
Cytokines as Biomarkers in Systemic Lupus Erythematosus: Value for Diagnosis and Drug Therapy


Abstract
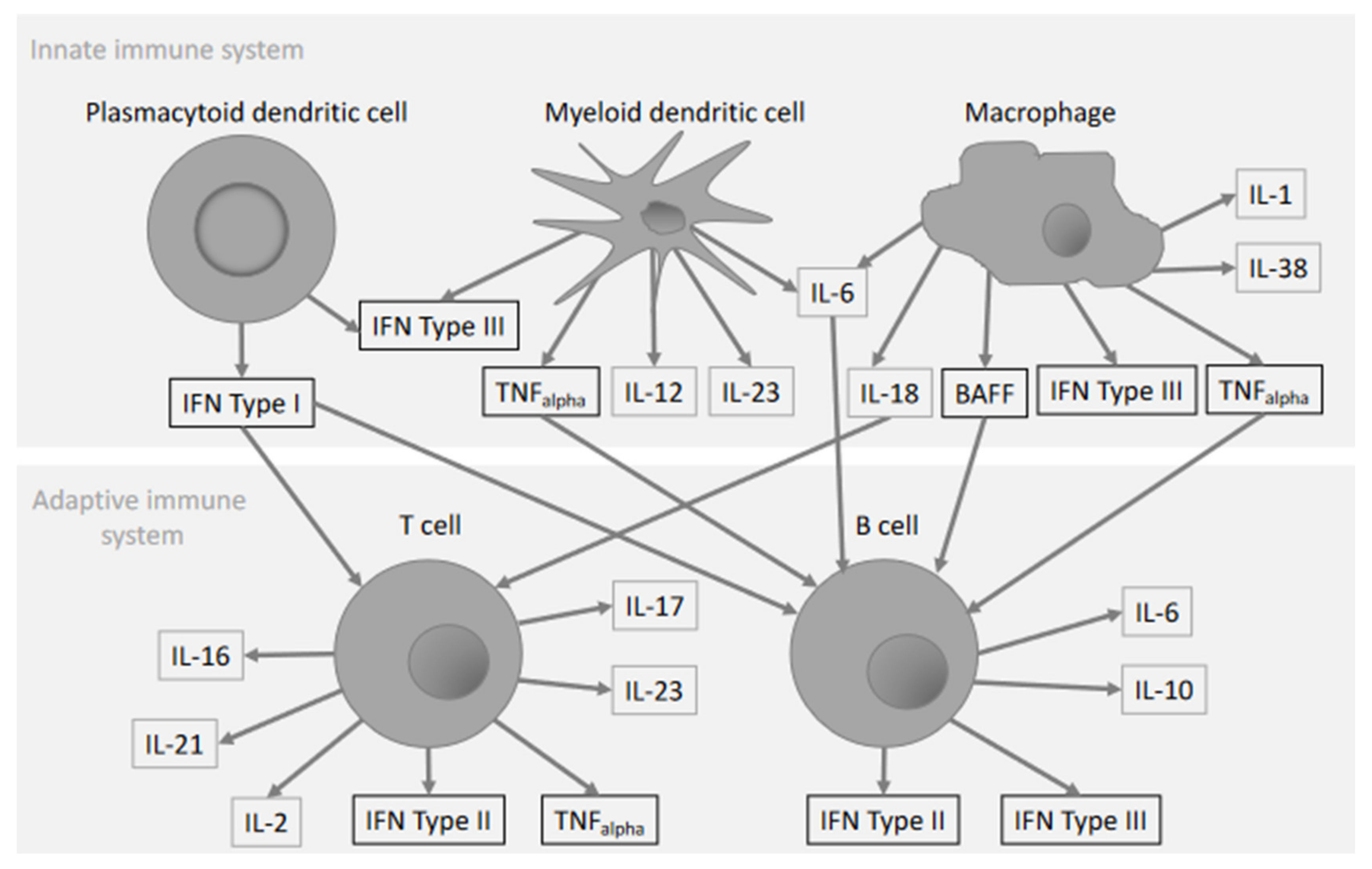
:1. Introduction
2. Interferons
2.1. Type I IFNs in SLE
2.1.1. IFN-α
2.1.2. IFN-β
2.1.3. IFN-κ
2.2. IFN Type II in SLE
2.3. IFN Type III in SLE
3. IFNs as Treatment Targets in SLE
3.1. IFNs Type I Targeting Therapy
3.2. IFN-γ Targeting Therapy
3.3. IFN-λ Targeting Therapy
4. TNF-α
5. BAFF/APRIL
6. IL-2
7. IL-6
8. IL-10
9. IL-16
10. IL-12, IL-17 and IL-23
11. IL-1, IL-18 and IL-38
12. IL-21
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- La Cava, A. Anticytokine therapies in systemic lupus erythematosus. Immunotherapy 2010, 2, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rönnblom, L.; Leonard, D. Interferon pathway in SLE: One key to unlocking the mystery of the disease. Lupus Sci. Med. 2019, 6, e000270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chasset, F.; Arnaud, L. Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun. Rev. 2018, 17, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Touma, Z.; Sayani, A.; Pineau, C.A.; Fortin, I.; Matsos, M.; Ecker, G.A.; Chow, A.; Iczkovitz, S. Belimumab use, clinical outcomes and glucocorticoid reduction in patients with systemic lupus erythematosus receiving belimumab in clinical practice settings: Results from the OBSErve Canada Study. Rheumatol. Int. 2017, 37, 865–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oke, V.; Gunnarsson, I.; Dorschner, J.; Eketjäll, S.; Zickert, A.; Niewold, T.B.; Svenungsson, E. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res. Ther. 2019, 21, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragón, C.C.; Tafúr, R.-A.; Suárez-Avellaneda, A.; Martínez, M.T.; de Salas, A.L.; Tobón, G.J. Urinary biomarkers in lupus nephritis. J. Transl. Autoimmun. 2020, 3, 100042. [Google Scholar] [CrossRef]
- Boedigheimer, M.J.; Martin, D.A.; Amoura, Z.; Sánchez-Guerrero, J.; Romero-Diaz, J.; Kivitz, A.; Aranow, C.; Chan, T.M.; Chong, Y.B.; Chiu, K.; et al. Safety, pharmacokinetics and pharmacodynamics of AMG 811, an anti-interferon-γ monoclonal antibody, in SLE subjects without or with lupus nephritis. Lupus Sci. Med. 2017, 4, e000226. [Google Scholar] [CrossRef]
- Aringer, M.; Smolen, J.S. Therapeutic blockade of TNF in patients with SLE-promising or crazy? Autoimmun. Rev. 2012, 11, 321–325. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Crow, M.K.; Rönnblom, L. Type I interferons in host defence and inflammatory diseases. Lupus Sci. Med. 2019, 6, e000336. [Google Scholar] [CrossRef] [Green Version]
- Rönnblom, L.E.; Alm, G.V.; Oberg, K.E. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J. Intern. Med. 1990, 227, 207–210. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rönnblom, L.; Elkon, K.B. Cytokines as therapeutic targets in SLE. Nat. Rev. Rheumatol. 2010, 6, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Rönnblom, L.E.; Perers, A.; Vallin, H.S.; Eriksson, I.; Osterlind, A.; Cederblad, B.; Alm, G. Detection of serum interferon-alpha by dissociation-enhanced lanthanide fluoroimmunoassay. Studies of patients with acute viral and bacterial infections. APMIS 1997, 105, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Kirou, K.; Lee, C.; Crow, M.K. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006, 54, 1906–1916. [Google Scholar] [CrossRef]
- Bengtsson, A.A.; Sturfelt, G.; Truedsson, L.; Blomberg, J.; Alm, G.; Vallin, H.; Rönnblom, L. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus 2000, 9, 664–671. [Google Scholar] [CrossRef]
- Mathian, A.; Mouries-Martin, S.; Dorgham, K.; Devilliers, H.; Barnabei, L.; Ben Salah, E.; Cohen-Aubart, F.; Garrido Castillo, L.; Haroche, J.; Hie, M.; et al. Monitoring disease activity in systemic lupus erythematosus with single-molecule array digital ELISA quantification of serum interferon-α. Arthritis Rheumatol. 2018, 71, 759–765. [Google Scholar]
- Oke, V.; Brauner, S.; Larsson, A.; Gustafsson, J.; Zickert, A.; Gunnarsson, I.; Svenungsson, E. IFN-λ1 with Th17 axis cytokines and IFN-α define different subsets in systemic lupus erythematosus (SLE). Arthritis Res. Ther. 2017, 19, 139. [Google Scholar] [CrossRef] [Green Version]
- Niewold, T.B.; Hua, J.; Lehman, T.J.A.; Harley, J.B.; Crow, M.K. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007, 8, 492–502. [Google Scholar] [CrossRef]
- Weckerle, C.E.; Franek, B.S.; Kelly, J.A.; Kumabe, M.; Mikolaitis, R.A.; Green, S.L.; Utset, T.O.; Jolly, M.; James, J.A.; Harley, J.B.; et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 1044–1053. [Google Scholar] [CrossRef]
- Paradowska-Gorycka, A.; Wajda, A.; Stypinska, B.; Walczuk, E.; Rzeszotarska, E.; Walczyk, M.; Haladyj, E.; Romanowska-Prochnicka, K.; Felis-Giemza, A.; Lewandowska, A.; et al. Variety of endosomal TLRs and Interferons (IFN-α, IFN-β, IFN-γ) expression profiles in patients with SLE, SSc and MCTD. Clin. Exp. Immunol. 2021, 204, 49–63. [Google Scholar] [CrossRef]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Zickert, A.; Oke, V.; Parodis, I.; Svenungsson, E.; Sundström, Y.; Gunnarsson, I. Interferon (IFN)-λ is a potential mediator in lupus nephritis. Lupus Sci. Med. 2016, 3, e000170. [Google Scholar] [CrossRef] [Green Version]
- Graninger, W.B.; Hassfeld, W.; Pesau, B.B.; Machold, K.P.; Zielinski, C.C.; Smolen, J.S. Induction of systemic lupus erythematosus by interferon-gamma in a patient with rheumatoid arthritis. J. Rheumatol. 1991, 18, 1621–1622. [Google Scholar] [PubMed]
- Munroe, M.E.; Lu, R.; Zhao, Y.D.; Fife, D.A.; Robertson, J.M.; Guthridge, J.M.; Niewold, T.B.; Tsokos, G.C.; Keith, M.P.; Harley, J.B.; et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann. Rheum. Dis. 2016, 75, 2014–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, M.K.; Hile, G.A.; Tsoi, L.C.; Xing, X.; Liu, J.; Liang, Y.; Berthier, C.C.; Swindell, W.R.; Patrick, M.T.; Shao, S.; et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann. Rheum. Dis. 2018, 77, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Machold, K.P.; Smolen, J.S. Interferon-gamma induced exacerbation of systemic lupus erythematosus. J. Rheumatol. 1990, 17, 831–832. [Google Scholar] [PubMed]
- Amezcua-Guerra, L.M.; Márquez-Velasco, R.; Chávez-Rueda, A.K.; Castillo-Martínez, D.; Massó, F.; Páez, A.; Colín-Fuentes, J.; Bojalil, R. Type III Interferons in Systemic Lupus Erythematosus: Association Between Interferon λ3, Disease Activity, and Anti-Ro/SSA Antibodies. J. Clin. Rheumatol. 2017, 23, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Thomason, J.L.; Obih, U.M.; Koelle, D.M.; Lood, C.; Hughes, A.G. An interferon-gamma release assay as a novel biomarker in systemic lupus erythematosus. Rheumatology 2020, 59, 3479–3487. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Wang, C.-M.; Chen, T.-D.; Jan Wu, Y.-J.; Lin, J.-C.; Lu, L.Y.; Wu, J. Interferon-λ3/4 genetic variants and interferon-λ3 serum levels are biomarkers of lupus nephritis and disease activity in Taiwanese. Arthritis Res. Ther. 2018, 20, 193. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Yang, Q.; Lourenco, E.; Sun, H.; Zhang, Y. Interferon-lambda1 induces peripheral blood mononuclear cell-derived chemokines secretion in patients with systemic lupus erythematosus: Its correlation with disease activity. Arthritis Res. Ther. 2011, 13, R88. [Google Scholar] [CrossRef] [Green Version]
- Zahn, S.; Rehkämper, C.; Kümmerer, B.M.; Ferring-Schmidt, S.; Bieber, T.; Tüting, T.; Wenzel, J. Evidence for a pathophysiological role of keratinocyte-derived type III interferon (IFNλ) in cutaneous lupus erythematosus. J. Investig. Dermatol. 2011, 131, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W.; CD1067 Study Investigators. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 1909–1916. [Google Scholar] [CrossRef] [Green Version]
- Chatham, W.W.; Furie, R.; Saxena, A.; Brohawn, P.; Schwetje, E.; Abreu, G.; Tummala, R. Long-Term Safety and Efficacy of Anifrolumab in Adults With Systemic Lupus Erythematosus: Results of a Phase II Open-Label Extension Study. Arthritis Rheumatol. 2021, 73, 816–825. [Google Scholar] [CrossRef]
- FDA Approves AstraZeneca’s Anifrolumab for Lupus. Available online: https://www.nature.com/articles/d41573-021-00139-y (accessed on 6 August 2021).
- Werth, V.P.; Fiorentino, D.; Sullivan, B.A.; Boedigheimer, M.J.; Chiu, K.; Wang, C.; Arnold, G.E.; Damore, M.A.; Bigler, J.; Welcher, A.A.; et al. Brief Report: Pharmacodynamics, Safety, and Clinical Efficacy of AMG 811, a Human Anti-Interferon-γ Antibody, in Patients With Discoid Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 1028–1034. [Google Scholar] [CrossRef]
- Cesaroni, M.; Seridi, L.; Loza, M.J.; Schreiter, J.; Sweet, K.; Franks, C.; Ma, K.; Orillion, A.; Campbell, K.; Gordon, R.M.; et al. Suppression of Serum Interferon-γ Levels as a Potential Measure of Response to Ustekinumab Treatment in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2021, 73, 472–477. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [Green Version]
- Cigni, A.; Pileri, P.V.; Faedda, R.; Gallo, P.; Sini, A.; Satta, A.E.; Marras, R.; Carta, E.; Argiolas, D.; Rum, I.; et al. Interleukin 1, interleukin 6, interleukin 10, and tumor necrosis factor α in active and quiescent systemic lupus erythematosus. J. Investig. Med. 2014, 62, 825–829. [Google Scholar] [CrossRef]
- Idborg, H.; Eketjäll, S.; Pettersson, S.; Gustafsson, J.T.; Zickert, A.; Kvarnström, M.; Oke, V.; Jakobsson, P.-J.; Gunnarsson, I.; Svenungsson, E. TNF-α and plasma albumin as biomarkers of disease activity in systemic lupus erythematosus. Lupus Sci. Med. 2018, 5, e000260. [Google Scholar] [CrossRef] [PubMed]
- Weckerle, C.E.; Mangale, D.; Franek, B.S.; Kelly, J.A.; Kumabe, M.; James, J.A.; Moser, K.L.; Harley, J.B.; Niewold, T.B. Large-scale analysis of tumor necrosis factor α levels in systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2947–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkauskaite, V.; Ek, M.; Popovic, K.; Harris, H.E.; Wahren-Herlenius, M.; Nyberg, F. Translocation of the novel cytokine HMGB1 to the cytoplasm and extracellular space coincides with the peak of clinical activity in experimentally UV-induced lesions of cutaneous lupus erythematosus. Lupus 2007, 16, 794–802. [Google Scholar] [CrossRef]
- Zampieri, S.; Alaibac, M.; Iaccarino, L.; Rondinone, R.; Ghirardello, A.; Sarzi-Puttini, P.; Peserico, A.; Doria, A. Tumour necrosis factor alpha is expressed in refractory skin lesions from patients with subacute cutaneous lupus erythematosus. Ann. Rheum. Dis. 2006, 65, 545–548. [Google Scholar] [CrossRef] [PubMed]
- De Bandt, M. Lessons for lupus from tumour necrosis factor blockade. Lupus 2006, 15, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Alves, P.; Bashir, M.M.; Wysocka, M.; Zeidi, M.; Feng, R.; Werth, V.P. Quinacrine Suppresses Tumor Necrosis Factor-α and IFN-α in Dermatomyositis and Cutaneous Lupus Erythematosus. J. Investig. Dermatol. Symp. Proc. 2017, 18, S57–S63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacre, K.; Criswell, L.A.; McCune, J.M. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther. 2012, 14, R155. [Google Scholar] [CrossRef] [Green Version]
- Hegazy, M.; Darwish, H.; Darweesh, H.; El-Shehaby, A.; Emad, Y. Raised serum level of APRIL in patients with systemic lupus erythematosus: Correlations with disease activity indices. Clin. Immunol. 2010, 135, 118–124. [Google Scholar] [CrossRef]
- Boghdadi, G.; Elewa, E.A. Increased serum APRIL differentially correlates with distinct cytokine profiles and disease activity in systemic lupus erythematosus patients. Rheumatol. Int. 2014, 34, 1217–1223. [Google Scholar] [CrossRef]
- Zhang, J.; Roschke, V.; Baker, K.P.; Wang, Z.; Alarcón, G.S.; Fessler, B.J.; Bastian, H.; Kimberly, R.P.; Zhou, T. Cutting edge: A role for B lymphocyte stimulator in systemic lupus erythematosus. J. Immunol. 2001, 166, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.M.; Bacon, P.A.; Gordon, C.; Isenberg, D.A.; Maddison, P.; Snaith, M.L.; Symmons, D.P.; Viner, N.; Zoma, A. The BILAG index: A reliable and valid instrument for measuring clinical disease activity in systemic lupus erythematosus. Q. J. Med. 1993, 86, 447–458. [Google Scholar]
- Vincent, F.B.; Kandane-Rathnayake, R.; Hoi, A.Y.; Slavin, L.; Godsell, J.D.; Kitching, A.R.; Harris, J.; Nelson, C.L.; Jenkins, A.J.; Chrysostomou, A.; et al. Urinary B-cell-activating factor of the tumour necrosis factor family (BAFF) in systemic lupus erythematosus. Lupus 2018, 27, 2029–2040. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Tziolos, N.; Bertsias, G.; Boumpas, D.T. Update οn the diagnosis and management of systemic lupus erythematosus. Ann. Rheum. Dis. 2021, 80, 14–25. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.-M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. BLISS-52 Study Group Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sánchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. BLISS-76 Study Group A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Dall’Era, M.; Chakravarty, E.; Wallace, D.; Genovese, M.; Weisman, M.; Kavanaugh, A.; Kalunian, K.; Dhar, P.; Vincent, E.; Pena-Rossi, C.; et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: Results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007, 56, 4142–4150. [Google Scholar] [CrossRef]
- Tocut, M.; Shoenfeld, Y.; Zandman-Goddard, G. Systemic lupus erythematosus: An expert insight into emerging therapy agents in preclinical and early clinical development. Expert Opin. Investig. Drugs 2020, 29, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.; Wofsy, D.; Wax, S.; Li, Y.; Pena-Rossi, C.; Isenberg, D. Post Hoc Analysis of the Phase II/III APRIL-SLE Study: Association Between Response to Atacicept and Serum Biomarkers Including BLyS and APRIL. Arthritis Rheumatol. 2017, 69, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Zickert, A.; Sundelin, B.; Axelsson, M.; Gerhardsson, J.; Svenungsson, E.; Malmström, V.; Gunnarsson, I. Evaluation of B lymphocyte stimulator and a proliferation inducing ligand as candidate biomarkers in lupus nephritis based on clinical and histopathological outcome following induction therapy. Lupus Sci. Med. 2015, 2, e000061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Lin, J.-X.; Leonard, W.J. IL-2 family cytokines: New insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol. 2011, 23, 598–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, M.; He, J.; Zhang, R.; Zhang, X.; Yang, Y.; Li, C.; Liu, X.; Sun, X.; Li, Z. Interleukin-2 Deficiency Associated with Renal Impairment in Systemic Lupus Erythematosus. J. Interferon Cytokine Res. 2019, 39, 117–124. [Google Scholar] [CrossRef]
- Shao, M.; Sun, X.-L.; Sun, H.; He, J.; Zhang, R.-J.; Zhang, X.; Li, Z.-G. Clinical Relevance of Autoantibodies against Interleukin-2 in Patients with Systemic Lupus Erythematosus. Chin. Med. J. 2018, 131, 1520–1526. [Google Scholar] [CrossRef]
- He, J.; Zhang, R.; Shao, M.; Zhao, X.; Miao, M.; Chen, J.; Liu, J.; Zhang, X.; Zhang, X.; Jin, Y.; et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: A randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2020, 79, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.S.; Hutcheson, J.; Mohan, C. The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J. Interferon Cytokine Res. 2011, 31, 781–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanadetsuntorn, C.; Ngamjanyaporn, P.; Setthaudom, C.; Hodge, K.; Saengpiya, N.; Pisitkun, P. The model of circulating immune complexes and interleukin-6 improves the prediction of disease activity in systemic lupus erythematosus. Sci. Rep. 2018, 8, 2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Lei, C.-T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illei, G.G.; Shirota, Y.; Yarboro, C.H.; Daruwalla, J.; Tackey, E.; Takada, K.; Fleisher, T.; Balow, J.E.; Lipsky, P.E. Tocilizumab in systemic lupus erythematosus: Data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010, 62, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.J.; Strand, V.; Merrill, J.T.; Popa, S.; Spindler, A.J.; Eimon, A.; Petri, M.; Smolen, J.S.; Wajdula, J.; Christensen, J.; et al. Efficacy and safety of an interleukin 6 monoclonal antibody for the treatment of systemic lupus erythematosus: A phase II dose-ranging randomised controlled trial. Ann. Rheum. Dis. 2017, 76, 534–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, M.N.; Tassiulas, I.; Hu, Y.; Mecklenbräuker, I.; Tarakhovsky, A.; Ivashkiv, L.B. IFN-alpha priming results in a gain of proinflammatory function by IL-10: Implications for systemic lupus erythematosus pathogenesis. J. Immunol. 2004, 172, 6476–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Wang, W.; Zhou, M.; Li, R.; Pan, H.-F.; Ye, D.-Q. Role of interleukin-10 and interleukin-10 receptor in systemic lupus erythematosus. Clin. Rheumatol. 2013, 32, 1255–1266. [Google Scholar] [CrossRef]
- Llorente, L.; Richaud-Patin, Y.; García-Padilla, C.; Claret, E.; Jakez-Ocampo, J.; Cardiel, M.H.; Alcocer-Varela, J.; Grangeot-Keros, L.; Alarcón-Segovia, D.; Wijdenes, J.; et al. Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheum. 2000, 43, 1790–1800. [Google Scholar] [CrossRef]
- Klavdianou, K.; Lazarini, A.; Fanouriakis, A. Targeted Biologic Therapy for Systemic Lupus Erythematosus: Emerging Pathways and Drug Pipeline. BioDrugs 2020, 34, 133–147. [Google Scholar] [CrossRef]
- Ludwiczek, O.; Kaser, A.; Koch, R.O.; Vogel, W.; Cruikshank, W.W.; Tilg, H. Activation of caspase-3 by interferon alpha causes interleukin-16 secretion but fails to modulate activation induced cell death. Eur. Cytokine Netw. 2001, 12, 478–486. [Google Scholar]
- Richmond, J.; Tuzova, M.; Cruikshank, W.; Center, D. Regulation of cellular processes by interleukin-16 in homeostasis and cancer. J. Cell. Physiol. 2014, 229, 139–147. [Google Scholar] [CrossRef]
- Lard, L.R.; Roep, B.O.; Verburgh, C.A.; Zwinderman, A.H.; Huizinga, T.W.J. Elevated IL-16 levels in patients with systemic lupus erythematosus are associated with disease severity but not with genetic susceptibility to lupus. Lupus 2002, 11, 181–185. [Google Scholar] [CrossRef]
- Niewold, T.B.; Meves, A.; Lehman, J.S.; Popovic-Silwerfeldt, K.; Häyry, A.; Söderlund-Matell, T.; Charlesworth, C.M.; Madden, B.; Lundberg, I.E.; Wahren-Herlenius, M.; et al. Proteome study of cutaneous lupus erythematosus (CLE) and dermatomyositis skin lesions reveals IL-16 is differentially upregulated in CLE. Arthritis Res. Ther. 2021, 23, 132. [Google Scholar] [CrossRef]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolk, K.; Witte, K.; Witte, E.; Raftery, M.; Kokolakis, G.; Philipp, S.; Schönrich, G.; Warszawska, K.; Kirsch, S.; Prösch, S.; et al. IL-29 is produced by T(H)17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci. Transl. Med. 2013, 5, 204ra129. [Google Scholar] [CrossRef] [PubMed]
- Zickert, A.; Amoudruz, P.; Sundström, Y.; Rönnelid, J.; Malmström, V.; Gunnarsson, I. IL-17 and IL-23 in lupus nephritis-association to histopathology and response to treatment. BMC Immunol. 2015, 16, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.-Q.; Zhang, Y.-P.; Yin, J.; Tang, Z.-Q.; Han, Y.-F.; Shi, Z.-R.; Tan, G.-Z.; Wang, L. Characterization of autoantibodies and cytokines related to cutaneous lupus erythematosus. Lupus 2021, 30, 315–319. [Google Scholar] [CrossRef]
- Tanasescu, C.; Balanescu, E.; Balanescu, P.; Olteanu, R.; Badea, C.; Grancea, C.; Vagu, C.; Bleotu, C.; Ardeleanu, C.; Georgescu, A. IL-17 in cutaneous lupus erythematosus. Eur. J. Intern. Med. 2010, 21, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Sippl, N.; Faustini, F.; Rönnelid, J.; Turcinov, S.; Chemin, K.; Gunnarsson, I.; Malmström, V. Arthritis in systemic lupus erythematosus is characterized by local IL-17A and IL-6 expression in synovial fluid. Clin. Exp. Immunol. 2021, 205, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Mathias, L.M.; Stohl, W. Systemic lupus erythematosus (SLE): Emerging therapeutic targets. Expert Opin. Ther. Targets 2020, 24, 1283–1302. [Google Scholar] [CrossRef]
- Van Vollenhoven, R.F.; Hahn, B.H.; Tsokos, G.C.; Wagner, C.L.; Lipsky, P.; Touma, Z.; Werth, V.P.; Gordon, R.M.; Zhou, B.; Hsu, B.; et al. Efficacy and safety of ustekinumab, an IL-12 and IL-23 inhibitor, in patients with active systemic lupus erythematosus: Results of a multicentre, double-blind, phase 2, randomised, controlled study. Lancet 2018, 392, 1330–1339. [Google Scholar] [CrossRef]
- Kinoshita, K.; Yamagata, T.; Nozaki, Y.; Sugiyama, M.; Ikoma, S.; Funauchi, M.; Kanamaru, A. Blockade of IL-18 receptor signaling delays the onset of autoimmune disease in MRL-Faslpr mice. J. Immunol. 2004, 173, 5312–5318. [Google Scholar] [CrossRef] [Green Version]
- Mende, R.; Vincent, F.B.; Kandane-Rathnayake, R.; Koelmeyer, R.; Lin, E.; Chang, J.; Hoi, A.Y.; Morand, E.F.; Harris, J.; Lang, T. Analysis of Serum Interleukin (IL)-1β and IL-18 in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1250. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Drenker, M.; Eiz-Vesper, B.; Werfel, T.; Wittmann, M. Evidence for a pathogenetic role of interleukin-18 in cutaneous lupus erythematosus. Arthritis Rheum. 2008, 58, 3205–3215. [Google Scholar] [CrossRef]
- Xiang, M.; Feng, Y.; Wang, Y.; Wang, J.; Zhang, Z.; Liang, J.; Xu, J. Correlation between circulating interleukin-18 level and systemic lupus erythematosus: A meta-analysis. Sci. Rep. 2021, 11, 4707. [Google Scholar] [CrossRef]
- Xu, W.-D.; Su, L.-C.; Liu, X.-Y.; Wang, J.-M.; Yuan, Z.-C.; Qin, Z.; Zhou, X.-P.; Huang, A.-F. IL-38: A novel cytokine in systemic lupus erythematosus pathogenesis. J. Cell. Mol. Med. 2020, 24, 12379–12389. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Chen, Y.; Wu, H.; Zhao, M.; Lu, Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J. Autoimmun. 2019, 99, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nakou, M.; Papadimitraki, E.D.; Fanouriakis, A.; Bertsias, G.K.; Choulaki, C.; Goulidaki, N.; Sidiropoulos, P.; Boumpas, D.T. Interleukin-21 is increased in active systemic lupus erythematosus patients and contributes to the generation of plasma B cells. Clin. Exp. Rheumatol. 2013, 31, 172–179. [Google Scholar]
- Pan, H.-F.; Wu, G.-C.; Fan, Y.-G.; Leng, R.-X.; Peng, H.; Zhou, M.; Li, B.-Z.; Zhu, Y.; Tao, J.-H.; Li, X.-P.; et al. Decreased serum level of IL-21 in new-onset systemic lupus erythematosus patients. Rheumatol. Int. 2013, 33, 2337–2342. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Kumar, V.; Karnell, J.L.; Naiman, B.; Gross, P.S.; Rahman, S.; Zerrouki, K.; Hanna, R.; Morehouse, C.; et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat. Commun. 2018, 9, 1758. [Google Scholar] [CrossRef]

{kind=link}
| Cytokine | Immunity 1 | Levels in Circulation 2 | DAS 3 | SLE Target Organ Involvement 4 | Where Cytokine Has Been Detected | ||||
|---|---|---|---|---|---|---|---|---|---|
| LN | Arthritis | CLE | Serositis | CNS | |||||
| IFN Type I | I | ↑ | + | ↑ | ↑ | ↑ | ↑ | ↑ | circulation |
| IFN Type II | A | ↑ | + | ↑ | ↑ | ↑ | NN | NN | circulation |
| IFN Type III | I | ↑ | + | ↑ | ↑ | ↑ | NN | NN | circulation, skin lesions |
| BAFF/APRIL | I | ↑ | + | ↑ | ↑ | NN | NN | ↑ | circulation |
| IL-2 | A | NN | NN | ↑ | NN | NN | NN | NN | renal tissue |
| IL-6 | I | ↑ | + | ↑ | ↑ | ↑ | NN | ↑ | circulation, CSF, urine, serum |
| IL-10 | I | ↑ | + | ↑ | NN | ↑ | NN | NN | circulation |
| IL-16 | A/I | ↑ | + | ↑ | NN | ↑ | NN | NN | circulation, urine, skin lesions |
| IL-12, IL-23 | A/I | ↑ | + | ↑ | NN | ↑ | NN | NN | circulation, kidney |
| IL-17 | A | ↑ | + | ↑ | ↑ | ↑ | NN | NN | circulation, kidney, skin, synovial fluid |
| IL-1 | I | ↑ | NN | ↑ | ↑ | ↑ | NN | NN | circulation, skin |
| IL-18 | I | ↑ | + | ↑ | NN | ↑ | NN | NN | circulation, skin lesions |
| IL-38 | I | ↑ | + | ↑ | ↑ | NN | ↑ | NN | circulation |
| Cytokine Target | Drug/Molecule and Results from Clinical Trials |
|---|---|
| IFN Type I |
|
| IFN Type II |
|
| IFN Type III | No trials identified |
| General IFN system: Target plasmacytoid dendritic cells |
|
| TNF-α |
|
| Blys/BAFF/APRIL |
|
| IL-2 |
|
| IL-6 |
|
| IL-10 | BT 063, Phase 2 completed, results unavailable, NCT02554019 |
| IL-16 | Not identified |
| IL-17 | Sekucinumab, Phase 3 recruiting, NCT04181762 |
| IL-12, IL-23 | Ustekinumab, primary endpoints met in phase 2a, NCT02349061. Phase 3 is ongoing, NCT03517722 |
| IL-21 |
|
| IL-1 | Anakinra, only case reports |
| IL-18 | Not identified |
| IL-38 | Not identified |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idborg, H.; Oke, V. Cytokines as Biomarkers in Systemic Lupus Erythematosus: Value for Diagnosis and Drug Therapy. Int. J. Mol. Sci. 2021, 22, 11327. https://doi.org/10.3390/ijms222111327
Idborg H, Oke V. Cytokines as Biomarkers in Systemic Lupus Erythematosus: Value for Diagnosis and Drug Therapy. International Journal of Molecular Sciences. 2021; 22(21):11327. https://doi.org/10.3390/ijms222111327
Chicago/Turabian StyleIdborg, Helena, and Vilija Oke. 2021. "Cytokines as Biomarkers in Systemic Lupus Erythematosus: Value for Diagnosis and Drug Therapy" International Journal of Molecular Sciences 22, no. 21: 11327. https://doi.org/10.3390/ijms222111327