Boecker, W.; Tiemann, M. Characterization of mast cell populations using different methods for their identification. Histochem. Cell Biol. 2017, 147, 683–694. [Google Scholar] [CrossRef]
- Sprinzl, B.; Greiner, G.; Uyanik, G.; Arock, M.; Haferlach, T.; Sperr, W.; Valent, P.; Hoermann, G. Genetic Regulation of Tryptase Production and Clinical Impact: Hereditary Alpha Tryptasemia, Mastocytosis and Beyond. Int. J. Mol. Sci. 2021, 22, 2458. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Nedoszytko, B.; Bonadonna, P.; Hartmann, K.; Niedoszytko, M.; Brockow, K.; Siebenhaar, F.; Triggiani, M.; Arock, M.; et al. Diagnosis, Classification and Management of Mast Cell Activation Syndromes (MCAS) in the Era of Personalized Medicine. Int. J. Mol. Sci. 2020, 21, 9030. [Google Scholar] [CrossRef]
- Martinez-Anton, A.; Gras, D.; Bourdin, A.; Dubreuil, P.; Chanez, P. KIT as a therapeutic target for non-oncological diseases. Pharmacol. Ther. 2019, 197, 11–37. [Google Scholar] [CrossRef]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; Román-Gil, M.S.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; Nilsson, G.; Reiter, A.; Hermine, O.; Sotlar, K.; Sperr, W.R.; Escribano, L.; George, T.; et al. Advances in the Classification and Treatment of Mastocytosis: Current Status and Outlook toward the Future. Cancer Res. 2017, 77, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Horny, H.P.; Parwaresch, M.R.; Lennert, K. Bone marrow findings in systemic mastocytosis. Hum. Pathol. 1985, 16, 808–814. [Google Scholar] [CrossRef]
- Hartmann, K.; Escribano, L.; Grattan, C.; Brockow, K.; Carter, M.C.; Alvarez-Twose, I.; Matito, A.; Broesby-Olsen, S.; Siebenhaar, F.; Lange, M.; et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J. Allergy Clin. Immunol. 2016, 137, 35–45. [Google Scholar] [PubMed]
- Méni, C.; Bruneau, J.; Georgin-Lavialle, S.; Peufeilhoux, L.L.S.D.; Damaj, G.; Hadj-Rabia, S.; Fraitag, S.; Dubreuil, P.; Hermine, O.; Bodemer, C. Paediatric mastocytosis: A systematic review of 1747 cases. Br. J. Dermatol. 2015, 172, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Ben-Amitai, D.; Metzker, A.; Cohen, H.A. Pediatric cutaneous mastocytosis: A review of 180 patients. Isr. Med. Assoc. J. 2005, 7, 320–322. [Google Scholar]
- Kiszewski, A.E.; Durán-Mckinster, C.; Orozco-Covarrubias, L.; Gutiérrez-Castrellón, P.; Ruiz-Maldonado, R. Cutaneous mastocytosis in children: A clinical analysis of 71 cases. J. Eur. Acad. Dermatol. Venereol. 2004, 18, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Scherber, R.M.; Borate, U. How we diagnose and treat systemic mastocytosis in adults. Br. J. Haematol. 2018, 180, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Lim, K.-H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.-Y.; Pardanani, A. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef] [Green Version]
- Rossini, M.; Zanotti, R.; Viapiana, O.; Tripi, G.; Orsolini, G.; Idolazzi, L.; Bonadonna, P.; Schena, D.; Escribano, L.; Adami, S.; et al. Bone involvement and osteoporosis in mastocytosis. Immunol. Allergy Clin. N. Am. 2014, 34, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Georgin-Lavialle, S.; Gaillard, R.; Moura, D.; Hermine, O. Mastocytosis in adulthood and neuropsychiatric disorders. Transl. Res. 2016, 174, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Brockow, K.; Jofer, C.; Behrendt, H.; Ring, J. Anaphylaxis in patients with mastocytosis: A study on history, clinical features and risk factors in 120 patients. Allergy 2008, 63, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage). Blood 2013, 121, 3085–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, A.; George, T.I.; Gotlib, J. New developments in diagnosis, prognostication, and treatment of advanced systemic mastocytosis. Blood 2020, 135, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Nagata, H.; Worobec, A.S.; Oh, C.K.; Chowdhury, B.A.; Tannenbaum, S.; Suzuki, Y.; Metcalfe, D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA 1995, 92, 10560–10564. [Google Scholar] [CrossRef] [Green Version]
- Arock, M.; Sotlar, K.; Akin, C.; Broesby-Olsen, S.; Hoermann, G.; Escribano, L.; Kluin-Nelemans, H.C.; Hermine, O.; Du-breuil, P.; Sperr, W.R.; et al. KIT mutation analysis in mast cell neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia 2015, 29, 1223–1232. [Google Scholar] [CrossRef] [Green Version]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.-P.; et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013, 122, 2460–2466. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Lasho, T.; Elala, Y.; Wassie, E.; Finke, C.; Reichard, K.K.; Chen, D.; Hanson, C.A.; Ketterling, R.P.; Tefferi, A. Next-generation sequencing in systemic mastocytosis: Derivation of a mutation-augmented clinical prognostic model for survival. Am. J. Hematol. 2016, 91, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggendorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.-P.; Metzgeroth, G.; Kluger, S.; Naumann, N.; et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia 2016, 30, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Naumann, N.; Jawhar, M.; Schwaab, J.; Kluger, S.; Lübke, J.; Metzgeroth, G.; Popp, H.D.; Khaled, N.; Horny, H.-P.; Sotlar, K.; et al. Incidence and prognostic impact of cytogenetic aberrations in patients with systemic mastocytosis. Genes Chromosomes Cancer 2018, 57, 252–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-González, J.I.; Jara-Acevedo, M.; Alvarez-Twose, I.; Merker, J.D.; Teodosio, C.; Hou, Y.; Henriques, A.; Roskin, K.M.; Sanchez-Muñoz, L.; Tsai, A.; et al. Impact of somatic and germline mutations on the outcome of systemic mastocytosis. Blood Adv. 2018, 2, 2814–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traina, F.; Visconte, V.; Jankowska, A.M.; Makishima, H.; O’Keefe, C.L.; Elson, P.; Han, Y.; Hsieh, F.H.; Sekeres, M.A.; Mali, R.S.; et al. Single nucleotide polymorphism array lesions, TET2, DNMT3A, ASXL1 and CBL mutations are present in systemic mastocytosis. PLoS ONE 2012, 7, e43090. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Naumann, N.; Horny, H.-P.; Sotlar, K.; Haferlach, T.; Metzgeroth, G.; Fabarius, A.; Valent, P.; Hofmann, W.-K.; et al. Response and progression on midostaurin in advanced systemic mastocytosis: KIT D816V and other molecular markers. Blood 2017, 130, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Leguit, R.; Hebeda, K.; Kremer, M.; Van Der Walt, J.; Gianelli, U.; Tzankov, A.; Orazi, A. The Spectrum of Aggressive Mastocytosis: A Workshop Report and Literature Review. Pathobiology 2020, 87, 2–19. [Google Scholar] [CrossRef]
- Monnier, J.; Georgin-Lavialle, S.; Canioni, D.; Lhermitte, L.; Soussan, M.; Arock, M.; Bruneau, J.; Dubreuil, P.; Bodemer, C.; Chandesris, M.-O.; et al. Mast cell sarcoma: New cases and literature review. Oncotarget 2016, 7, 66299–66309. [Google Scholar] [CrossRef] [Green Version]
- Weiler, C.R.; Austen, K.F.; Akin, C.; Barkoff, M.S.; Bernstein, J.A.; Bonadonna, P.; Butterfield, J.H.; Carter, M.; Fox, C.C.; Maitland, A.; et al. AAAAI Mast Cell Disorders Committee Work Group Report: Mast cell activation syndrome (MCAS) diagnosis and management. J. Allergy Clin. Immunol. 2019, 144, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Giannetti, A.; Filice, E.; Caffarelli, C.; Ricci, G.; Pession, A. Mast Cell Activation Disorders. Medicina 2021, 57, 124. [Google Scholar] [CrossRef]
- Krishnaswamy, G. Critical Care Management of the Patient with Anaphylaxis: A Concise Definitive Review. Crit. Care Med. 2021, 49, 838–857. [Google Scholar] [CrossRef]
- LoVerde, D.; Iweala, O.I.; Eginli, A.; Krishnaswamy, G. Anaphylaxis. Chest 2018, 153, 528–543. [Google Scholar] [CrossRef]
- Gotlib, J.; George, T.I.; Carter, M.C.; Austen, K.F.; Bochner, B.; Dwyer, D.F.; Lyons, J.J.; Hamilton, M.J.; Butterfield, J.; Bonadonna, P.; et al. Proceedings from the Inaugural American Initiative in Mast Cell Diseases (AIM) Investigator Conference. J. Allergy Clin. Immunol. 2021, 147, 2043–2052. [Google Scholar] [CrossRef]
- Ravi, A.; Butterfield, J.; Weiler, C.R. Mast cell activation syndrome: Improved identification by combined determinations of serum tryptase and 24-h urine 11β-prostaglandin2α. J. Allergy Clin. Immunol. Pract. 2014, 2, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Gulen, T.; Akin, C. Idiopathic Anaphylaxis: A Perplexing Diagnostic Challenge for Allergists. Curr. Allergy Asthma Rep. 2021, 21, 11. [Google Scholar] [CrossRef]
- Giannetti, M.P.; Akin, C.; Castells, M. Idiopathic Anaphylaxis: A Form of Mast Cell Activation Syndrome. J. Allergy Clin. Immunol. Pract. 2020, 8, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Grammer, L.C.; Shaughnessy, M.A.; Harris, K.E.; Goolsby, C.L. Lymphocyte subsets and activation markers in patients with acute episodes of idiopathic anaphylaxis. Ann. Allergy Asthma Immunol. 2000, 85, 368–371. [Google Scholar] [CrossRef]
- Ditto, A.M.; Harris, K.E.; Krasnick, J.; Miller, M.A.; Patterson, R. Idiopathic anaphylaxis: A series of 335 cases. Ann. Allergy Asthma Immunol. 1996, 77, 285–291. [Google Scholar] [CrossRef]
- Vos, B.J.P.R.; van Anrooij, B.; van Doormaal, J.J.; Dubois, A.E.J.; Oude Elberink, J.N.G. Fatal Anaphylaxis to Yellow Jacket Stings in Mastocytosis: Options for Identification and Treatment of At-Risk Patients. J. Allergy Clin. Immunol. Pract. 2017, 5, 1264–1271. [Google Scholar] [CrossRef]
- Bonadonna, P.; Scaffidi, L. Hymenoptera Anaphylaxis as a Clonal Mast Cell Disorder. Immunol. Allergy Clin. N. Am. 2018, 38, 455–468. [Google Scholar] [CrossRef]
- Frieri, M. Mast Cell Activation Syndrome. Clin. Rev. Allergy Immunol. 2018, 54, 353–365. [Google Scholar] [CrossRef]
- Bonadonna, P.; Perbellini, O.; Passalacqua, G.; Caruso, B.; Colarossi, S.; Dal Fior, D.; Castellani, L.; Bonetto, C.; Frattini, F.; Dama, A.; et al. Clonal mast cell disorders in patients with systemic reactions to Hymenoptera stings and increased serum tryptase levels. J. Allergy Clin. Immunol. 2009, 123, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Rueff, F.; Placzek, M.; Przybilla, B. Mastocytosis and Hymenoptera venom allergy. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Blank, S.; Grosch, J.; Ollert, M.; Bilo, M.B. Precision Medicine in Hymenoptera Venom Allergy: Diagnostics, Biomarkers, and Therapy of Different Endotypes and Phenotypes. Front. Immunol. 2020, 11, 579409. [Google Scholar] [CrossRef] [PubMed]
- Kosnik, M.; Korosec, P. Importance of basophil activation testing in insect venom allergy. Allergy Asthma Clin. Immunol. 2009, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Zanotti, R.; Tanasi, I.; Bernardelli, A.; Orsolini, G.; Bonadonna, P. Bone Marrow Mastocytosis: A Diagnostic Challenge. J. Clin. Med. 2021, 10, 1420. [Google Scholar] [CrossRef]
- Reichard, K.K.; Chen, D.; Pardanani, A.; McClure, R.F.; Howard, M.T.; Kurtin, P.J.; Wood, A.J.; Ketterling, R.P.; King, R.L.; He, R.; et al. Morphologically occult systemic mastocytosis in bone marrow: Clinicopathologic features and an algorithmic approach to diagnosis. Am. J. Clin. Pathol. 2015, 144, 493–502. [Google Scholar] [CrossRef]
- Luskin, K.T.; White, A.A.; Lyons, J.J. The Genetic Basis and Clinical Impact of Hereditary Alpha-Tryptasemia. J. Allergy Clin. Immunol. Pract. 2021, 9, 2235–2242. [Google Scholar] [CrossRef]
- Wu, R.; Lyons, J.J. Hereditary Alpha-Tryptasemia: A Commonly Inherited Modifier of Anaphylaxis. Curr. Allergy Asthma Rep. 2021, 21, 33. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, M.; Sala-Cunill, A.; Luengo, O.; Labrador-Horrillo, M.; Cardona, V. The Mast Cell, Contact, and Coagulation System Connection in Anaphylaxis. Front. Immunol. 2017, 8, 846. [Google Scholar] [CrossRef] [Green Version]
- Cunill, A.S.; Björkqvist, J.; Senter, R.; Guilarte, M.; Cardona, V.; Labrador-Horrillo, M.; Nickel, K.F.; Butler, L.; Luengo, O.; Kumar, P.; et al. Plasma contact system activation drives anaphylaxis in severe mast cell-mediated allergic reactions. J. Allergy Clin. Immunol. 2015, 135, 1031–1043. [Google Scholar] [CrossRef]
- Elieh Ali, K.D.; Shafaghat, F.; Kovanen, P.T.; Meri, S. Mast cells and complement system: Ancient interactions between components of innate immunity. Allergy 2020, 75, 2818–2828. [Google Scholar] [CrossRef] [PubMed]
- Giannetti, M.P.; Weller, E.; Bormans, C.; Novak, P.; Hamilton, M.J.; Castells, M. Hereditary alpha-tryptasemia in 101 patients with mast cell activation-related symptomatology including anaphylaxis. Ann. Allergy Asthma Immunol. 2021, 126, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.; Hollingsworth, P.; Lucas, M. Successful treatment of idiopathic mast cell activation syndrome with low-dose Omalizumab. Clin. Transl. Immunol. 2019, 8, e01075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cafarotti, A.; Fiocchi, A.; Arasi, S. Biologics as treatment options for anaphylaxis. Curr. Opin. Allergy Clin. Immunol. 2021, 21, 455–464. [Google Scholar] [CrossRef]
- Carter, M.C.; Robyn, J.A.; Bressler, P.B.; Walker, J.C.; Shapiro, G.G.; Metcalfe, D.D. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J. Allergy Clin. Immunol. 2007, 119, 1550–1551. [Google Scholar] [CrossRef]
- Constantine, G.M.; Bressler, P.B.; Petroni, D.; Metcalfe, D.D.; Carter, M.C. Twelve-year follow-up of omalizumab therapy for anaphylaxis in 2 patients with systemic mastocytosis. J. Allergy Clin. Immunol. Pract. 2019, 7, 1314–1316. [Google Scholar] [CrossRef]
- Broesby-Olsen, S.; Vestergaard, H.; Mortz, C.G.; Jensen, B.; Havelund, T.; Hermann, A.P.; Siebenhaar, F.; Møller, M.B.; Kris-tensen, T.K.; Bindslev-Jensen, C. Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. Allergy 2018, 73, 230–238. [Google Scholar] [CrossRef]
- Giannetti, M.; Silver, J.; Hufdhi, R.; Castells, M. One-day ultrarush desensitization for Hymenoptera venom anaphylaxis in patients with and without mast cell disorders with adjuvant omalizumab. J. Allergy Clin. Immunol. Pract. 2020, 8, 1431–1435. [Google Scholar] [CrossRef]
- Kaminsky, L.W.; Aukstuolis, K.; Petroni, D.H.; Al-Shaikhly, T. Use of omalizumab for management of idiopathic anaphylaxis: A systematic review and retrospective case series. Ann. Allergy Asthma Immunol. 2021, 127, 481–487. [Google Scholar] [CrossRef]
- Lemal, R.; Fouquet, G.; Terriou, L.; Vaes, M.; Livideanu, C.B.; Frenzel, L.; Barete, S.; Canioni, D.; Lhermitte, L.; Rossignol, J.; et al. Omalizumab Therapy for Mast Cell-Mediator Symptoms in Patients with ISM, CM, MMAS, and MCAS. J. Allergy Clin. Immunol. Pract. 2019, 7, 2387–2395. [Google Scholar] [CrossRef]
- Alvarez, L.B.M.; Barker, R.; Nelson, C.; DiMaggio, T.; Stone, K.D.; Milner, J.D.; Rosenthal, J.; Petroni, D.H.; Glover, S.C.; Lyons, J.J. Clinical response to omalizumab in patients with hereditary α-tryptasemia. Ann. Allergy Asthma Immunol. 2020, 124, 99–100. [Google Scholar] [CrossRef] [Green Version]
- Carter, M.C.; Metcalfe, D.D.; Komarow, H.D. Mastocytosis. Immunol. Allergy Clin. N. Am. 2014, 34, 181–196. [Google Scholar] [CrossRef] [Green Version]
- Castells, M.; Butterfield, J. Mast Cell Activation Syndrome and Mastocytosis: Initial Treatment Options and Long-Term Management. J. Allergy Clin. Immunol. Pract. 2019, 7, 1097–1106. [Google Scholar] [CrossRef]
- Butterfield, J.H. Survey of aspirin administration in systemic mastocytosis. Prostaglandins Other Lipid Mediat. 2009, 88, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.J.; Metcalfe, D.D. Targeting Mast Cells with Biologics. Immunol. Allergy Clin. N. Am. 2020, 40, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Verstovsek, S. Avapritinib for Systemic Mastocytosis. Expert Rev. Hematol. 2021, 14, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Li, T.; Wisell, J.; Schowinsky, J.; Robinson, W.A. Avapritinib for Cutaneous Mastocytosis. Acta Derm. Venereol. 2021, 101, adv00362. [Google Scholar]
- Gilreath, J.A.; Tchertanov, L.; Deininger, M.W. Novel approaches to treating advanced systemic mastocytosis. Clin. Pharmacol. 2019, 11, 77–92. [Google Scholar] [CrossRef] [Green Version]
- Gotlib, J.; Kluin-Nelemans, J.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef]
- Shomali, W.; Gotlib, J. Response Criteria in Advanced Systemic Mastocytosis: Evolution in the Era of KIT Inhibitors. Int. J. Mol. Sci. 2021, 22, 2983. [Google Scholar] [CrossRef] [PubMed]
- Piris-Villaespesa, M.; Alvarez-Twose, I. Systemic Mastocytosis: Following the Tyrosine Kinase Inhibition Roadmap. Front. Pharmacol. 2020, 11, 443. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Baccarani, M.; Giles, F.J.; Le Coutre, P.D.; Müller, M.C.; Reiter, A.; Santanastasio, H.; Leung, M.; Novick, S.; Kantarjian, H.M. Nilotinib in patients with systemic mastocytosis: Analysis of the phase 2, open-label, single-arm nilotinib registration study. J. Cancer Res. Clin. Oncol. 2015, 141, 2047–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstovsek, S.; Tefferi, A.; Cortes, J.; O’Brien, S.; Garcia-Manero, G.; Pardanani, A.; Akin, C.; Faderl, S.; Manshouri, T.; Thomas, D.; et al. Phase II study of dasatinib in Philadelphia chromosome-negative acute and chronic myeloid diseases, including systemic mastocytosis. Clin. Cancer Res. 2008, 14, 3906–3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
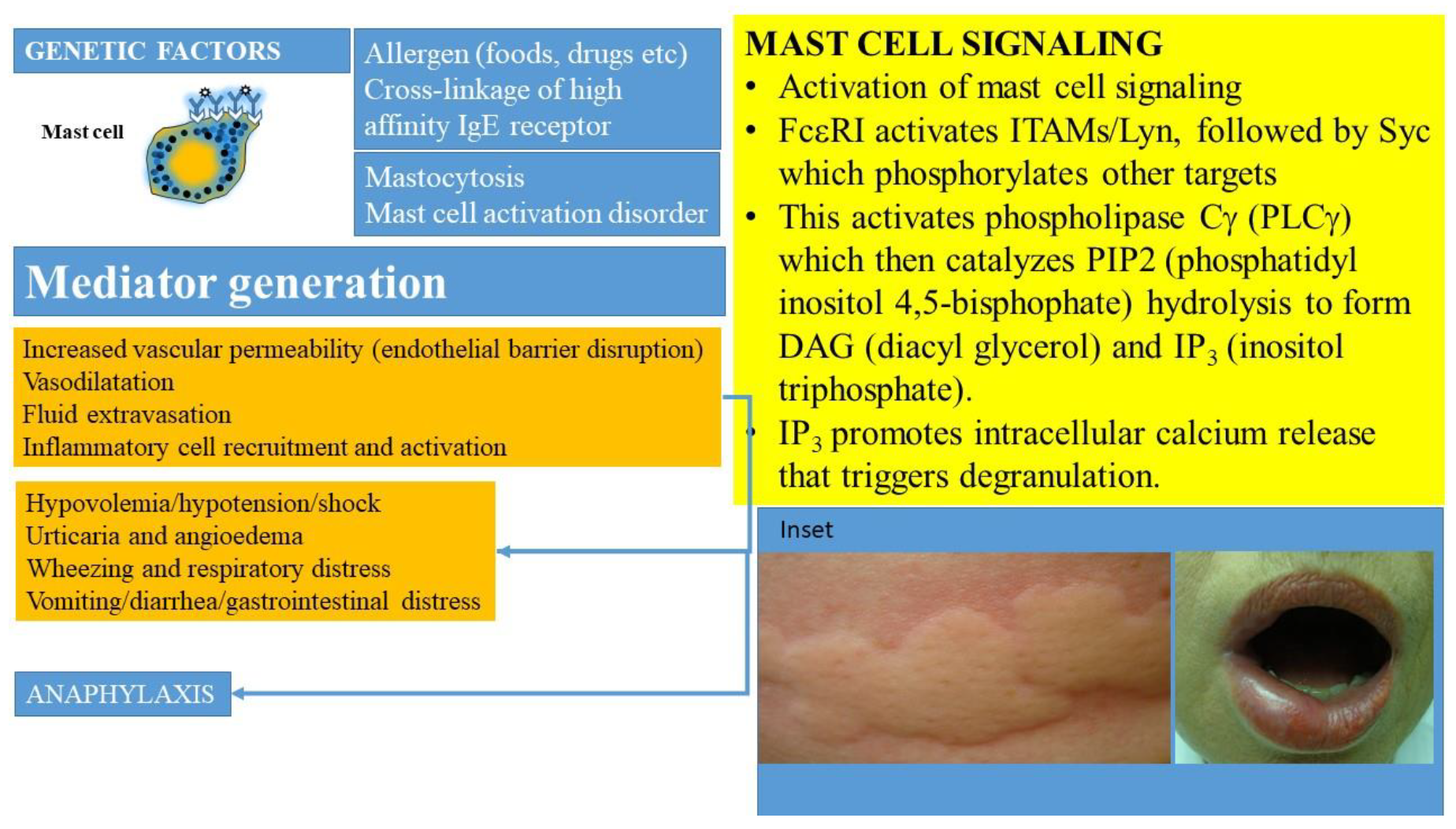



{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
| Drug Class | Medication and Dosing | Primary Indication and Comments |
|---|---|---|
| H1-receptor antagonist | Long-acting:
Short-acting:
| Histamine-related symptoms (i.e., flushing, pruritis) |
| H2-receptor antagonist | Ranitidine (150 mg orally, twice/day) Famotidine (10 mg orally twice/day) | Gastrointestinal symptoms |
| Proton-Pump Inhibitor (PPI) | Omeprazole (20–40 mg orally daily Pantoprazole (20–40 mg orally daily) | Gastrointestinal symptoms |
| Leukotriene receptor antagonist | Montelukast (10 mg orally daily Zafirlukast (20 mg orally daily) | Histamine-related symptoms (i.e., flushing, pruritis) |
| Mast cell stabilizer | Cromolyn sodium (four times a day maximum daily dose 40 mg/kg/day) | Gastrointestinal symptoms Histamine-related symptoms (i.e., flushing, pruritis) |
| Non-steroidal anti-inflammatory drug (NSAID) | Aspirin (variable dosing) | Histamine-related symptoms (i.e., flushing, pruritis) |
| Anti-IgE monoclonal antibody | Omalizumab (150–300 mg administered subcutaneously every 2–4 weeks) | Histamine-related symptoms (i.e., flushing, pruritis, recurrent anaphylaxis) not responsive to conservative measures |
| Immediate administration for anaphylaxis | Epinephrine (0.3–0.5 mg intramuscularly every 5–15 min in the lateral thigh) | Anaphylaxis |
| Cytoreductive therapy | Interferon-α (IFN-α)
Cladribine (2-CdA)
| IFN-α: often co-administer with prednisone to improve tolerability Flu-like syndrome 2-CdA: myelosuppression and lymphopenia common. Prophylaxis for Pneumocystis jirovecii infection needed |
| Drug | Drug Target | Dosing | Comments |
|---|---|---|---|
| Midostaurin * | Multi-kinase inhibitor of KIT, VEGFR, FLT3, PDGFR | 100 mg PO BID | Multi-kinase inhibitor with activity against KIT-D816V |
| Avapritinib * | Selective KIT-D816V inhibitor | 200 mg PO daily | Selective KIT-D816V inhibition; Effective in patients having received prior Midostaurin therapy |
| Imatinib | Multi-kinase inhibitor of KIT, PDGFR, Bcr-Abl | 400 mg PO daily | Resistance observed in KIT-D816V |
| Masitinib | Multi-kinase inhibitor of KIT, PDGFR, Lyn, FGFR3 | 6 mg/kg PO divided into two daily doses | Resistance observed in KIT-D816V |
| Dasatinib | Multi-kinase inhibitor of KIT, PDGFR, Bcr-Abl, Src | 140 mg PO daily | Limited therapeutic activity in SM |
| Nilotinib | Multi-kinase inhibitor of KIT, PDGFR, Bcr-Abl | 400 mg PO BID | Limited therapeutic activity in SM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, C.W.; Pratt, C.M.; Rupprecht, C.P.; Pattanaik, D.; Krishnaswamy, G. Mastocytosis and Mast Cell Activation Disorders: Clearing the Air. Int. J. Mol. Sci. 2021, 22, 11270. https://doi.org/10.3390/ijms222011270
Jackson CW, Pratt CM, Rupprecht CP, Pattanaik D, Krishnaswamy G. Mastocytosis and Mast Cell Activation Disorders: Clearing the Air. International Journal of Molecular Sciences. 2021; 22(20):11270. https://doi.org/10.3390/ijms222011270
Chicago/Turabian StyleJackson, Clayton Webster, Cristina Marie Pratt, Chase Preston Rupprecht, Debendra Pattanaik, and Guha Krishnaswamy. 2021. "Mastocytosis and Mast Cell Activation Disorders: Clearing the Air" International Journal of Molecular Sciences 22, no. 20: 11270. https://doi.org/10.3390/ijms222011270
APA StyleJackson, C. W., Pratt, C. M., Rupprecht, C. P., Pattanaik, D., & Krishnaswamy, G. (2021). Mastocytosis and Mast Cell Activation Disorders: Clearing the Air. International Journal of Molecular Sciences, 22(20), 11270. https://doi.org/10.3390/ijms222011270