
Primary Amenorrhea Due to Anatomical Abnormalities of the Reproductive Tract: Molecular Insight


Abstract
:1. Introduction
2. Embryogenesis of the Female Genital Tract
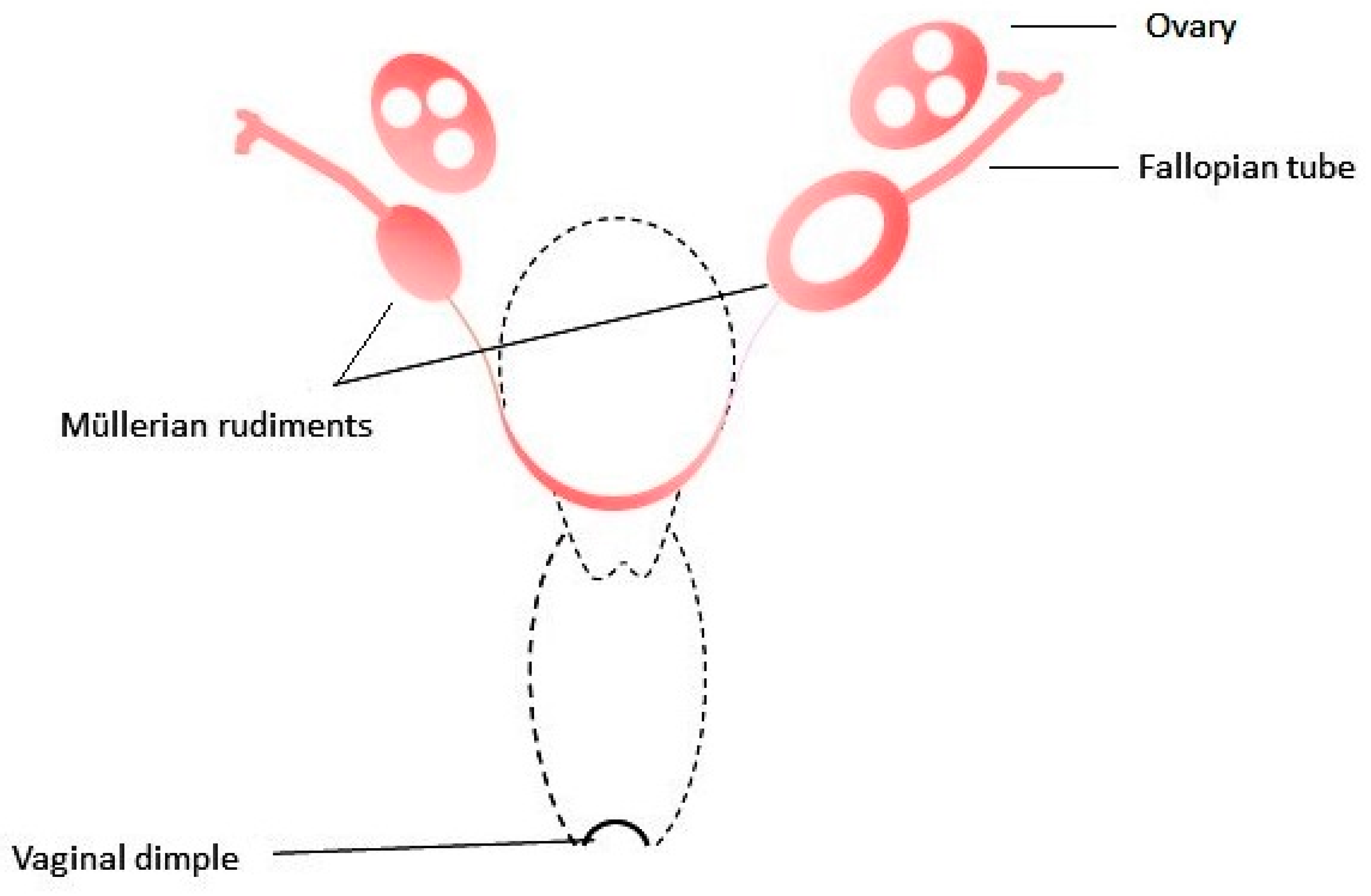
3. MRKH Syndrome
4. 46,XY Phenotypical Females with CAUV
4.1. Complete Androgen Insensitivity Syndrome
4.2. 5α-Reductase Type 2 Deficiency
4.3. 17β-Hydroxysteroid Dehydrogenase Type 3 Deficiency
4.4. Leydig Cells Hypoplasia
4.5. Obstructive Uterovaginal Anomalies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grimbizis, G.F.; Gordts, S.; Sardo, A.D.S.; Brucker, S.; DE Angelis, C.; Gergolet, M.; Vasilios, T.; Tanos, V.; Brölmann, H.; Gianaroli, L.; et al. The ESHRE–ESGE consensus on the classification of female genital tract congenital anomalies. Gynecol. Surg. 2013, 10, 199–212. [Google Scholar] [CrossRef] [Green Version]
- AlSubaihin, A.; Vandermeulen, J.; Harris, K.; Duck, J.; McCready, E. Müllerian Agenesis in Cat Eye Syndrome and 22q11 Chromosome Abnormalities: A Case Report and Literature Review. J. Pediatr. Adolesc. Gynecol. 2018, 31, 158–161. [Google Scholar] [CrossRef]
- Ledig, S.; Schippert, C.; Strick, R.; Beckmann, M.W.; Oppelt, P.G.; Wieacker, P. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Küster-Hauser syndrome. Fertil. Steril. 2011, 95, 1589–1594. [Google Scholar] [CrossRef]
- Brabbing-Goldstein, D.; Yaron, Y.; Reches, A. Familial Beckwith-Wiedemann syndrome: Prenatal manifestation and a possible expansion of the phenotype. Eur. J. Med. Genet. 2021, 64, 104137. [Google Scholar] [CrossRef] [PubMed]
- Al-Qattan, M. Molecular basis of the clinical features of Al-Awadi-Raas-Rothschild (limb/pelvis/uterus-hypoplasia/aplasia) syndrome (AARRS) and Fuhrmann syndrome. Am. J. Med. Genet. Part A 2013, 161, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Pope, S. Association of Mayer–Rokitansky–Küster–Hauser Syndrome with Thrombocytopenia Absent Radii syndrome: A rare presentation. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 139, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Jacquinet, A.; Millar, D.; Lehman, A. Etiologies of uterine malformations. Am. J. Med. Genet. Part A 2016, 170, 2141–2172. [Google Scholar] [CrossRef]
- Rall, K.; Eisenbeis, S.; Henninger, V.; Henes, M.; Wallwiener, D.; Bonin, M.; Brucker, S. Typical and Atypical Associated Findings in a Group of 346 Patients with Mayer-Rokitansky-Kuester-Hauser Syndrome. J. Pediatr. Adolesc. Gynecol. 2015, 28, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Kapczuk, K.; Iwaniec, K.; Friebe, Z.; Kędzia, W. Congenital malformations and other comorbidities in 125 women with Mayer-Rokitansky-Küster-Hauser syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 207, 45–49. [Google Scholar] [CrossRef]
- Tewes, A.-C.; Rall, K.K.; Römer, T.; Hucke, J.; Kapczuk, K.; Brucker, S.; Wieacker, P.; Ledig, S. Variations in RBM8A and TBX6 are associated with disorders of the müllerian ducts. Fertil. Steril. 2015, 103, 1313–1318. [Google Scholar] [CrossRef]
- Guioli, S.; Sekido, R.; Lovell-Badge, R. The origin of the Mullerian duct in chick and mouse. Dev. Biol. 2007, 302, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Roly, Z.Y.; Backhouse, B.; Cutting, A.; Tan, T.Y.; Sinclair, A.H.; Ayers, K.L.; Major, A.T.; Smith, C.A. The cell biology and molecular genetics of Müllerian duct development. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7, e310. [Google Scholar] [CrossRef] [PubMed]
- Atsuta, Y.; Takahashi, Y. Early formation of the Müllerian duct is regulated by sequential actions of BMP/Pax2- and FGF/Lim1 signaling. Development 2016, 143, 3549–3559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, T.J.; Park, J.-S.; Hayashi, S.; Majumdar, A.; McMahon, A.P. Wnt9b Plays a Central Role in the Regulation of Mesenchymal to Epithelial Transitions Underlying Organogenesis of the Mammalian Urogenital System. Dev. Cell 2005, 9, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Cunha, G.R.; Robboy, S.J.; Kurita, T.; Isaacson, D.; Shen, J.; Cao, M.; Baskin, L.S. Development of the human female reproductive tract. Differentiation 2018, 103, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Robboy, S.J.; Kurita, T.; Baskin, L.; Cunha, G.R. New insights into human female reproductive tract development. Differentiation 2017, 97, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Taylor, H.S. The Role of Hox Genes in Female Reproductive Tract Development, Adult Function, and Fertility. Cold Spring Harb. Perspect. Med. 2015, 6, a023002. [Google Scholar] [CrossRef] [Green Version]
- Herlin, M.; Bjørn, A.-M.B.; Rasmussen, M.; Trolle, B.; Petersen, M.B. Prevalence and patient characteristics of Mayer–Rokitansky–Küster–Hauser syndrome: A nationwide registry-based study. Hum. Reprod. 2016, 31, 2384–2390. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Pan, H.; Luo, G.; Wang, P.; Xie, Z.; Hua, K.; Luo, X.; Huang, X.; Liu, Q.; Sun, L.; et al. Clinical characteristics of 1,055 Chinese patients with Mayer-Rokitansky-Küster-Hauser syndrome: A nationwide multicentric study. Fertil. Steril. 2021, 116, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Białka, A.; Gawlik, A.; Drosdzol-Cop, A.; Wilk, K.; Małecka-Tendera, E.; Skrzypulec-Plinta, V. Coexistence of Mayer-Rokitansky-Küster-Hauser Syndrome and Turner Syndrome: A Case Report. J. Pediatr. Adolesc. Gynecol. 2016, 29, e35–e38. [Google Scholar] [CrossRef]
- Fontana, L.; Gentilin, B.; Fedele, L.; Gervasini, C.; Miozzo, M. Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Clin. Genet. 2017, 91, 233–246. [Google Scholar] [CrossRef]
- Ledig, S.; Wieacker, P. Clinical and genetic aspects of Mayer-Rokitansky-Küster-Hauser syndrome. Med. Genet. 2018, 30, 3–11. [Google Scholar] [CrossRef]
- Maniglio, P.; Ricciardi, E.; Laganà, A.S.; Triolo, O.; Caserta, D. Epigenetic modifications of primordial reproductive tract: A common etiologic pathway for Mayer-Rokitansky-Kuster-Hauser Syndrome and endometriosis? Med. Hypotheses 2016, 90, 4–5. [Google Scholar] [CrossRef]
- Rall, K.; Eisenbeis, S.; Barresi, G.; Rückner, D.; Walter, M.; Poths, S.; Wallwiener, D.; Riess, O.; Bonin, M.; Brucker, S. Mayer-Rokitansky-Küster-Hauser syndrome discordance in monozygotic twins: Matrix metalloproteinase 14, low-density lipoprotein receptor–related protein 10, extracellular matrix, and neoangiogenesis genes identified as candidate genes in a tissue-specific mosaicism. Fertil. Steril. 2015, 103, 494–502.e3. [Google Scholar] [CrossRef] [PubMed]
- Herlin, M.; Højland, A.T.; Petersen, M.B. Familial occurrence of Mayer-Rokitansky-Küster-Hauser syndrome: A case report and review of the literature. Am. J. Med. Genet. Part A 2014, 164, 2276–2286. [Google Scholar] [CrossRef] [PubMed]
- Biason-Lauber, A.; Konrad, D.; Navratil, F.; Schoenle, E.J. A WNT4 Mutation Associated with Müllerian-Duct Regression and Virilization in a 46, XX Woman. N. Engl. J. Med. 2004, 351, 792–798. [Google Scholar] [CrossRef] [Green Version]
- Oppelt, P.G.; Müller, A.; Stephan, L.; Dittrich, R.; Lermann, J.; Büttner, C.; Ekici, A.B.; Conzelmann, G.; Seeger, H.; Schöller, D.; et al. Hyperandrogenemia and high prolactin in congenital utero–vaginal aplasia patients. Reproduction 2017, 153, 555–563. [Google Scholar] [CrossRef]
- Philibert, P.; Biason-Lauber, A.; Rouzier, R.; Pienkowski, C.; Paris, F.; Konrad, D.; Schoenle, E.; Sultan, C. Identification and Functional Analysis of a New WNT4 Gene Mutation among 28 Adolescent Girls with Primary Amenorrhea and Müllerian Duct Abnormalities: A French Collaborative Study. J. Clin. Endocrinol. Metab. 2008, 93, 895–900. [Google Scholar] [CrossRef]
- Philibert, P.; Biason-Lauber, A.; Gueorguieva, I.; Stuckens, C.; Pienkowski, C.; Lebon-Labich, B.; Paris, F.; Sultan, C. Molecular analysis of WNT4 gene in four adolescent girls with mullerian duct abnormality and hyperandrogenism (atypical Mayer-Rokitansky-Küster-Hauser syndrome). Fertil. Steril. 2011, 95, 2683–2686. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhao, S.; Jolly, A.; Wang, L.; Pan, H.; Yuan, J.; Chen, S.; Koch, A.; Ma, C.; Tian, W.; et al. Perturbations of genes essential for Müllerian duct and Wölffian duct development in Mayer-Rokitansky-Küster-Hauser syndrome. Am. J. Hum. Genet. 2021, 108, 337–345. [Google Scholar] [CrossRef]
- Smol, T.; Ribero-Karrouz, W.; Edery, P.; Gorduza, D.B.; Catteau-Jonard, S.; Manouvrier-Hanu, S.; Ghoumid, J. Mayer-Rokitansky-Künster-Hauser syndrome due to 2q12.1q14.1 deletion: PAX8 the causing gene? Eur. J. Med Genet. 2020, 63, 103812. [Google Scholar] [CrossRef] [PubMed]
- Mikhael, S.; Dugar, S.; Morton, M.; Chorich, L.P.; Tam, K.B.; Lossie, A.C.; Kim, H.-G.; Knight, J.; Taylor, H.S.; Mukherjee, S.; et al. Genetics of agenesis/hypoplasia of the uterus and vagina: Narrowing down the number of candidate genes for Mayer–Rokitansky–Küster–Hauser Syndrome. Hum. Genet. 2021, 140, 667–680. [Google Scholar] [CrossRef]
- Backhouse, B.; Hanna, C.; Robevska, G.; Bergen, J.V.D.; Pelosi, E.; Simons, C.; Koopman, P.; Juniarto, A.Z.; Grover, S.; Faradz, S.; et al. Identification of Candidate Genes for Mayer-Rokitansky-Küster-Hauser Syndrome Using Genomic Approaches. Sex. Dev. 2018, 13, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Fu, X.; Dong, S.; Tang, S.; Song, C.; Yang, N.; Zhang, L.; Wang, H.; Shi, H.; et al. Joint utilization of genetic analysis and semi-cloning technology reveals a digenic etiology of Müllerian anomalies. Cell Res. 2020, 30, 91–94. [Google Scholar] [CrossRef]
- Ledig, S.; Brucker, S.; Barresi, G.; Schomburg, J.; Rall, K.; Wieacker, P. Frame shift mutation of LHX1 is associated with Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Hum. Reprod. 2012, 27, 2872–2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhou, X.; Liu, L.; Zhu, Y.; Liu, C.; Pan, H.; Xing, Q.; Wang, J.; Wang, X.; Zhang, X.; et al. Identification and functional analysis of a novel LHX1 mutation associated with congenital absence of the uterus and vagina. Oncotarget 2017, 8, 8785–8790. [Google Scholar] [CrossRef] [Green Version]
- Jacquinet, A.; Boujemla, B.; Fasquelle, C.; Thiry, J.; Josse, C.; Lumaka, A.; Brischoux-Boucher, E.; Dubourg, C.; David, V.; Pasquier, L.; et al. GREB1L variants in familial and sporadic hereditary urogenital adysplasia and Mayer-Rokitansky-Kuster-Hauser syndrome. Clin. Genet. 2020, 98, 126–137. [Google Scholar] [CrossRef]
- Brucker, S.Y.; Frank, L.; Eisenbeis, S.; Henes, M.; Wallwiener, D.; Riess, O.; Van Eijck, B.; Schöller, D.; Bonin, M.; Rall, K.K. Sequence variants in ESR1 and OXTR are associated with Mayer-Rokitansky-Küster-Hauser syndrome. Acta Obstet. Gynecol. Scand. 2017, 96, 1338–1346. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, K.S. The Mayer-Rokitansky-Küster syndrome. An analysis of its morphology and embryology. Part I: Morphology. Arch. Gynecol. Obstet. 1998, 262, 1–26. [Google Scholar] [CrossRef]
- Rall, K.; Barresi, G.; Walter, M.; Poths, S.; Haebig, K.; Schaeferhoff, K.; Schoenfisch, B.; Riess, O.; Wallwiener, D.; Bonin, M.; et al. A combination of transcriptome and methylation analyses reveals embryologically-relevant candidate genes in MRKH patients. Orphanet J. Rare Dis. 2011, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Jääskeläinen, J. Molecular biology of androgen insensitivity. Mol. Cell. Endocrinol. 2012, 352, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Hornig, N.C.; Holterhus, P.-M. Molecular basis of androgen insensitivity syndromes. Mol. Cell. Endocrinol. 2021, 523, 111146. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef]
- Mongan, N.; Cuccaro, R.T.; Bunch, T.; Hughes, I.A. Androgen insensitivity syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yao, F.; Tian, T.; Deng, S.; Luo, M.; Tian, Q. Clinical characteristics and molecular genetics of complete androgen insensitivity syndrome patients: A series study of 30 cases from a Chinese tertiary medical center. Fertil. Steril. 2021, 115, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Hornig, N.C.; De Beaufort, C.; Denzer, F.; Cools, M.; Wabitsch, M.; Ukat, M.; Kulle, A.E.; Schweikert, H.-U.; Werner, R.; Hiort, O.; et al. A Recurrent Germline Mutation in the 5′UTR of the Androgen Receptor Causes Complete Androgen Insensitivity by Activating Aberrant uORF Translation. PLoS ONE 2016, 11, e0154158. [Google Scholar] [CrossRef] [Green Version]
- Känsäkoski, J.; Jääskeläinen, J.; Jääskeläinen, T.; Tommiska, J.; Saarinen, L.; Lehtonen, R.J.; Hautaniemi, S.; Frilander, M.J.; Palvimo, J.; Toppari, J.; et al. Complete androgen insensitivity syndrome caused by a deep intronic pseudoexon-activating mutation in the androgen receptor gene. Sci. Rep. 2016, 6, 32819. [Google Scholar] [CrossRef]
- Ramos, L.; Vilchis, F.; Chávez, B.; Mares, L. Mutational analysis of SRD5A2: From gene to functional kinetics in individuals with steroid 5α-reductase 2 deficiency. J. Steroid Biochem. Mol. Biol. 2020, 200, 105691. [Google Scholar] [CrossRef]
- Maimoun, L.L.; Philibert, P.; Cammas, B.B.; Audran, F.F.; Bouchard, P.; Fenichel, P.P.; Cartigny, M.M.; Pienkowski, C.C.; Polak, M.M.; Skordis, N.N.; et al. Phenotypical, Biological, and Molecular Heterogeneity of 5α-Reductase Deficiency: An Extensive International Experience of 55 Patients. J. Clin. Endocrinol. Metab. 2011, 96, 296–307. [Google Scholar] [CrossRef] [Green Version]
- Avendaño, A.; Paradisi, I.; Cammarata-Scalisi, F.; Callea, M. 5-α-Reductase type 2 deficiency: Is there a genotype-phenotype correlation? A review. Hormones 2018, 17, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Fenichel, P.; Paris, F.; Philibert, P.; Hieronimus, S.; Gaspari, L.; Kurzenne, J.-Y.; Chevallier, P.; Bermon, S.; Chevalier, N.; Sultan, C. Molecular Diagnosis of 5α-Reductase Deficiency in 4 Elite Young Female Athletes Through Hormonal Screening for Hyperandrogenism. J. Clin. Endocrinol. Metab. 2013, 98, E1055–E1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, H.M.; Mohnach, L.H.; Fechner, P.Y.; Chen, M.; Thomas, I.H.; Ramsdell, L.A.; Shnorhavorian, M.; McCauley, E.A.; Oelschlager, A.-M.E.A.; Park, J.M.; et al. Unexpected ethical dilemmas in sex assignment in 46,XY DSD due to 5-alpha reductase type 2 deficiency. Am. J. Med Genet. Part C Semin. Med Genet. 2017, 175, 260–267. [Google Scholar] [CrossRef]
- Akcay, T.; Cancio, M.F.; Turan, S.; Guran, T.; Audi, L.; Bereket, A. AR and SRD5A2 gene mutations in a series of 51 Turkish 46,XY DSD children with a clinical diagnosis of androgen insensitivity. Andrology 2014, 2, 572–578. [Google Scholar] [CrossRef]
- Berra, M.; Williams, E.L.; Muroni, B.; Creighton, S.M.; Honour, J.W.; Rumsby, G.; Conway, G.S. Recognition of 5α-reductase-2 deficiency in an adult female 46XY DSD clinic. Eur. J. Endocrinol. 2011, 164, 1019–1025. [Google Scholar] [CrossRef] [Green Version]
- Ata, A.; Özen, S.; Onay, H.; Uzun, S.; Gökşen, D.; Özkınay, F.; Özbaran, N.B.; Ulman, I.; Darcan, Ş. A large cohort of disorders of sex development and their genetic characteristics: 6 novel mutations in known genes. Eur. J. Med. Genet. 2021, 64, 104154. [Google Scholar] [CrossRef]
- George, M.M.; New, M.I.; Ten, S.; Sultan, C.; Bhangoo, A. The Clinical and Molecular Heterogeneity of 17βHSD-3 Enzyme Deficiency. Horm. Res. Paediatr. 2010, 74, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Faienza, M.F.; Baldinotti, F.; Marrocco, G.; Tyutyusheva, N.; Peroni, D.; Baroncelli, G.I.; Bertelloni, S. 17β-hydroxysteroid dehydrogenase type 3 deficiency: Female sex assignment and follow-up. J. Endocrinol. Investig. 2020, 43, 1711–1716. [Google Scholar] [CrossRef]
- Mendonca, B.B.; Gomes, N.L.; Costa, E.M.; Inacio, M.; Martin, R.M.; Nishi, M.Y.; Carvalho, F.M.; Tibor, F.D.; Domenice, S. 46,XY disorder of sex development (DSD) due to 17β-hydroxysteroid dehydrogenase type 3 deficiency. J. Steroid Biochem. Mol. Biol. 2017, 165, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ye, L.; Wang, W.; Zhao, Y.; Wang, W.; Jia, H.; Dong, Z.; Chen, Y.; Wang, W.; Ning, G.; et al. 17β-Hydroxysteroid dehydrogenase 3 deficiency: Three case reports and a systematic review. J. Steroid Biochem. Mol. Biol. 2017, 174, 141–145. [Google Scholar] [CrossRef]
- Phelan, N.; Williams, E.L.; Cardamone, S.; Lee, M.; Creighton, S.M.; Rumsby, G.; Conway, G.S. Screening for mutations in 17β-hydroxysteroid dehydrogenase and androgen receptor in women presenting with partially virilised 46,XY disorders of sex development. Eur. J. Endocrinol. 2015, 172, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Hmida, I.B.H.; Mougou-Zerelli, S.; Hadded, A.; Dimassi, S.; Kammoun, M.; Bignon-Topalovic, J.; Bibi, M.; Saad, A.; Bashamboo, A.; McElreavey, K. Novel homozygous nonsense mutations in the luteinizing hormone receptor (LHCGR) gene associated with 46,XY primary amenorrhea. Fertil. Steril. 2016, 106, 225–229.e11. [Google Scholar] [CrossRef] [Green Version]
- Hassan, H.A.; Essawi, M.L.; Mekkawy, M.K.; Mazen, I. Novel mutations of the LHCGR gene in two families with 46,XY DSD causing Leydig cell hypoplasia I. Hormones 2020, 19, 573–579. [Google Scholar] [CrossRef]
- Newton, C.; Anderson, R.; Katz, A.A.; Millar, R.P. Loss-of-Function Mutations in the Human Luteinizing Hormone Receptor Predominantly Cause Intracellular Retention. Endocrinology 2016, 157, 4364–4377. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Dilihuma, J.; Luo, Y.; Reyilanmu, B.; Shen, Y.; Mireguli, M. Novel Compound Heterozygous Variants in the LHCGR Gene in a Genetically Male Patient with Female External Genitalia. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Eggers, S.; Sadedin, S.; Bergen, J.A.V.D.; Robevska, G.; Ohnesorg, T.; Hewitt, J.; Lambeth, L.; Bouty, A.; Knarston, I.; Tan, T.Y.; et al. Disorders of sex development: Insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016, 17, 243. [Google Scholar] [CrossRef] [Green Version]
- Ledig, S.; Tewes, A.; Hucke, J.; Römer, T.; Kapczuk, K.; Schippert, C.; Hillemanns, P.; Wieacker, P. Array-comparative genomic hybridization analysis in patients with Müllerian fusion anomalies. Clin. Genet. 2018, 93, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Sakalkale, R.; Samarakkody, U. Familial Occurrence of Imperforate Hymen. J. Pediatr. Adolesc. Gynecol. 2005, 18, 427–429. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Disease | OMIM | Karyotype | Genetic Etiology | Gonads |
|---|---|---|---|---|
| MRKHS type 1 | 277000 | 46,XX | Largely unknown | Normal ovaries |
| MRKHS type 2 (including MURCS association) | 601076 | 46,XX | Largely unknown | Normal ovaries (rarely ovarian agenesis/dysgenesis) |
| MRKHS and hyperandrogenism | 158330 | 46,XX | WNT4 mutations | Normal ovaries, hyperandrogenism |
| CAIS | 300068 | 46,XY | AR mutations | Testes, high T |
| 5α-reductase type 2 deficiency | 607306 | 46,XY | SRD5A2 mutations | Testes, high T |
| 17β-hydroxysteroid dehydrogenase type 3 deficiency | 264300 | 46,XY | HSD17B3 mutations | Testes, low T |
| Leydig cells hypoplasia type 1 | 238320 | 46,XY | LHCGR mutations | Testes, low T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapczuk, K.; Kędzia, W. Primary Amenorrhea Due to Anatomical Abnormalities of the Reproductive Tract: Molecular Insight. Int. J. Mol. Sci. 2021, 22, 11495. https://doi.org/10.3390/ijms222111495
Kapczuk K, Kędzia W. Primary Amenorrhea Due to Anatomical Abnormalities of the Reproductive Tract: Molecular Insight. International Journal of Molecular Sciences. 2021; 22(21):11495. https://doi.org/10.3390/ijms222111495
Chicago/Turabian StyleKapczuk, Karina, and Witold Kędzia. 2021. "Primary Amenorrhea Due to Anatomical Abnormalities of the Reproductive Tract: Molecular Insight" International Journal of Molecular Sciences 22, no. 21: 11495. https://doi.org/10.3390/ijms222111495
APA StyleKapczuk, K., & Kędzia, W. (2021). Primary Amenorrhea Due to Anatomical Abnormalities of the Reproductive Tract: Molecular Insight. International Journal of Molecular Sciences, 22(21), 11495. https://doi.org/10.3390/ijms222111495