
p53 Transactivation Domain Mediates Binding and Phase Separation with Poly-PR/GR


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
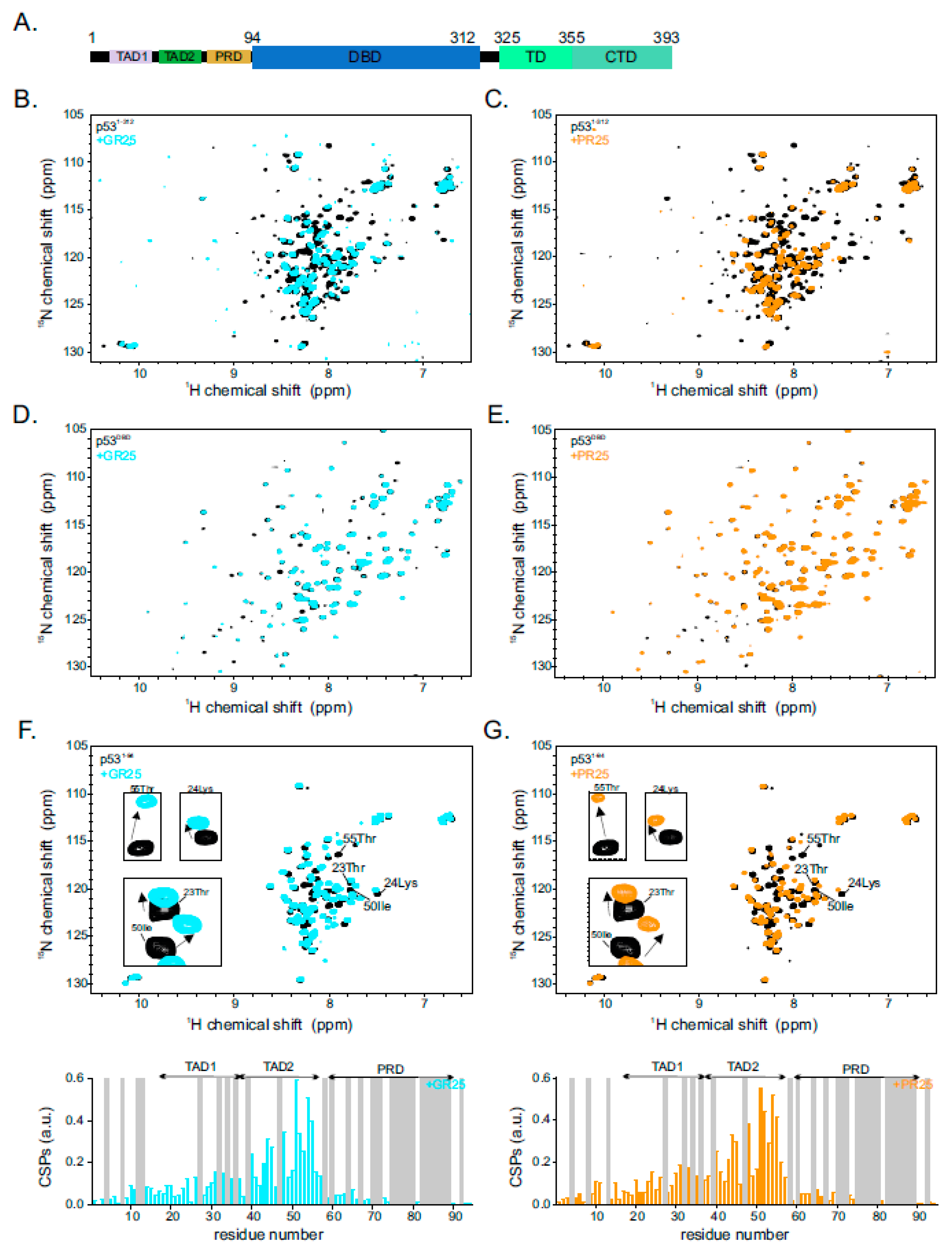
2.1. Poly(PR)25/Poly(GR)25 Interact Directly with p53
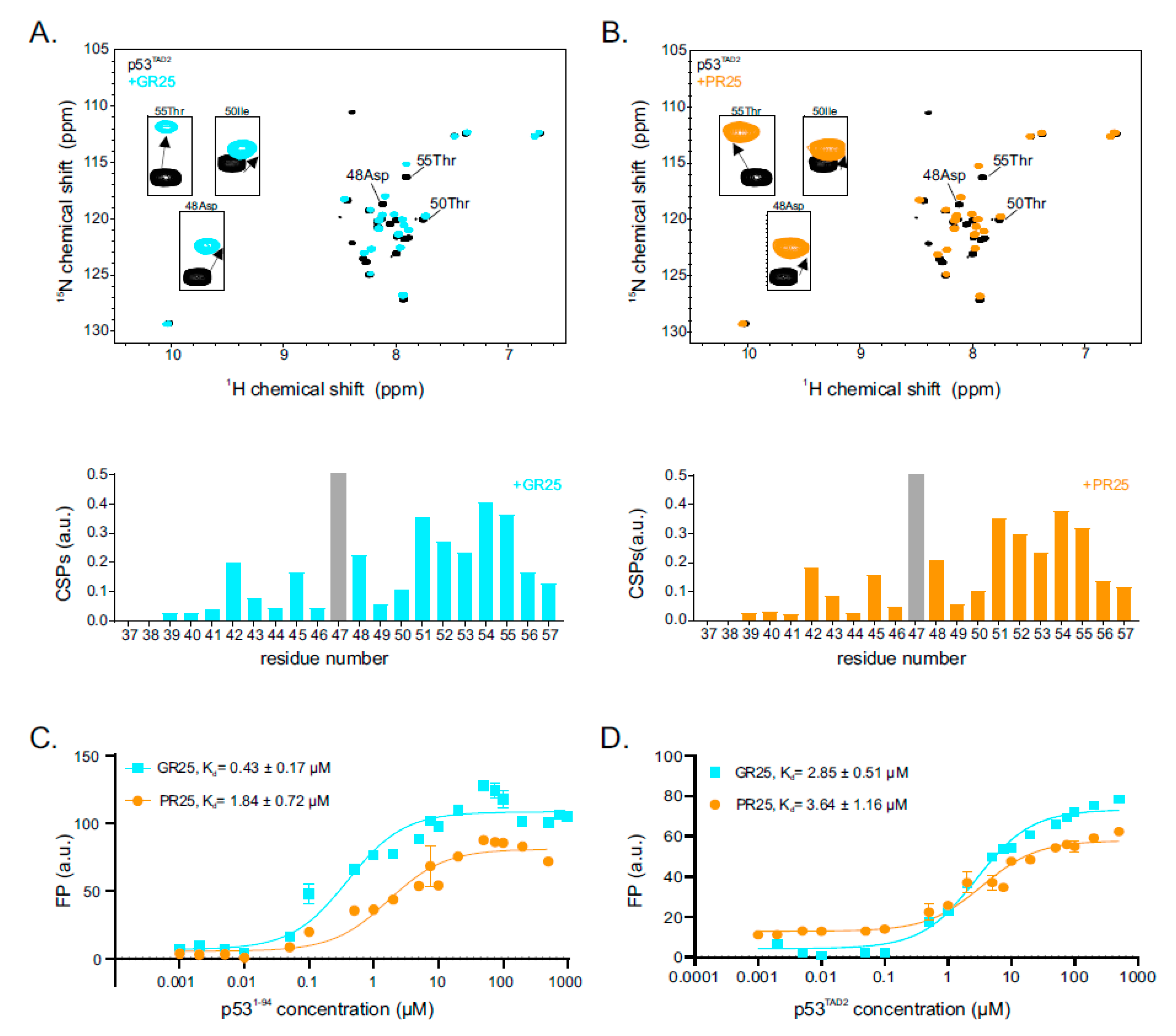
2.2. p53 Transactivation Domain 2 Is the Main PR25/GR25 Binding Site
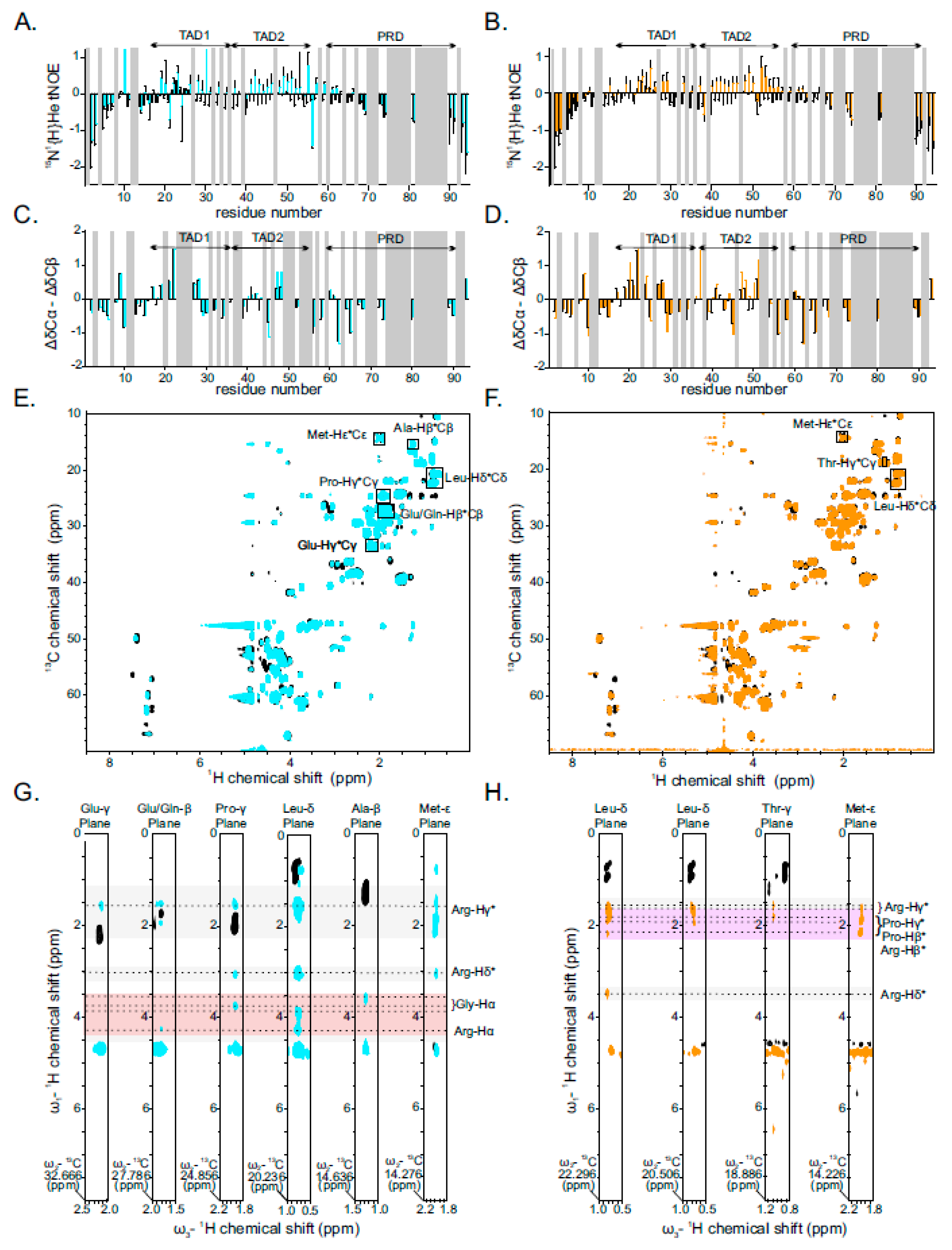
2.3. Binding of PR25/GR25 to p531−94 Increases Its Rigidity and α-Helical Secondary Structure Propensity
2.4. Interaction between PR25/GR25 and p531−94 Is Mediated by Electrostatic and/or Hydrophobic Interactions
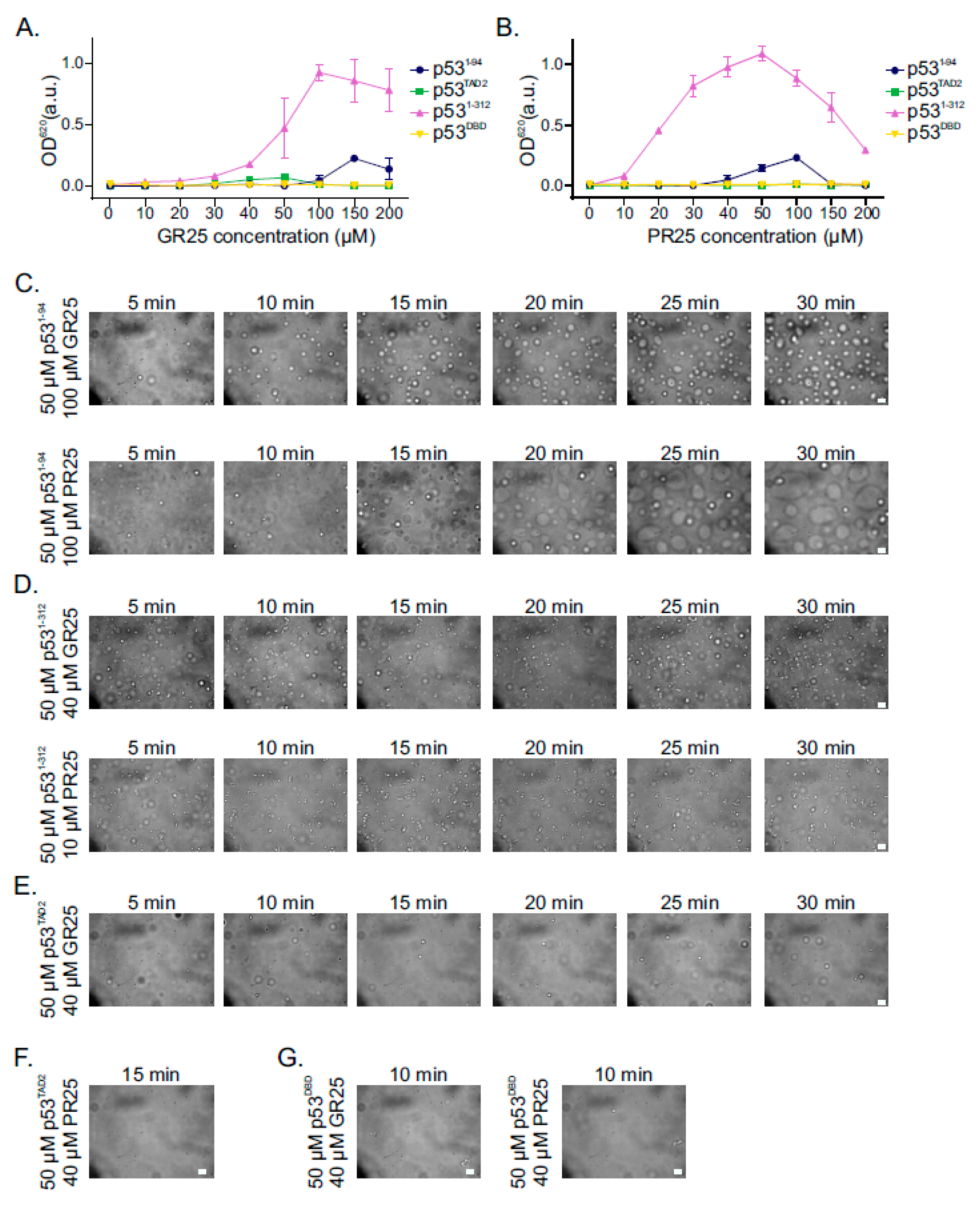
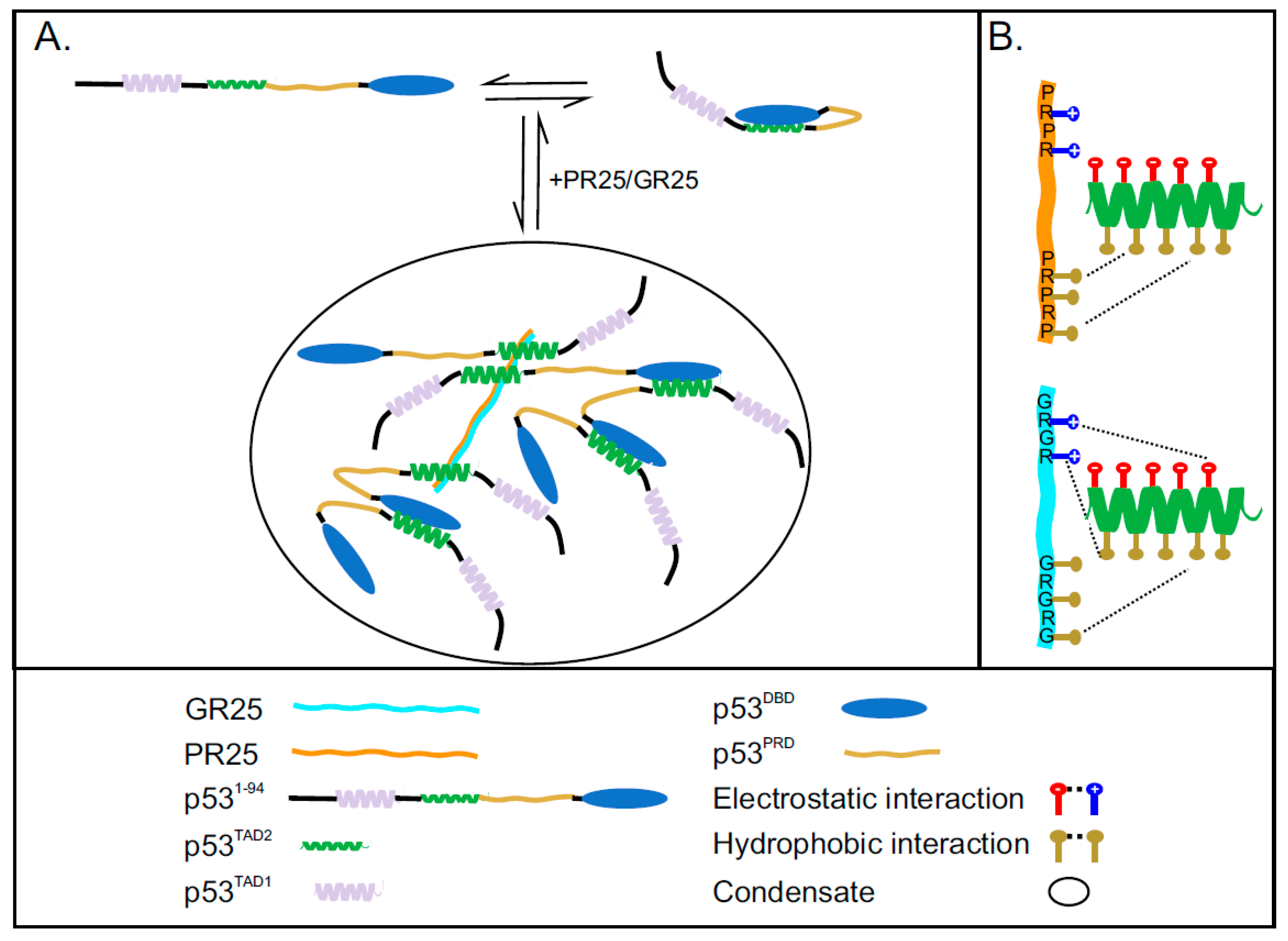
2.5. PR25/GR25 Induce Phase Separation of p531−94 and p531−312
3. Discussion
4. Materials and Methods
4.1. Plasmids and Synthetic Peptides
4.2. Protein Expression and Purification
4.3. NMR Spectroscopy
4.3.1. Binding Assays
4.3.2. Resonance Assignment and NOE Experiments
4.4. Fluorescence Polarization Measurements
4.5. Turbidity Assay
4.6. Differential Interference Contrast Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, B.; Robberecht, W.; Van Den Bosch, L. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 2020, 39, e101112. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Taylor, J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Stalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Coyne, A.N.; Zaepfel, B.L.; Hayes, L.; Fitchman, B.; Salzberg, Y.; Luo, E.C.; Bowen, K.; Trost, H.; Aigner, S.; Rigo, F.; et al. G4C2 Repeat RNA Initiates a POM121-Mediated Reduction in Specific Nucleoporins in C9orf72 ALS/FTD. Neuron 2020, 107, 1124–1140 e1111. [Google Scholar] [CrossRef]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; Dejesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W., III; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Lammich, S.; Mackenzie, I.R.; Forne, I.; Zilow, S.; Kretzschmar, H.; Edbauer, D.; Janssens, J.; Kleinberger, G.; Cruts, M.; et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013, 125, 413–423. [Google Scholar] [CrossRef]
- Zu, T.; Liu, Y.; Banez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Author Correction: Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 644. [Google Scholar] [CrossRef]
- Gitler, A.D.; Tsuiji, H. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 2016, 1647, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizielinska, S.; Gronke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Sun, Y.; Eshov, A.; Zhou, J.; Isiktas, A.U.; Guo, J.U. C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat. Commun. 2020, 11, 3354. [Google Scholar] [CrossRef] [PubMed]
- Jovicic, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W., III; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [Green Version]
- Boeynaems, S.; Bogaert, E.; Michiels, E.; Gijselinck, I.; Sieben, A.; Jovicic, A.; De Baets, G.; Scheveneels, W.; Steyaert, J.; Cuijt, I.; et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 2016, 6, 20877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Gendron, T.F.; Ebbert, M.T.W.; O’Raw, A.D.; Yue, M.; Jansen-West, K.; Zhang, X.; Prudencio, M.; Chew, J.; Cook, C.N.; et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 2018, 24, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Liu, L.; Tao, Z.; Wang, R.; Ren, H.; Sun, H.; Lin, Z.; Zhang, Z.; Mu, C.; Zhou, J.; et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat. Commun. 2019, 10, 2906. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, N.J.; Haney, M.S.; Morgens, D.W.; Jovicic, A.; Couthouis, J.; Li, A.; Ousey, J.; Ma, R.; Bieri, G.; Tsui, C.K.; et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 2018, 50, 603–612. [Google Scholar] [CrossRef]
- Shi, K.Y.; Mori, E.; Nizami, Z.F.; Lin, Y.; Kato, M.; Xiang, S.; Wu, L.C.; Ding, M.; Yu, Y.; Gall, J.G.; et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 2017, 114, E1111–E1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, L.R.; Duan, L.; Bowen, K.; Kalab, P.; Rothstein, J.D. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. eLife 2020, 9, e51685. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Conlon, E.G.; Lu, L.; Sharma, A.; Yamazaki, T.; Tang, T.; Shneider, N.A.; Manley, J.L. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. eLife 2016, 5, e17820. [Google Scholar] [CrossRef]
- Yin, S.; Lopez-Gonzalez, R.; Kunz, R.C.; Gangopadhyay, J.; Borufka, C.; Gygi, S.P.; Gao, F.B.; Reed, R. Evidence that C9ORF72 Dipeptide Repeat Proteins Associate with U2 snRNP to Cause Mis-splicing in ALS/FTD Patients. Cell Rep. 2017, 19, 2244–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanekura, K.; Yagi, T.; Cammack, A.J.; Mahadevan, J.; Kuroda, M.; Harms, M.B.; Miller, T.M.; Urano, F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum. Mol. Genet. 2016, 25, 1803–1813. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788 e717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Mori, E.; Kato, M.; Xiang, S.; Wu, L.; Kwon, I.; McKnight, S.L. Toxic PR Poly-Dipeptides Encoded by the C9orf72 Repeat Expansion Target LC Domain Polymers. Cell 2016, 167, 789–802 e712. [Google Scholar] [CrossRef] [Green Version]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.H.; et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol. Cell 2017, 65, 1044–1055 e1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.R.; Mitrea, D.M.; Zhang, P.; Stanley, C.B.; Cassidy, D.E.; Nourse, A.; Phillips, A.H.; Tolbert, M.; Taylor, J.P.; Kriwacki, R.W. C9orf72 Poly(PR) Dipeptide Repeats Disturb Biomolecular Phase Separation and Disrupt Nucleolar Function. Mol. Cell 2019, 74, 713–728 e716. [Google Scholar] [CrossRef]
- Solomon, D.A.; Smikle, R.; Reid, M.J.; Mizielinska, S. Altered Phase Separation and Cellular Impact in C9orf72-Linked ALS/FTD. Front. Cell. Neurosci. 2021, 15, 664151. [Google Scholar] [CrossRef]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. [Google Scholar] [CrossRef]
- Maor-Nof, M.; Shipony, Z.; Lopez-Gonzalez, R.; Nakayama, L.; Zhang, Y.J.; Couthouis, J.; Blum, J.A.; Castruita, P.A.; Linares, G.R.; Ruan, K.; et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 2021, 184, 689–708 e620. [Google Scholar] [CrossRef]
- Lee, C.W.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry 2010, 49, 9964–9971. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.W.; Lee, S.H.; Kim, D.H.; Ahn, M.J.; Kim, J.S.; Woo, J.Y.; Torizawa, T.; Kainosho, M.; Han, K.H. Structural details on mdm2-p53 interaction. J. Biol. Chem. 2005, 280, 38795–38802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartlmuller, C.; Spreitzer, E.; Gobl, C.; Falsone, F.; Madl, T. NMR characterization of solvent accessibility and transient structure in intrinsically disordered proteins. J. Biomol. NMR 2019, 73, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Akabayov, S.R.; Kessler, N.; Cohen, L.S.; Solanki, J.; Naider, F.; Kay, L.E.; Anglister, J. The methyl (13)C-edited/(13)C-filtered transferred NOE for studying protein interactions with short linear motifs. J. Biomol. NMR 2020, 74, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Odeh, H.M.; Shorter, J. Arginine-rich dipeptide-repeat proteins as phase disruptors in C9-ALS/FTD. Emerg. Top. Life Sci. 2020, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Kamagata, K.; Kanbayashi, S.; Honda, M.; Itoh, Y.; Takahashi, H.; Kameda, T.; Nagatsugi, F.; Takahashi, S. Liquid-like droplet formation by tumor suppressor p53 induced by multivalent electrostatic interactions between two disordered domains. Sci. Rep. 2020, 10, 580. [Google Scholar] [CrossRef] [PubMed]
- Orti, F.; Navarro, A.M.; Rabinovich, A.; Wodak, S.J.; Marino-Buslje, C. Insight into membraneless organelles and their associated proteins: Drivers, Clients and Regulators. Comput. Struct. Biotechnol. J. 2021, 19, 3964–3977. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Candau, R.; Scolnick, D.M.; Darpino, P.; Ying, C.Y.; Halazonetis, T.D.; Berger, S.L. Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene 1997, 15, 807–816. [Google Scholar] [CrossRef] [Green Version]
- Raj, N.; Attardi, L.D. The Transactivation Domains of the p53 Protein. Cold Spring Harb. Perspect. Med. 2017, 7, a026047. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Bochkareva, E.; Kaustov, L.; Ayed, A.; Yi, G.S.; Lu, Y.; Pineda-Lucena, A.; Liao, J.C.; Okorokov, A.L.; Milner, J.; Arrowsmith, C.H.; et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. USA 2005, 102, 15412–15417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lello, P.; Jenkins, L.M.M.; Jones, T.N.; Nguyen, B.D.; Hara, T.; Yamaguchi, H.; Dikeakos, J.D.; Appella, E.; Legault, P.; Omichinski, J.G. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell 2006, 22, 731–740. [Google Scholar] [CrossRef]
- Krois, A.S.; Ferreon, J.C.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Recognition of the disordered p53 transactivation domain by the transcriptional adapter zinc finger domains of CREB-binding protein. Proc. Natl. Acad. Sci. USA 2016, 113, E1853–E1862. [Google Scholar] [CrossRef] [Green Version]
- Ecsedi, P.; Gogl, G.; Hof, H.; Kiss, B.; Harmat, V.; Nyitray, L. Structure Determination of the Transactivation Domain of p53 in Complex with S100A4 Using Annexin A2 as a Crystallization Chaperone. Structure 2020, 28, 943–953 e944. [Google Scholar] [CrossRef] [PubMed]
- Orre, L.M.; Panizza, E.; Kaminskyy, V.O.; Vernet, E.; Graslund, T.; Zhivotovsky, B.; Lehtio, J. S100A4 interacts with p53 in the nucleus and promotes p53 degradation. Oncogene 2013, 32, 5531–5540. [Google Scholar] [CrossRef]
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.S.; Desterro, J.M.; Lain, S.; Lane, D.P.; Hay, R.T. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell. Biol. 2000, 20, 8458–8467. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Luo, J.; Brooks, C.L.; Gu, W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 2002, 277, 50607–50611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.L.; Gu, W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Miller Jenkins, L.M.; Feng, H.; Durell, S.R.; Tagad, H.D.; Mazur, S.J.; Tropea, J.E.; Bai, Y.; Appella, E. Characterization of the p300 Taz2-p53 TAD2 complex and comparison with the p300 Taz2-p53 TAD1 complex. Biochemistry 2015, 54, 2001–2010. [Google Scholar] [CrossRef]
- Rowell, J.P.; Simpson, K.L.; Stott, K.; Watson, M.; Thomas, J.O. HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure 2012, 20, 2014–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, L.F.; Cerqueira, F.M.; Macedo, A.F.; Garcia, C.C.; Angeli, J.P.; Schumacher, R.I.; Sogayar, M.C.; Augusto, O.; Carri, M.T.; Di Mascio, P.; et al. Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochim. Biophys. Acta 2010, 1802, 462–471. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Borcherds, W.; Song, T.; Wei, X.; Das, M.; Chen, L.; Daughdrill, G.W.; Chen, J. Interaction between p53 N terminus and core domain regulates specific and nonspecific DNA binding. Proc. Natl. Acad. Sci. USA 2019, 116, 8859–8868. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Dyson, H.J.; Wright, P.E. A phosphorylation-dependent switch in the disordered p53 transactivation domain regulates DNA binding. Proc. Natl. Acad. Sci. USA 2021, 118, e2021456118. [Google Scholar] [CrossRef]
- Krois, A.S.; Dyson, H.J.; Wright, P.E. Long-range regulation of p53 DNA binding by its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2018, 115, E11302–E11310. [Google Scholar] [CrossRef] [Green Version]
- March, Z.M.; King, O.D.; Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016, 1647, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, M.T.; Vlatkovic, N.; Rubbi, C.P. The nucleolus directly regulates p53 export and degradation. J. Cell Biol. 2011, 194, 689–703. [Google Scholar] [CrossRef] [Green Version]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855 e1816. [Google Scholar] [CrossRef] [Green Version]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sykes, B.D. The 13C chemical-shift index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Tamiola, K.; Acar, B.; Mulder, F.A. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 2010, 132, 18000–18003. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usluer, S.; Spreitzer, E.; Bourgeois, B.; Madl, T. p53 Transactivation Domain Mediates Binding and Phase Separation with Poly-PR/GR. Int. J. Mol. Sci. 2021, 22, 11431. https://doi.org/10.3390/ijms222111431
Usluer S, Spreitzer E, Bourgeois B, Madl T. p53 Transactivation Domain Mediates Binding and Phase Separation with Poly-PR/GR. International Journal of Molecular Sciences. 2021; 22(21):11431. https://doi.org/10.3390/ijms222111431
Chicago/Turabian StyleUsluer, Sinem, Emil Spreitzer, Benjamin Bourgeois, and Tobias Madl. 2021. "p53 Transactivation Domain Mediates Binding and Phase Separation with Poly-PR/GR" International Journal of Molecular Sciences 22, no. 21: 11431. https://doi.org/10.3390/ijms222111431
APA StyleUsluer, S., Spreitzer, E., Bourgeois, B., & Madl, T. (2021). p53 Transactivation Domain Mediates Binding and Phase Separation with Poly-PR/GR. International Journal of Molecular Sciences, 22(21), 11431. https://doi.org/10.3390/ijms222111431