
Increased Post-Hypoxic Oxidative Stress and Activation of the PERK Branch of the UPR in Trap1-Deficient Drosophila melanogaster Is Abrogated by Metformin

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
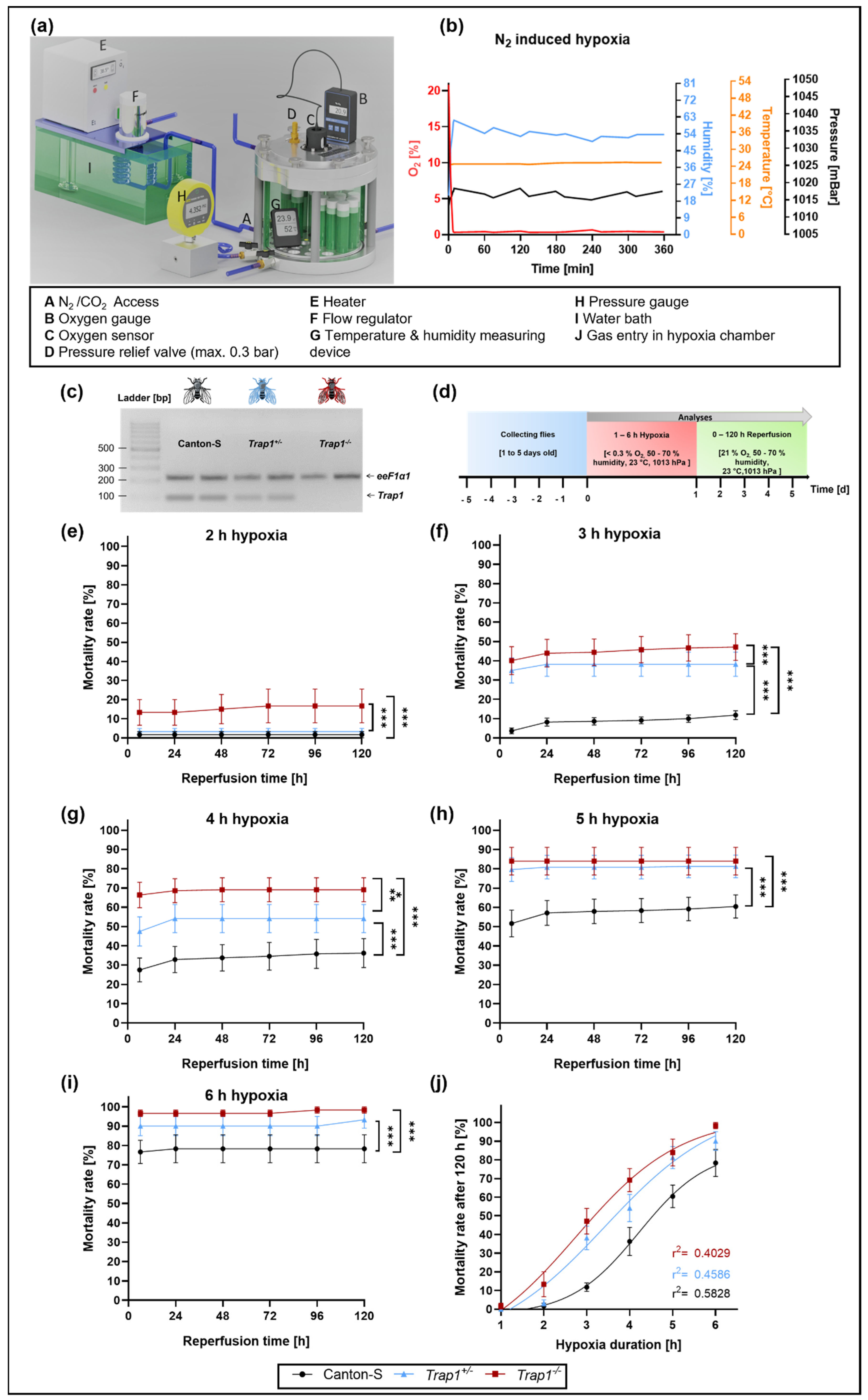
2.1. Trap1 Deficiency Leads to Increased Mortality Rates after Severe Hypoxia (<0.3% O2) in D. melanogaster
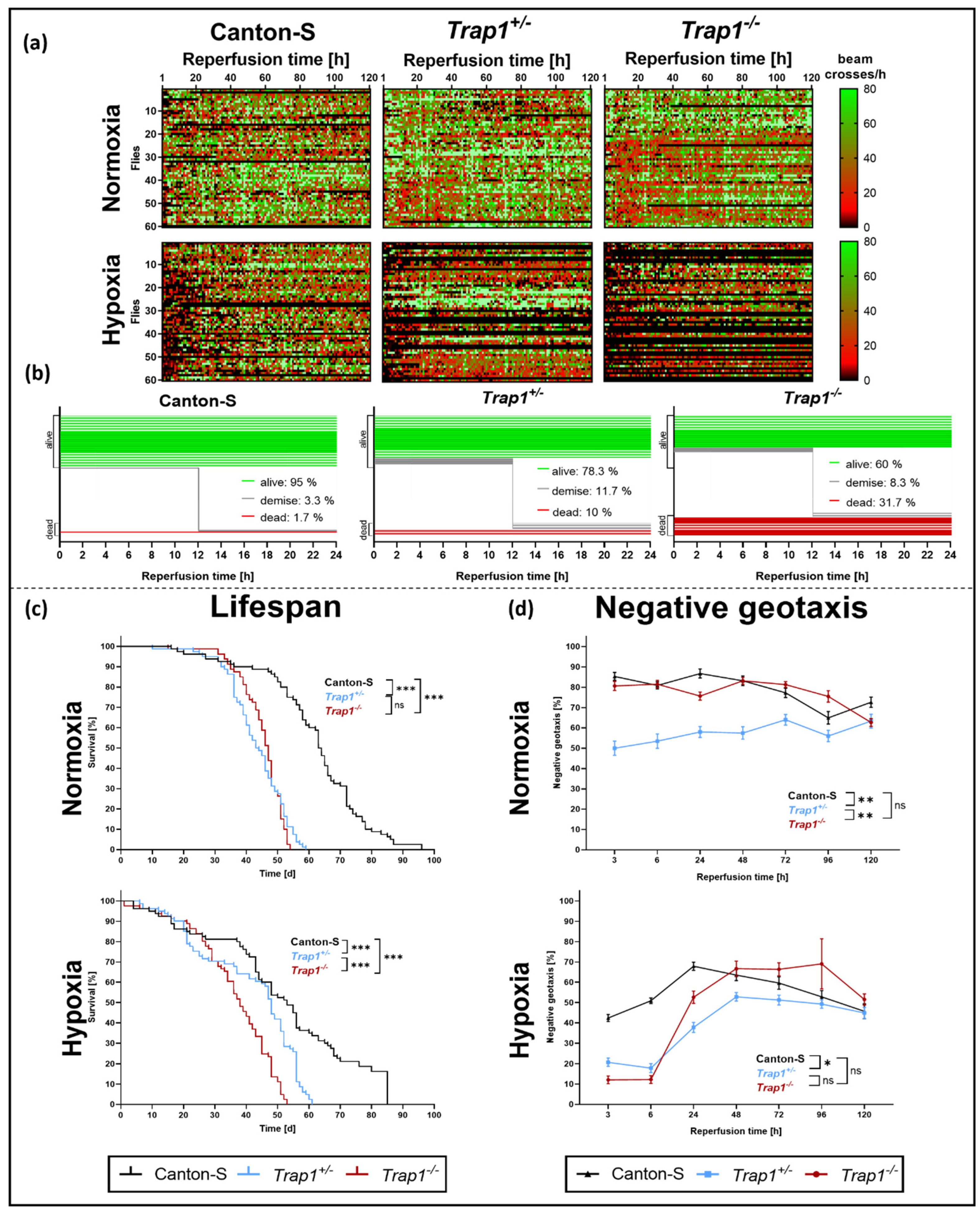
2.2. Trap1 Deficiency Impairs Lifespan, Activity Rates, and Negative Geotaxis after Hypoxia in D. melanogaster
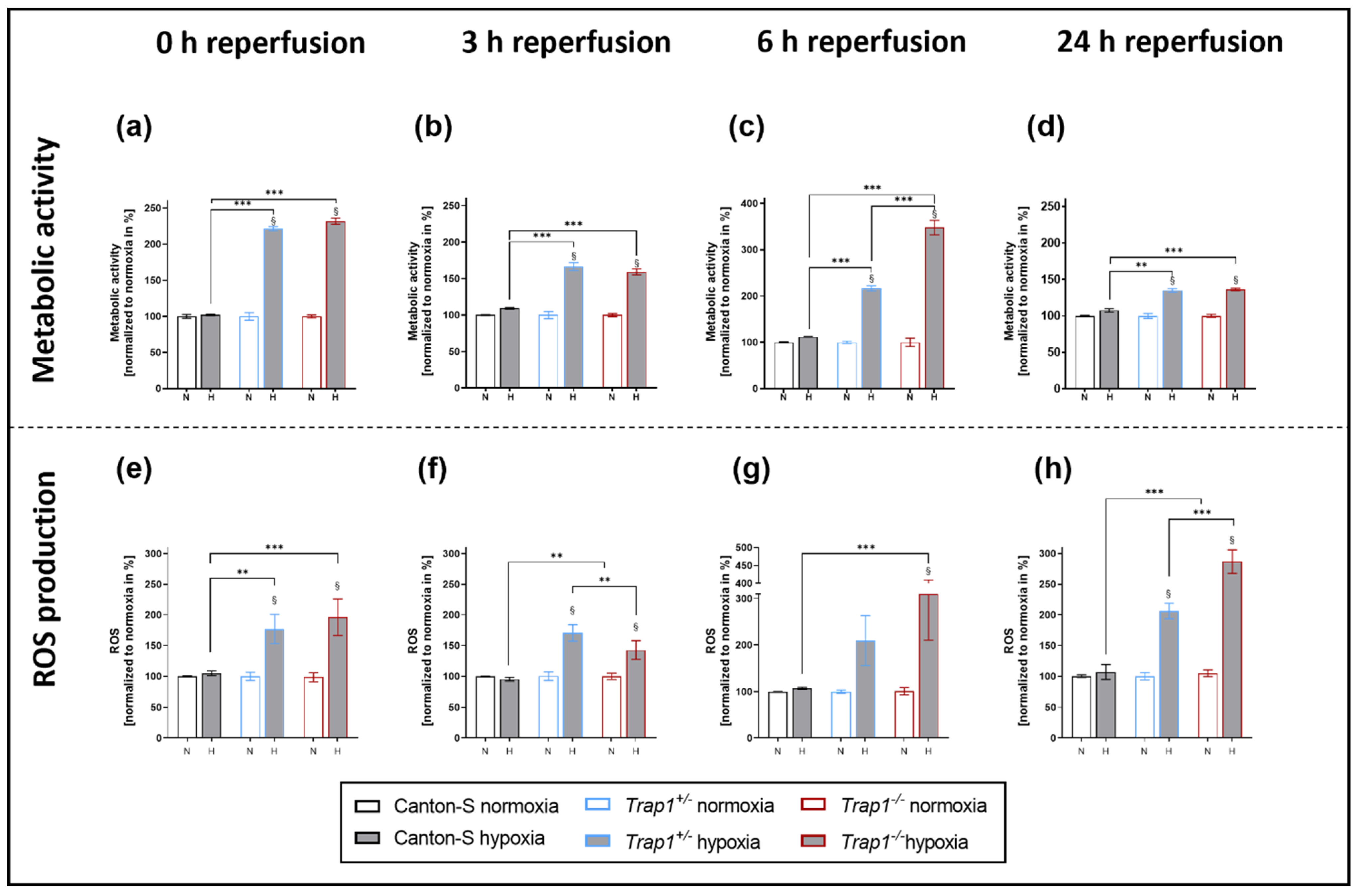
2.3. Hypoxia-Dependent Metabolic Activity and ROS Production Are Increased in Trap1-Deficient Flies
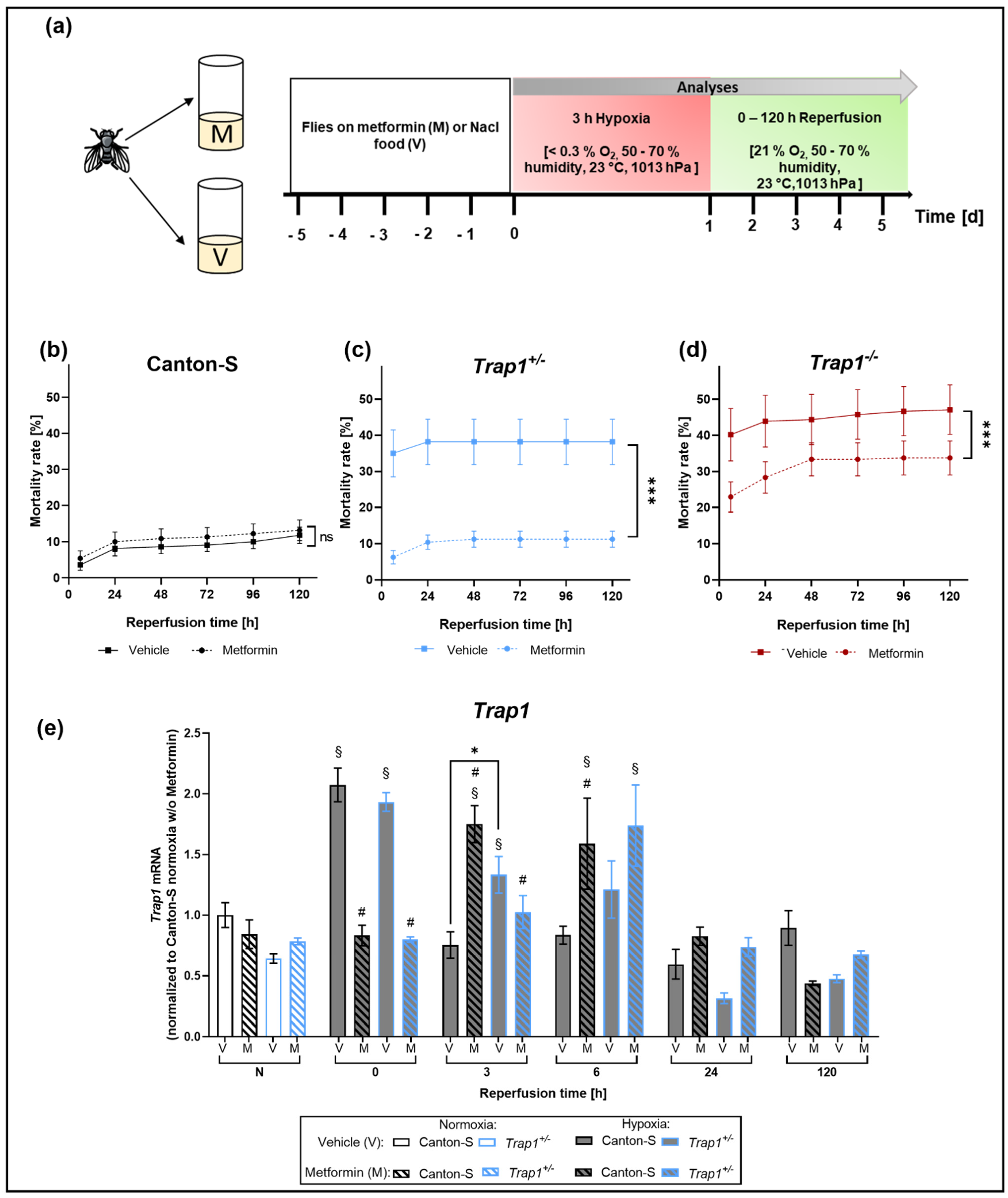
2.4. Metformin Rescues Hypoxia-Dependent Mortality in Trap1-Deficient Flies and Impairs the Expression Pattern of Trap1 mRNA
2.5. ROS Production and mRNAs of Antioxidant Proteins Sod, Hsp70 and Catalase Are Upregulated in Trap1-Deficient Flies after Hypoxia, Metformin Attenuates This Upregulation in the Early Reperfusion Period
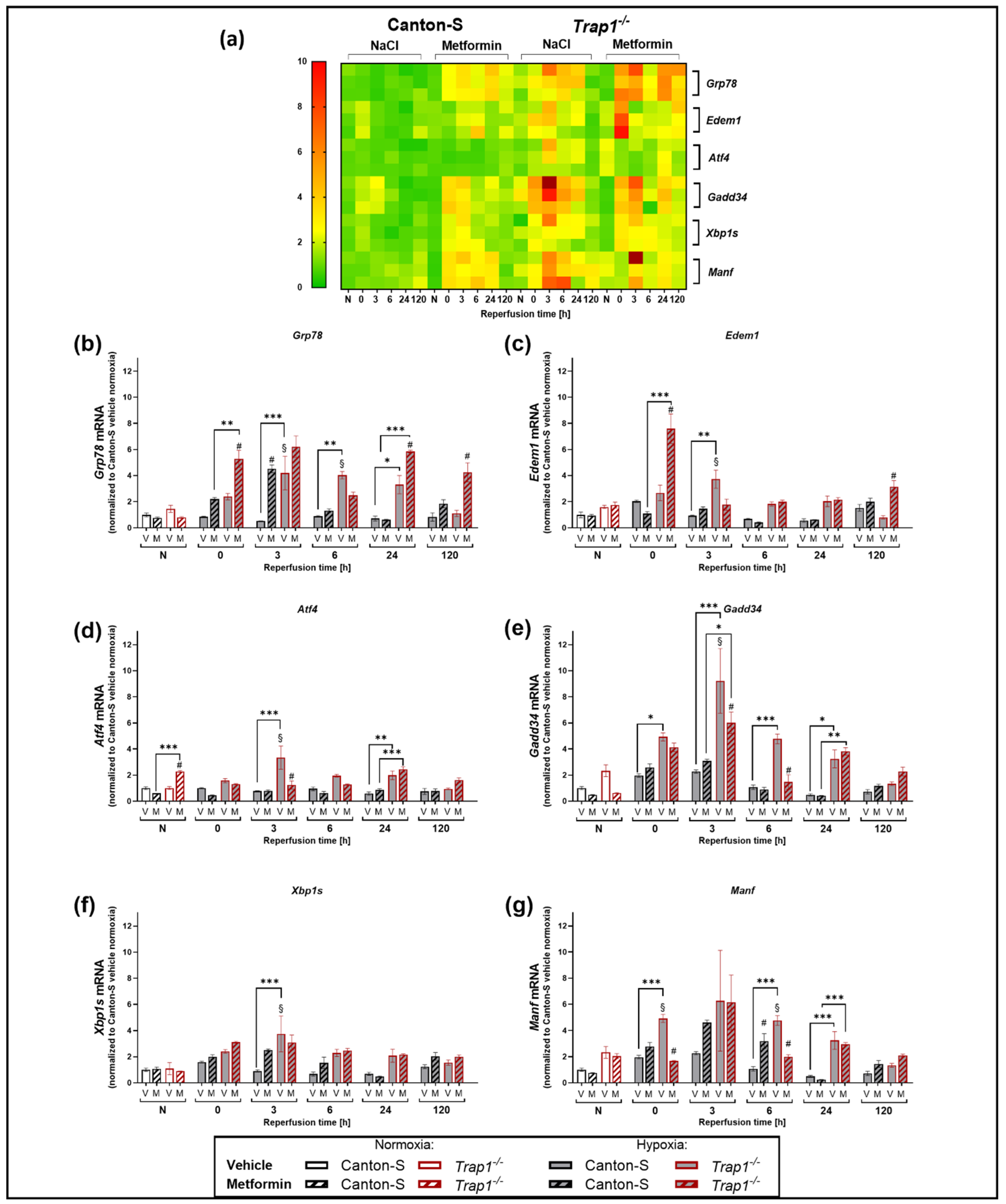
2.6. Trap1 Deficiency Enhances the UPR Activation after Hypoxia and Metformin Reduces the Activation of the PERK Branch of the UPR
3. Discussion
4. Materials and Methods
4.1. Drosophila melanogaster
4.2. Metformin Treatment
4.3. Hypoxia Chamber
4.4. Mortality Rate
4.5. Drosophila Activity Monitoring Assay (DAM)
4.6. Negative Geotaxis Assay
4.7. Semiquantitative PCR and RT-qPCR
4.8. Metabolic Activity Assay
4.9. ROS Assay
4.10. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Peacock, A. Pulmonary hypertension due to chronic hypoxia. BMJ Clin. Res. Ed. 1990, 300, 763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.W.; Haddad, G.G. Review: Hypoxic and oxidative stress resistance in Drosophila melanogaster. Placenta 2011, 32 (Suppl. 2), S104–S108. [Google Scholar] [CrossRef] [Green Version]
- Shekhar, S.; Liu, Y.; Wang, S.; Zhang, H.; Fang, X.; Zhang, J.; Fan, L.; Zheng, B.; Roman, R.J.; Wang, Z.; et al. Novel Mechanistic Insights and Potential Therapeutic Impact of TRPC6 in Neurovascular Coupling and Ischemic Stroke. Int. J. Mol. Sci. 2021, 22, 2074. [Google Scholar] [CrossRef]
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.E.; et al. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef] [Green Version]
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Fiskum, G. Mitochondrial participation in ischemic and traumatic neural cell death. J. Neurotrauma. 2000, 17, 843–855. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 2011, 300, C385–C393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Delgado-Esteban, M.; Bolaños, J.P.; Medina, J.M. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J. Neurochem. 2002, 81, 207–217. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Xin, Q.; Ji, B.; Cheng, B.; Wang, C.; Liu, H.; Chen, X.; Chen, J.; Bai, B. Endoplasmic reticulum stress in cerebral ischemia. Neurochem. Int. 2014, 68, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.p.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Hillary, R.F.; FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [Green Version]
- Felts, S.J.; Owen, B.A.; Nguyen, P.; Trepel, J.; Donner, D.B.; Toft, D.O. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 2000, 275, 3305–3312. [Google Scholar] [CrossRef] [Green Version]
- Sisinni, L.; Maddalena, F.; Lettini, G.; Condelli, V.; Matassa, D.S.; Esposito, F.; Landriscina, M. TRAP1 role in endoplasmic reticulum stress protection favors resistance to anthracyclins in breast carcinoma cells. Int. J. Oncol. 2014, 44, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, K.; Miyata, S.; Takamura, H.; Katayama, T.; Tohyama, M. Mitochondrial TRAP1 regulates the unfolded protein response in the endoplasmic reticulum. Neurochem. Int. 2011, 58, 880–887. [Google Scholar] [CrossRef]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef]
- Song, H.Y.; Dunbar, J.D.; Zhang, Y.X.; Guo, D.; Donner, D.B. Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. J. Biol. Chem. 1995, 270, 3574–3581. [Google Scholar] [CrossRef] [Green Version]
- Alimonti, J.B.; Shi, L.; Baijal, P.K.; Greenberg, A.H. Granzyme B induces BID-mediated cytochrome c release and mitochondrial permeability transition. J. Biol. Chem. 2001, 276, 6974–6982. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef]
- Hadari, Y.R.; Haring, H.U.; Zick, Y. p75, a member of the heat shock protein family, undergoes tyrosine phosphorylation in response to oxidative stress. J. Biol. Chem. 1997, 272, 657–662. [Google Scholar] [CrossRef] [Green Version]
- Carette, J.; Lehnert, S.; Chow, T.Y. Implication of PBP74/mortalin/GRP75 in the radio-adaptive response. Int. J. Radiat. Biol. 2002, 78, 183–190. [Google Scholar] [CrossRef]
- Massa, S.M.; Longo, F.M.; Zuo, J.; Wang, S.; Chen, J.; Sharp, F.R. Cloning of rat grp75, an hsp70-family member, and its expression in normal and ischemic brain. J. Neurosci. Res. 1995, 40, 807–819. [Google Scholar] [CrossRef]
- Hua, G.; Zhang, Q.; Fan, Z. Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme M-mediated apoptosis. J. Biol. Chem. 2007, 282, 20553–20560. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, J.C.; Zimprich, A.; Carvajal Berrio, D.A.; Schindler, K.M.; Maurer, B.; Schulte, C.; Bus, C.; Hauser, A.K.; Kubler, M.; Lewin, R.; et al. Metformin reverses TRAP1 mutation-associated alterations in mitochondrial function in Parkinson’s disease. Brain 2017, 140, 2444–2459. [Google Scholar] [CrossRef]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar] [CrossRef]
- Bailey, C.J.; Turner, R.C. Metformin. N. Engl. J. Med. 1996, 334, 574–579. [Google Scholar] [CrossRef]
- Nasri, H.; Rafieian-Kopaei, M. Metformin: Current knowledge. J. Res. Med. Sci 2014, 19, 658–664. [Google Scholar]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Cheng, J.; Ni, J.; Zhen, X. Neuropharmacological Actions of Metformin in Stroke. Curr. Neuropharmacol. 2015, 13, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Ouslimani, N.; Peynet, J.; Bonnefont-Rousselot, D.; Thérond, P.; Legrand, A.; Beaudeux, J.L. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism 2005, 54, 829–834. [Google Scholar] [CrossRef]
- Chukwunonso Obi, B.; Chinwuba Okoye, T.; Okpashi, V.E.; Nonye Igwe, C.; Olisah Alumanah, E. Comparative Study of the Antioxidant Effects of Metformin, Glibenclamide, and Repaglinide in Alloxan-Induced Diabetic Rats. J. Diabetes Res 2016, 2016, 1635361. [Google Scholar] [CrossRef] [Green Version]
- Bilen, J.; Bonini, N.M. Drosophila as a Model for Hu.uman Neurodegenerative Disease. Annu. Rev. Genet. 2005, 39, 153–171. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, T.E.; Taylor, J.P. Flightless flies: Drosophila models of neuromuscular disease. Ann. N. Y. Acad. Sci. 2010, 1184, e1–e20. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Xu, W.; Meng, S.; Lim, N.K.H.; Wang, W.; Huang, F.D. An Efficient and Reliable Assay for Investigating the Effects of Hypoxia/Anoxia on Drosophila. Neurosci. Bull. 2018, 34, 397–402. [Google Scholar] [CrossRef]
- Habib, P.; Jung, J.; Wilms, G.M.; Kokott-Vuong, A.; Habib, S.; Schulz, J.B.; Voigt, A. Posthypoxic behavioral impairment and mortality of Drosophila melanogaster are associated with high temperatures, enhanced predeath activity and oxidative stress. Exp. Mol. Med. 2021, 53, 264–280. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.C.; Loh, S.H.; Martins, L.M. Drosophila Trap1 protects against mitochondrial dysfunction in a PINK1/parkin model of Parkinson’s disease. Cell Death Dis 2013, 4, e467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.E.; Cook, K.R.; Hemenway, E.A.; Fang, V.; Miller, A.L.; Hales, K.G.; Hawley, R.S. The Molecular and Genetic Characterization of Second Chromosome Balancers in Drosophila melanogaster. G3 Genes Genomes Genet. 2018, 8, 1161–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018, 9, 924–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Li, F. Endoplasmic reticulum stress in brain ischemia. Int. J. Neurosci. 2016, 126, 681–691. [Google Scholar] [CrossRef]
- Voloboueva, L.A.; Duan, M.; Ouyang, Y.; Emery, J.F.; Stoy, C.; Giffard, R.G. Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J. Cereb. Blood Flow Metab. 2008, 28, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Agorreta, J.; Hu, J.; Liu, D.; Delia, D.; Turley, H.; Ferguson, D.J.P.; Iborra, F.; Pajares, M.J.; Larrayoz, M.; Zudaire, I.; et al. TRAP1 regulates proliferation, mitochondrial function, and has prognostic significance in NSCLC. Mol. Cancer Res. 2014, 12, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Li, X.; Jiang, Q.; Liu, Z.; Yang, H.; Wang, S.; Xie, S.; Liu, Q.; Liu, T.; Huang, J.; et al. Transcriptional patterns, biomarkers and pathways characterizing nasopharyngeal carcinoma of Southern China. J. Transl. Med. 2008, 6, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leav, I.; Plescia, J.; Goel, H.L.; Li, J.; Jiang, Z.; Cohen, R.J.; Languino, L.R.; Altieri, D.C. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am. J. Pathol. 2010, 176, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Wang, J.; Huang, Z.; Wei, P.; Liu, Y.; Hao, J.; Zhao, L.; Zhang, F.; Tu, Y.; Wei, T. Aberrantly upregulated TRAP1 is required for tumorigenesis of breast cancer. Oncotarget 2015, 6, 44495–44508. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, J.; Chen, Q.Z.; Zhu, N.; Jiang, D.Q.; Li, M.X.; Wang, Y. Overexpression of mitochondrial Hsp75 protects neural stem cells against microglia-derived soluble factor-induced neurotoxicity by regulating mitochondrial permeability transition pore opening in vitro. Int. J. Mol. Med. 2015, 36, 1487–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Lu, Y.; Yu, D.; Zhang, D.; Hu, W. TRAP1 Provides Protection Against Myocardial Ischemia-Reperfusion Injury by Ameliorating Mitochondrial Dysfunction. Cell Physiol. Biochem. 2015, 36, 2072–2082. [Google Scholar] [CrossRef]
- Li, J.; Benashski, S.E.; Venna, V.R.; McCullough, L.D. Effects of metformin in experimental stroke. Stroke 2010, 41, 2645–2652. [Google Scholar] [CrossRef] [Green Version]
- Arbeláez-Quintero, I.; Palacios, M. To Use or Not to Use Metformin in Cerebral Ischemia: A Review of the Application of Metformin in Stroke Rodents. Stroke Res. Treat. 2017, 2017, 9756429. [Google Scholar] [CrossRef]
- Abd-Elsameea, A.A.; Moustaf, A.A.; Mohamed, A.M. Modulation of the oxidative stress by metformin in the cerebrum of rats exposed to global cerebral ischemia and ischemia/reperfusion. Eur. Rev. Med. Pharm. Sci. 2014, 18, 2387–2392. [Google Scholar]
- Correia, S.; Carvalho, C.; Santos, M.S.; Proença, T.; Nunes, E.; Duarte, A.I.; Monteiro, P.; Seiça, R.; Oliveira, C.R.; Moreira, P.I. Metformin protects the brain against the oxidative imbalance promoted by type 2 diabetes. Med. Chem. 2008, 4, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Dinter, E.; Saridaki, T.; Nippold, M.; Plum, S.; Diederichs, L.; Komnig, D.; Fensky, L.; May, C.; Marcus, K.; Voigt, A.; et al. Rab7 induces clearance of α-synuclein aggregates. J. Neurochem. 2016, 138, 758–774. [Google Scholar] [CrossRef]
- Habib, P.; Stamm, A.S.; Zeyen, T.; Noristani, R.; Slowik, A.; Beyer, C.; Wilhelm, T.; Huber, M.; Komning, D.; Schulz, J.B.; et al. EPO regulates neuroprotective Transmembrane BAX Inhibitor-1 Motif-containing (TMBIM) family members GRINA and FAIM2 after cerebral ischemia-reperfusion injury. Exp. Neurol. 2019, 320, 112978. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.A.; da Rosa Avila, E.; da Silva, G.F.; Macedo, G.E.; Rodrigues, N.R.; de Brum Vieira, P.; Nascimento, M.S.; Picoloto, R.S.; Martins, I.K.; de Carvalho, N.R.; et al. Exposure of Drosophila melanogaster to Mancozeb Induces Oxidative Damage and Modulates Nrf2 and HSP70/83. Oxid. Med. Cell Longev. 2018, 2018, 5456928. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokott-Vuong, A.; Jung, J.; Fehr, A.T.; Kirschfink, N.; Noristani, R.; Voigt, A.; Reich, A.; Schulz, J.B.; Huber, M.; Habib, P. Increased Post-Hypoxic Oxidative Stress and Activation of the PERK Branch of the UPR in Trap1-Deficient Drosophila melanogaster Is Abrogated by Metformin. Int. J. Mol. Sci. 2021, 22, 11586. https://doi.org/10.3390/ijms222111586
Kokott-Vuong A, Jung J, Fehr AT, Kirschfink N, Noristani R, Voigt A, Reich A, Schulz JB, Huber M, Habib P. Increased Post-Hypoxic Oxidative Stress and Activation of the PERK Branch of the UPR in Trap1-Deficient Drosophila melanogaster Is Abrogated by Metformin. International Journal of Molecular Sciences. 2021; 22(21):11586. https://doi.org/10.3390/ijms222111586
Chicago/Turabian StyleKokott-Vuong, Alma, Jennifer Jung, Aaron T. Fehr, Nele Kirschfink, Rozina Noristani, Aaron Voigt, Arno Reich, Jörg B. Schulz, Michael Huber, and Pardes Habib. 2021. "Increased Post-Hypoxic Oxidative Stress and Activation of the PERK Branch of the UPR in Trap1-Deficient Drosophila melanogaster Is Abrogated by Metformin" International Journal of Molecular Sciences 22, no. 21: 11586. https://doi.org/10.3390/ijms222111586
APA StyleKokott-Vuong, A., Jung, J., Fehr, A. T., Kirschfink, N., Noristani, R., Voigt, A., Reich, A., Schulz, J. B., Huber, M., & Habib, P. (2021). Increased Post-Hypoxic Oxidative Stress and Activation of the PERK Branch of the UPR in Trap1-Deficient Drosophila melanogaster Is Abrogated by Metformin. International Journal of Molecular Sciences, 22(21), 11586. https://doi.org/10.3390/ijms222111586