
Hepatocarcinogenesis Prevention by Pirfenidone Is PPARγ Mediated and Involves Modification of Nuclear NF-kB p65/p50 Ratio

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. MRHM Design
2.3. Human Tissue Samples
2.4. Liver Histological Assessment
2.5. Protein Extraction and Western Blot
2.6. Cell Culture
2.7. Immunofluorescence of Human and Rat Tissues and Cells
2.8. Molecular Docking and Structure Viewing
2.9. Statistical Analysis
3. Results
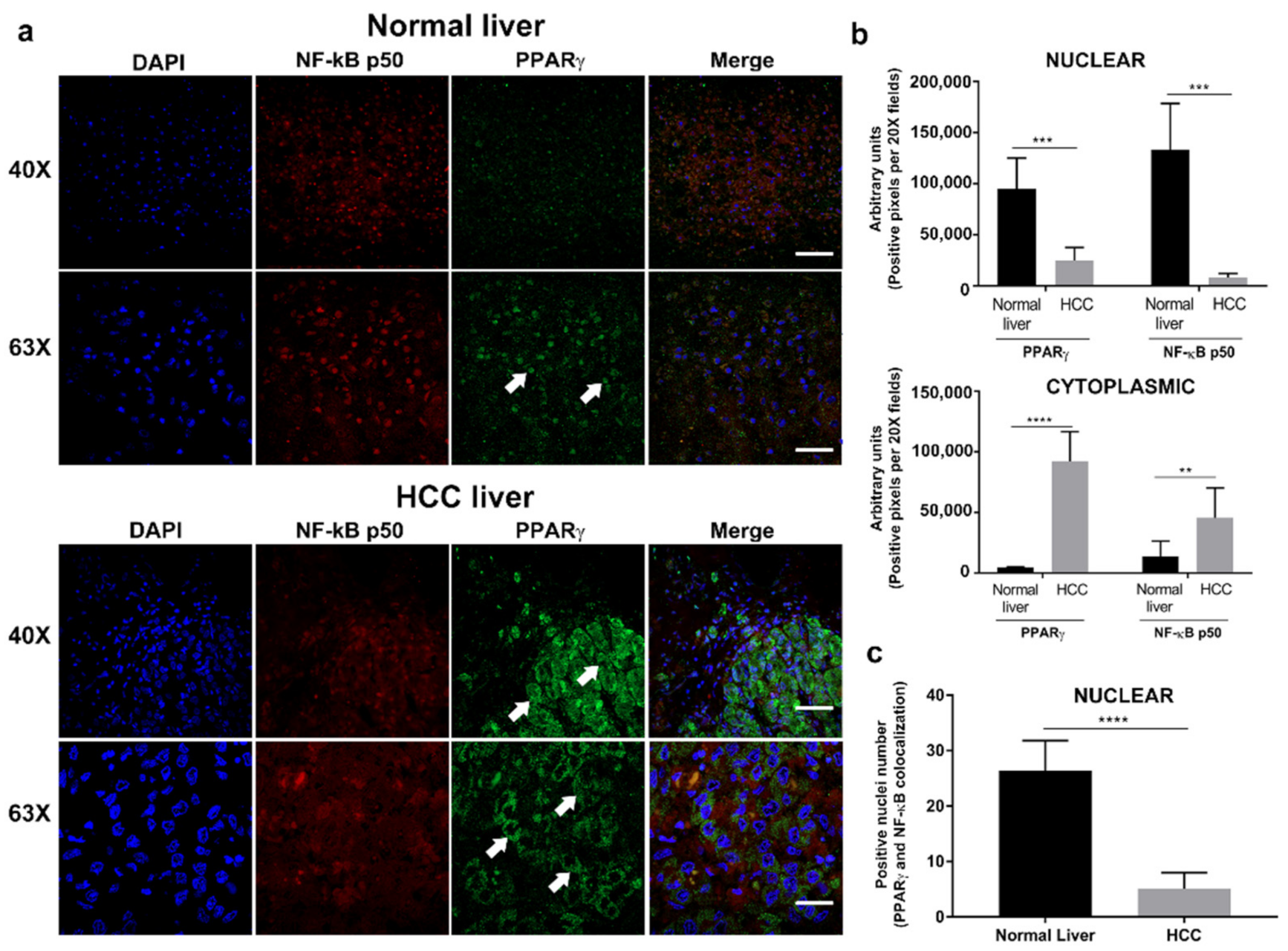
3.1. PPARγ Is Overexpressed in the Cytoplasm of Hepatocytes in Human HCC Tissue
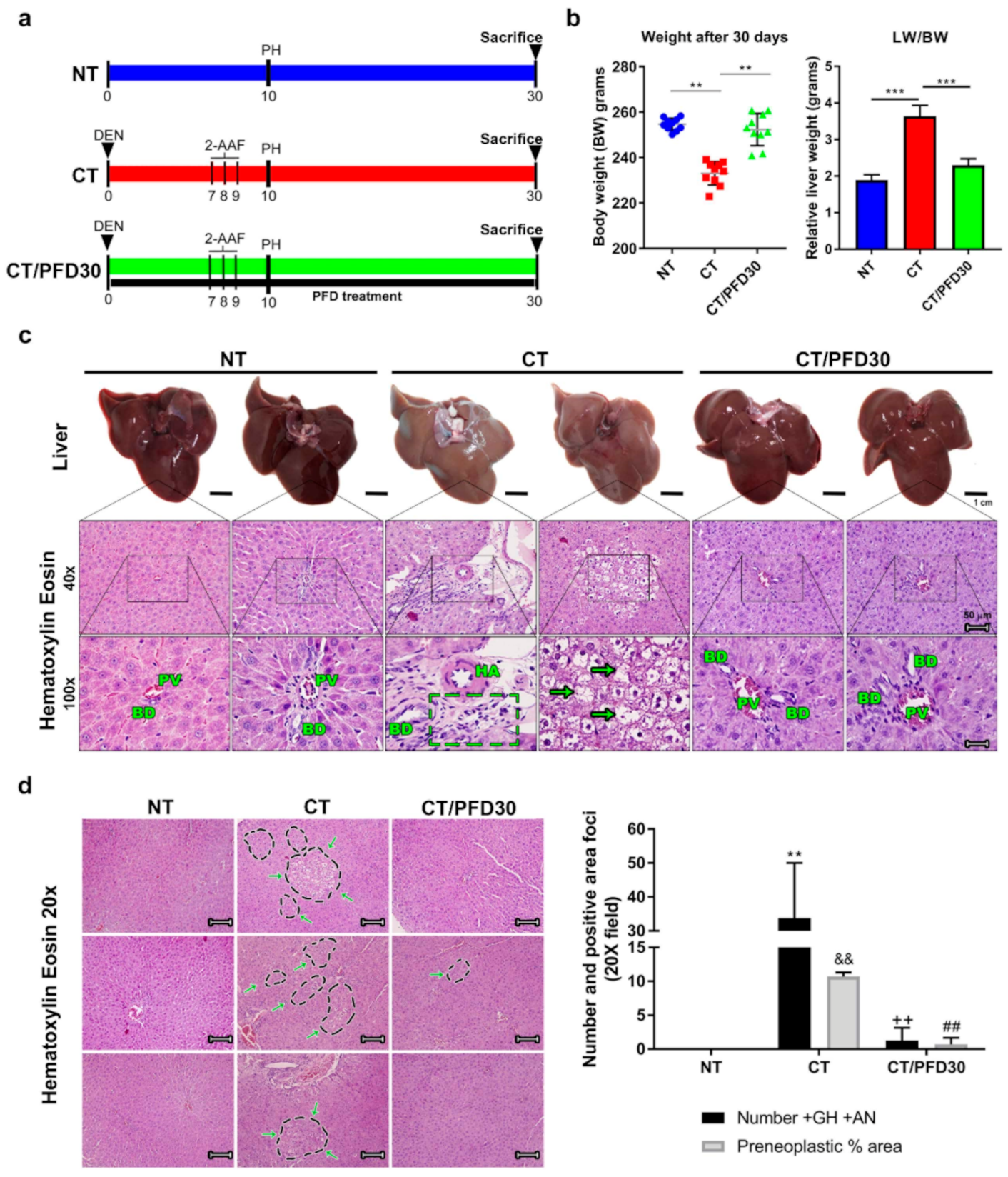
3.2. Pirfenidone Prevents Weight Loss Induced by Hepatotoxic Damage and Upholds Healthy Liver Morphology
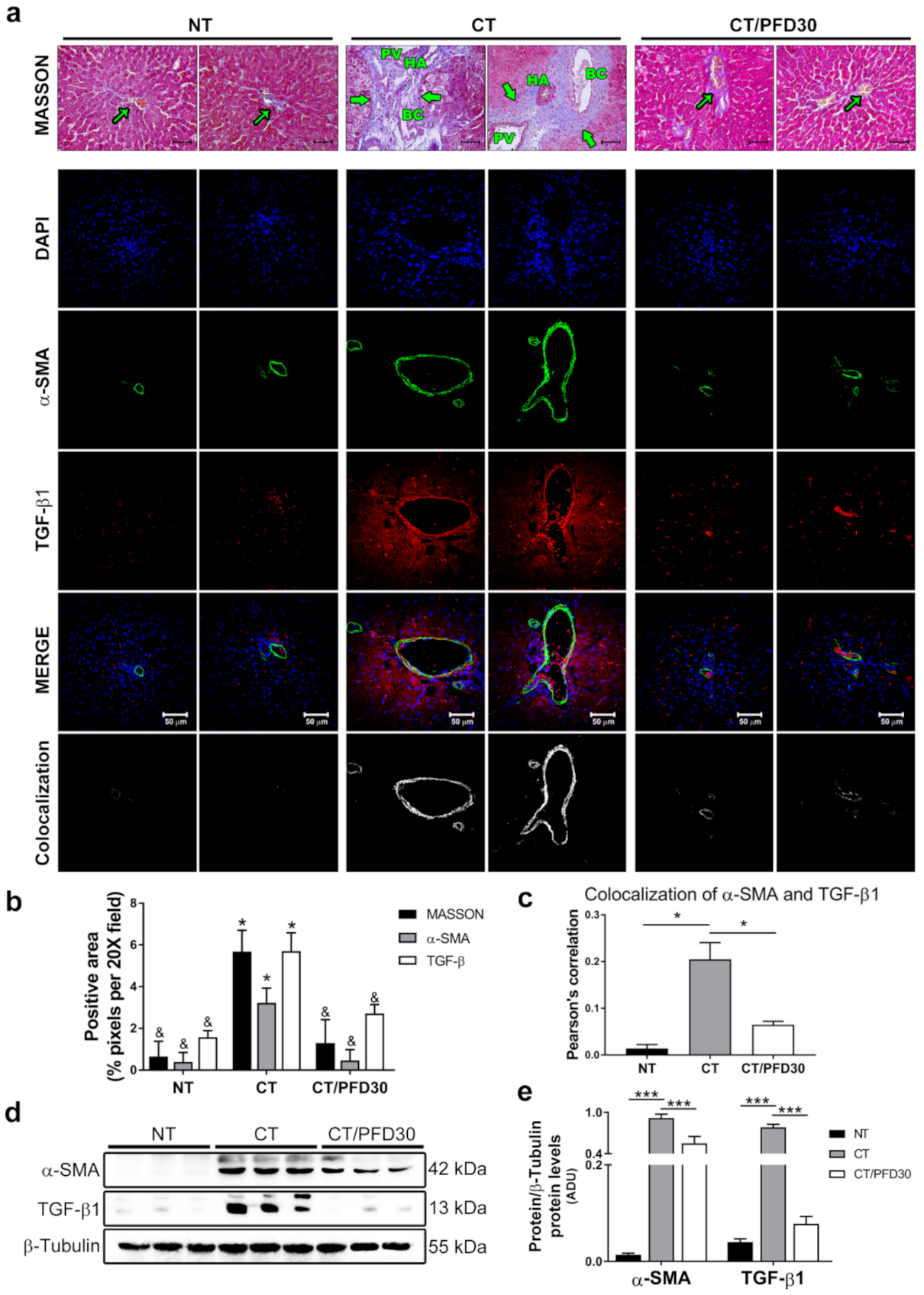
3.3. Fibrosis-Induction Is Prevented by PFD Administration in the MRHM
3.4. Inflammation Is Modulated by the Pirfenidone-Induced NF-kB p65/p50 Pathway
3.5. Increased Expression of Apoptotic Markers in Nuclear and Cytoplasmic Fractions by Pirfenidone
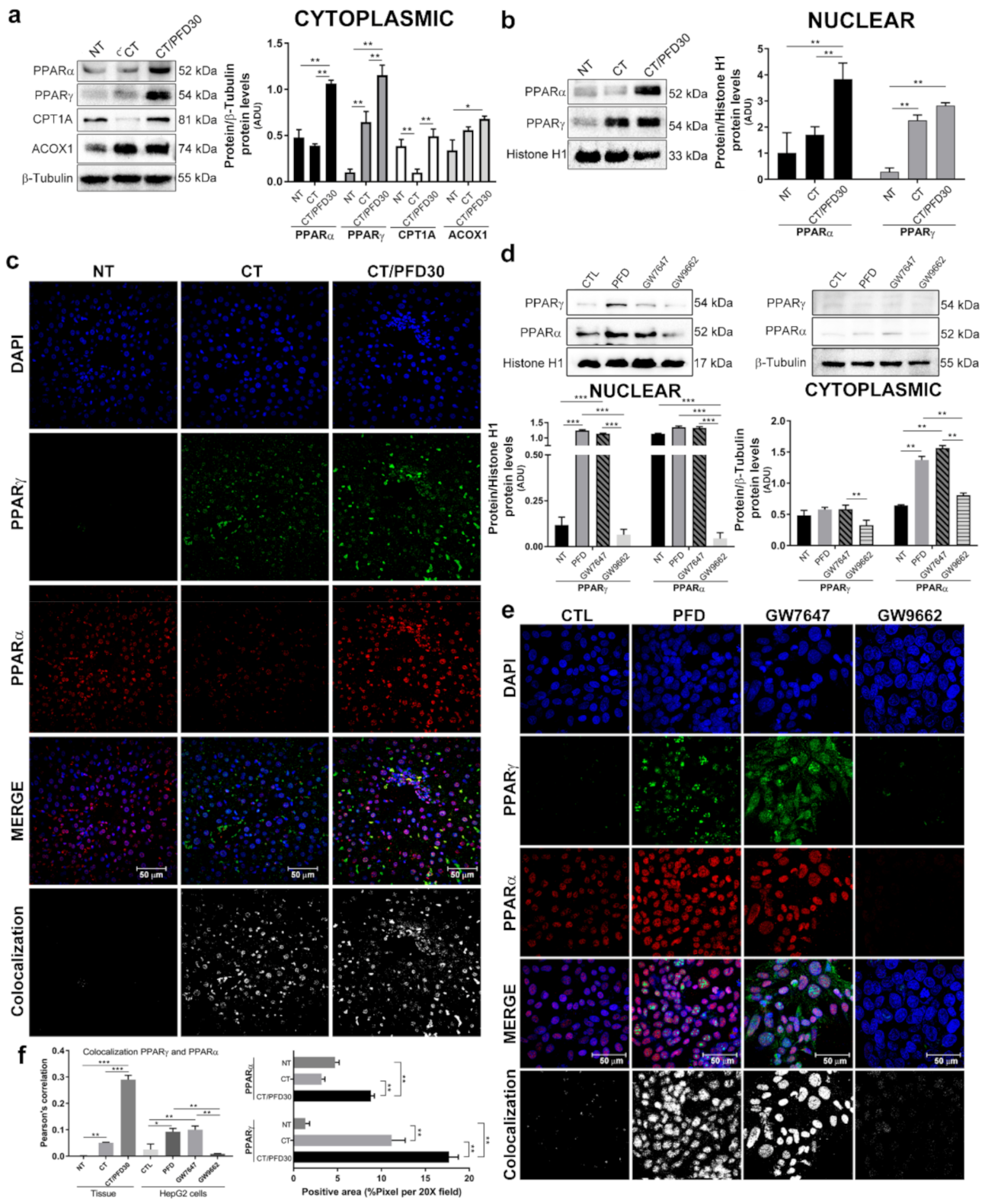
3.6. Pirfenidone Promotes PPARγ Expression and Its Translocation to Nuclei
3.7. In-Silico Assay Demonstrate That Pirfenidone Is a PPARγ Ligand
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Jeon, Y.T.; Kim, S.H.; Song, Y.S. NF-kappaB as a potential molecular target for cancer therapy. Biofactors 2007, 29, 19–35. [Google Scholar] [CrossRef]
- Jana, A.; Krett, N.L.; Guzman, G.; Khalid, A.; Ozden, O.; Staudacher, J.J.; Bauer, J.; Baik, S.H.; Carroll, T.; Yazici, C.; et al. NFkB is essential for activin-induced colorectal cancer migration via upregulation of PI3K-MDM2 pathway. Oncotarget 2017, 8, 37377–37393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbecki, J.; Bobiński, R.; Dutka, M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019, 68, 443–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panigrahy, D.; Shen, L.Q.; Kieran, M.W.; Kaipainen, A. Therapeutic potential of thiazolidinediones as anticancer agents. Expert Opin. Investig. Drugs 2003, 12, 1925–1937. [Google Scholar] [CrossRef]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative crosstalk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [Green Version]
- Zúñiga, J.; Cancino, M.; Medina, F.; Varela, P.; Vargas, R.; Tapia, G.; Videla, L.A.; Fernández, A. N-3 PUFA supplementation triggers PPAR-α activation and PPAR-α/NF-κB interaction: Anti-inflammatory implications in liver ischemia-reperfusion injury. PLoS ONE 2011, 6, e28502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.W.; Kang, B.Y.; Kim, S.H.; Pak, Y.K.; Cho, D.; Trinchieri, G.; Kim, T.S. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J. Biol. Chem. 2000, 275, 32681–32687. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Moreau, F.; Chadee, K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat. Commun. 2012, 3, 1300. [Google Scholar] [CrossRef] [Green Version]
- Khandekar, M.J.; Banks, A.S.; Laznik-Bogoslavski, D.; White, J.P.; Choi, J.H.; Kazak, L.; Lo, J.C.; Cohen, P.; Wong, K.-K.; Kamenecka, T.M.; et al. Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2018, 115, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Solt, D.B.; Medline, A.; Farber, E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am. J. Pathol. 1977, 88, 595–618. [Google Scholar]
- Carrasco-Torres, G.; Monroy-Ramírez, H.C.; Martínez-Guerra, A.A.; Baltiérrez-Hoyos, R.; Romero-Tlalolini, M.L.Á.; Villa-Treviño, S.; Sánchez-Chino, X.; Vásquez-Garzón, V.R. Quercetin Reverses Rat Liver Preneoplastic Lesions Induced by Chemical Carcinogenesis. Oxid. Med. Cell Longev. 2017, 2017, 4674918. [Google Scholar] [CrossRef]
- Vera, M.C.; Pisani, G.B.; Biancardi, M.E.; Bottai, H.; Alvarez, M.L.; Quintana, A.B. Comparison of two chemical models to induce hepatic preneoplasia in male Wistar rats. Ann. Hepatol. 2015, 14, 259–266. [Google Scholar] [CrossRef]
- Lopez-de la Mora, D.A.; Sanchez-Roque, C.; Montoya-Buelna, M.; Sanchez-Enriquez, S.; Lucano-Landeros, S.; Macias-Barragan, J.; Armendariz-Borunda, J. Role and New Insights of Pirfenidone in Fibrotic Diseases. Int. J. Med. Sci. 2015, 12, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, L.; Hernandez, I.; Sandoval, A.; Salazar, A.; Garcia, J.; Vera, J.; Grijalva, G.; Muriel, P.; Margolin, S.; Armendariz-Borunda, J. Pirfenidone effectively reverses experimental liver fibrosis. J. Hepatol. 2002, 37, 797–805. [Google Scholar] [CrossRef]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.F.; Ni, H.F.; Pan, M.M.; Liu, H.; Xu, M.; Zhang, M.H.; Liu, B.C. Pirfenidone inhibits macrophage infiltration in 5/6 nephrectomized rats. Am. J. Physiol. Renal. Physiol. 2013, 304, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ni, Y.; Nagata, N.; Xu, L.; Zhuge, F.; Nagashimada, M.; Kaneko, S.; Ota, T. Pirfenidone prevents and reverses hepatic insulin resistance and steatohepatitis by polarizing M2 macrophages. Lab Investig. 2019, 99, 1335–1348. [Google Scholar] [CrossRef]
- Zou, W.J.; Huang, Z.; Jiang, T.P.; Shen, Y.P.; Zhao, A.S.; Zhou, S.; Zhang, S. Pirfenidone Inhibits Proliferation and Promotes Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the Wnt/β-Catenin Signaling Pathway. Med. Sci. Monit. 2017, 23, 6107–6113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marwitz, S.; Turkowski, K.; Nitschkowski, D.; Weigert, A.; Brandenburg, J.; Reiling, N.; Thomas, M.; Reck, M.; Drömann, D.; Seeger, W.; et al. The Multi-Modal Effect of the Anti-fibrotic Drug Pirfenidone on NSCLC. Front. Oncol. 2020, 21, 1550–1563. [Google Scholar] [CrossRef]
- Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Meza-Rios, A.; Garcia-Bañuelos, J.; Vera-Cruz, J.; Gutiérrez-Cuevas, J.; Silva-Gomez, J.; Staels, B.; Dominguez-Rosales, J.; Galicia-Moreno, M.; et al. Pirfenidone is an agonistic ligand for PPARα and improves NASH by activation of SIRT1/LKB1/pAMPK. Hepatol. Commun. 2020, 4, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic. Acids. Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, K.L.; Wada, K.; Takahashi, H.; Matsuhashi, N.; Ohnishi, S.; Wolfe, M.M.; Turner, J.R.; Nakajima, A.; Borkan, S.C.; Saubermann, L.J.; et al. Peroxisome proliferator-activated receptor gamma inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res. 2005, 65, 2251–2259. [Google Scholar] [CrossRef] [Green Version]
- Afaloniati, H.; Angelopoulou, K.; Giakoustidis, A.; Hardas, A.; Pseftogas, A.; Makedou, K.; Gargavanis, A.; Goulopoulos, T.; Iliadis, S.; Papadopoulos, V.; et al. HDAC1/2 Inhibitor Romidepsin Suppresses DEN-Induced Hepatocellular Carcinogenesis in Mice. Onco. Targets Ther. 2020, 13, 5575–5588. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharm. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Vásquez-Garzón, V.R.; Beltrán-Ramírez, O.; Salcido-Neyoy, M.E.; Cervante-Anaya, N.; Villa-Treviño, S. Analysis of gene expression profiles as a tool to uncover tumor markers of liver cancer progression in a rat model. Biomed. Rep. 2015, 3, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Arrese, M.; Hernandez, A.; Astete, L.; Estrada, L.; Cabello-Verrugio, C.; Cabrera, D. TGF-β and Hepatocellular Carcinoma: When a Friend Becomes an Enemy. Curr. Protein. Pept. Sci. 2018, 19, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Sancho, P.; Bertran, E.; Fabregat, I. Dissecting the effect of targeting the epidermal growth factor receptor on TGF-β-induced-apoptosis in human hepatocellular carcinoma cells. J. Hepatol. 2011, 55, 351–358. [Google Scholar] [CrossRef]
- Murillo, M.M.; del Castillo, G.; Sánchez, A.; Fernández, M.; Fabregat, I. Involvement of EGF receptor and c-Src in the survival signals induced by TGF-beta1 in hepatocytes. Oncogene 2005, 24, 4580–4587. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Román, R.; Salazar-González, D.; Rosas, S.; Arellanes-Robledo, J.; Beltrán-Ramírez, O.; Fattel-Fazenda, S.; Villa-Treviño, S. The differential NF-kB modulation by S-adenosyl-L-methionine, N-acetylcysteine and quercetin on the promotion stage of chemical hepatocarcinogenesis. Free Radic. Res. 2008, 42, 331–343. [Google Scholar] [CrossRef]
- Buroker, N.E.; Barboza, J.; Huang, J.Y. The IkappaB alpha gene is a peroxisome proliferator-activated receptor cardiac target gene. FEBS J. 2009, 276, 3247–3255. [Google Scholar] [CrossRef]
- Wu, J.M.; Sheng, H.; Saxena, R.; Skill, N.J.; Bhat-Nakshatri, P.; Yu, M.; Nakshatri, H.; Maluccio, M.A. NF-kappaB inhibition in human hepatocellular carcinoma and its potential as adjunct to sorafenib based therapy. Cancer Lett. 2009, 278, 145–155. [Google Scholar] [CrossRef]
- Delerive, P.; De Bosscher, K.; Vanden Berghe, W.; Fruchart, J.C.; Haegeman, G.; Staels, B. DNA binding-independent induction of IkappaB alpha gene transcription by PPARalpha. Mol. Endocrinol. 2002, 16, 1029–1039. [Google Scholar] [CrossRef]
- Yu, Y.; Wan, Y.; Huang, C. The biological functions of NF-kappaB1 (p50) and its potential as an anti-cancer target. Curr. Cancer Drug Targets 2009, 9, 566–571. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. NF-kappaB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.C.; Lin, Y.J.; Lee, C.T.; Liu, H.S.; Lee, J.C. Cyclooxygenase-2 expression in the tumor environment is associated with poor prognosis in colorectal cancer patients. Oncol. Lett. 2013, 6, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Meng, X.; Franklin, D.A.; Dong, J.; Zhang, Y. MDM2-p53 pathway in hepatocellular carcinoma. Cancer Res. 2014, 74, 7161–7167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonofiglio, D.; Aquila, S.; Catalano, S.; Gabriele, S.; Belmonte, M.; Middea, E.; Qi, H.; Morelli, C.; Gentile, M.; Maggiolini, M.; et al. Peroxisome proliferator-activated receptor-gamma activates p53 gene promoter binding to the nuclear factor-kappaB sequence in human MCF7 breast cancer cells. Mol. Endocrinol. 2006, 20, 3083–3092. [Google Scholar] [CrossRef]
- Okura, T.; Nakamura, M.; Takata, Y.; Watanabe, S.; Kitami, Y.; Hiwada, K. Troglitazone induces apoptosis via the p53 and Gadd45 pathway in vascular smooth muscle cells. Eur. J. Pharmacol. 2000, 407, 227–235. [Google Scholar] [CrossRef]
- Cheung, K.F.; Zhao, J.; Hao, Y.; Li, X.; Lowe, A.W.; Cheng, A.S.; Sung, J.J.; Yu, J. CITED2 is a novel direct effector of peroxisome proliferator-activated receptor γ in suppressing hepatocellular carcinoma cell growth. Cancer 2013, 119, 1217–1226. [Google Scholar] [CrossRef]
- Venturi, A.; Piaz, F.D.; Giovannini, C.; Gramantieri, L.; Chieco, P.; Bolondi, L. Human hepatocellular carcinoma expresses specific PCNA isoforms: An in vivo and in vitro evaluation. Lab. Investig. 2008, 88, 995–1007. [Google Scholar] [CrossRef] [Green Version]
- Mun, K.S.; Cheah, P.L.; Baharudin, N.B.; Looi, L.M. Proliferating cell nuclear antigen (PCNA) activity in hepatocellular carcinoma, benign peri-neoplastic and normal liver. Malays. J. Pathol. 2006, 28, 73–77. [Google Scholar]
- Shu, Y.; Lu, Y.; Pang, X.; Zheng, W.; Huang, Y.; Li, J.; Ji, J.; Zhang, C.; Shen, P. Phosphorylation of PPARγ at Ser84 promotes glycolysis and cell proliferation in hepatocellular carcinoma by targeting PFKFB4. Oncotarget 2016, 7, 76984–76994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroker, A.J.; Bruning, J.B. Review of the Structural and Dynamic Mechanisms of PPARγ Partial Agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biochemical Marker | NT | CT | CT/PFD30 | p |
|---|---|---|---|---|
| AST (U/L) | 162.50 ± 11.42 | 123.50 ± 16.26 | 166.50 ± 15.76 | <0.01 |
| ALT (U/dL) | 74.50 ± 6.36 | 82.00 ± 1.41 | 81.00 ± 5.66 | NS |
| Alkaline Phosphatase (U/L) | 338 ± 9.90 | 442 ± 25.46 | 301 ± 65.05 | <0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-Gomez, J.A.; Galicia-Moreno, M.; Sandoval-Rodriguez, A.; Miranda-Roblero, H.O.; Lucano-Landeros, S.; Santos, A.; Monroy-Ramirez, H.C.; Armendariz-Borunda, J. Hepatocarcinogenesis Prevention by Pirfenidone Is PPARγ Mediated and Involves Modification of Nuclear NF-kB p65/p50 Ratio. Int. J. Mol. Sci. 2021, 22, 11360. https://doi.org/10.3390/ijms222111360
Silva-Gomez JA, Galicia-Moreno M, Sandoval-Rodriguez A, Miranda-Roblero HO, Lucano-Landeros S, Santos A, Monroy-Ramirez HC, Armendariz-Borunda J. Hepatocarcinogenesis Prevention by Pirfenidone Is PPARγ Mediated and Involves Modification of Nuclear NF-kB p65/p50 Ratio. International Journal of Molecular Sciences. 2021; 22(21):11360. https://doi.org/10.3390/ijms222111360
Chicago/Turabian StyleSilva-Gomez, Jorge Antonio, Marina Galicia-Moreno, Ana Sandoval-Rodriguez, Hipolito Otoniel Miranda-Roblero, Silvia Lucano-Landeros, Arturo Santos, Hugo Christian Monroy-Ramirez, and Juan Armendariz-Borunda. 2021. "Hepatocarcinogenesis Prevention by Pirfenidone Is PPARγ Mediated and Involves Modification of Nuclear NF-kB p65/p50 Ratio" International Journal of Molecular Sciences 22, no. 21: 11360. https://doi.org/10.3390/ijms222111360