
Pathogenic Variants in Selenoproteins and Selenocysteine Biosynthesis Machinery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pathogenic Variants in Selenoproteins
2.1. SELENON
2.2. GPX4
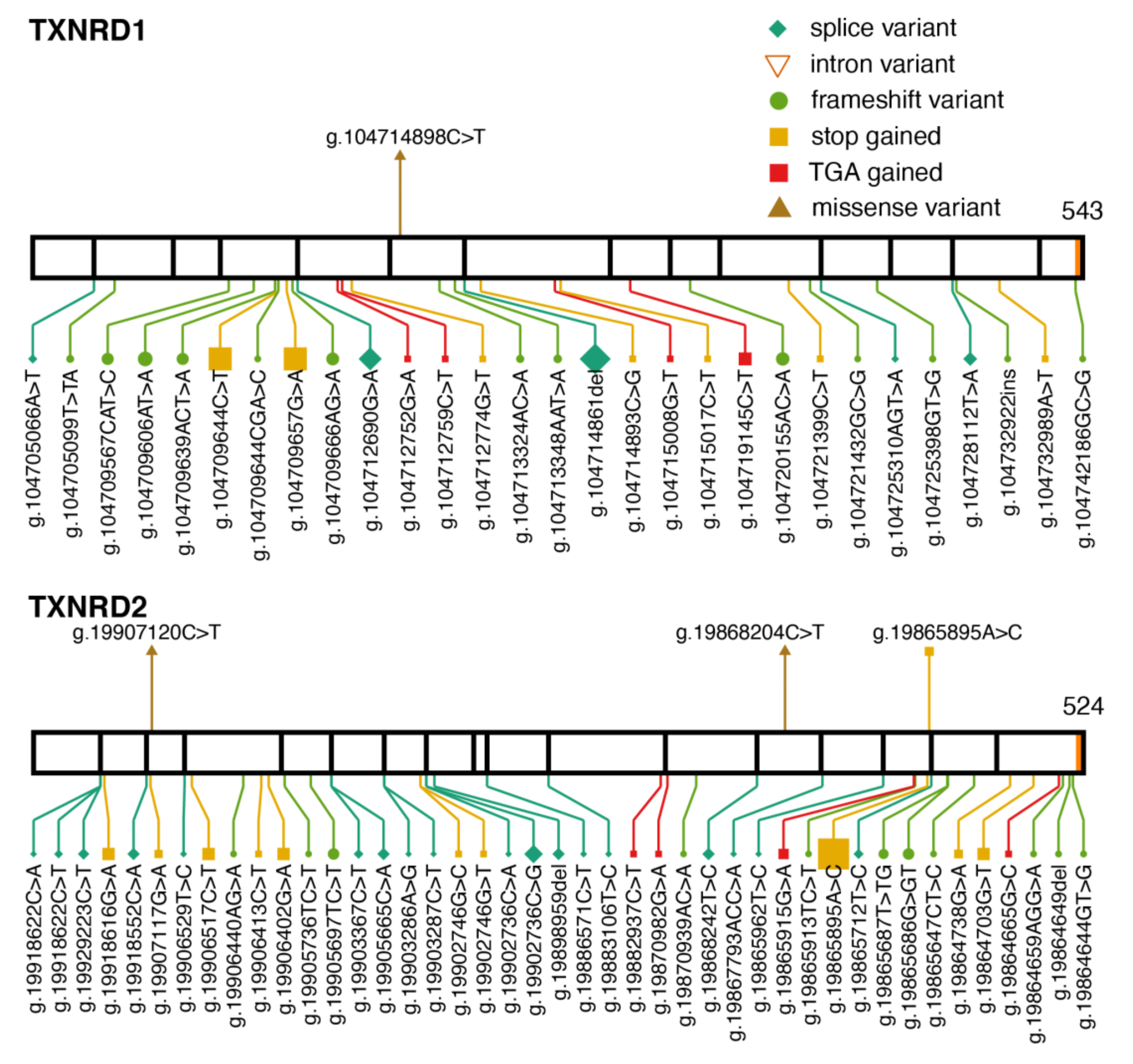
2.3. Thioredoxin Reductases
2.4. SELENOI
3. Pathogenic Variants Disrupting Selenoprotein Synthesis
3.1. TRU-TCA1-1
3.2. SECISBP2
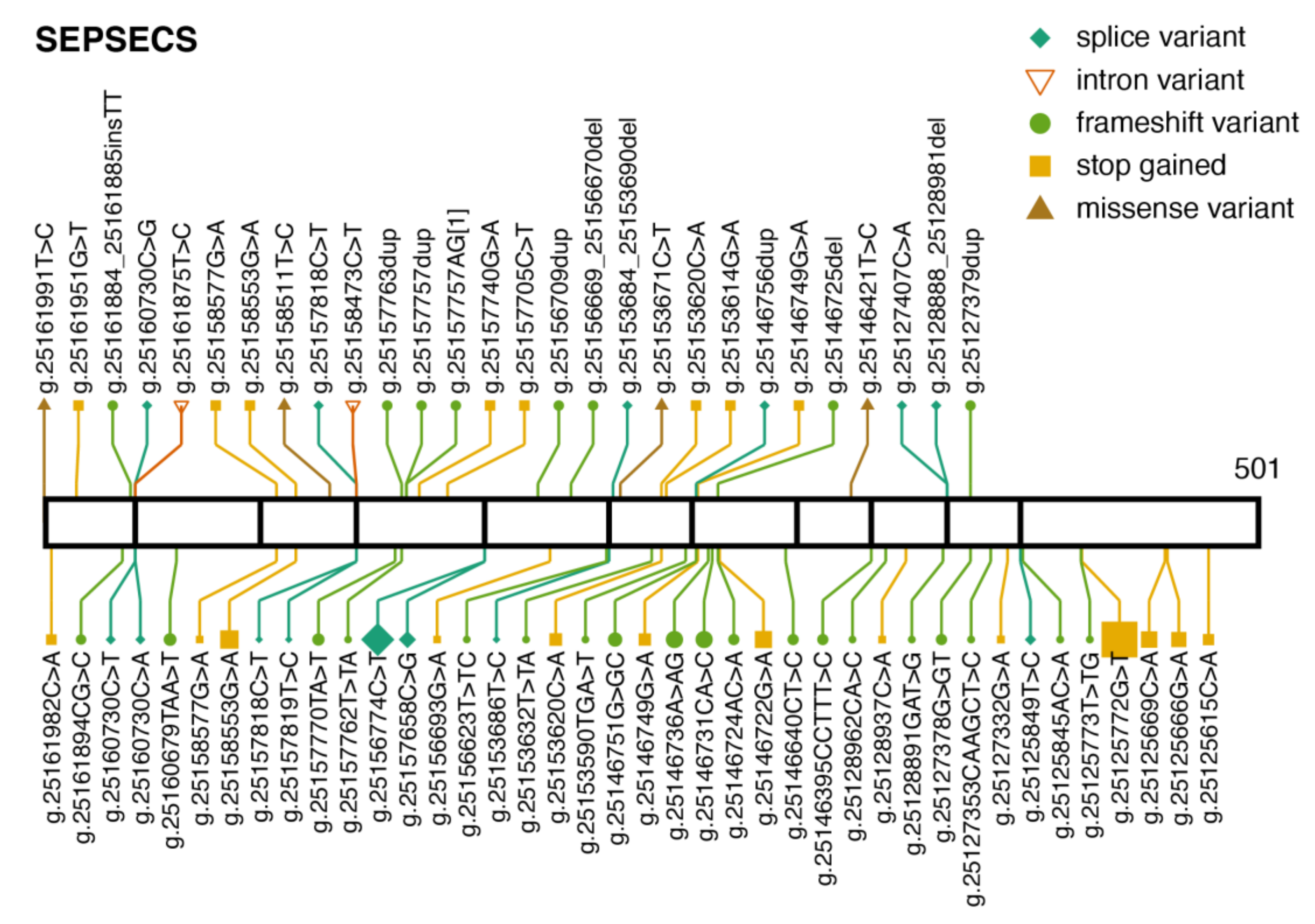
3.3. SEPSECS
4. Common Genetic Variance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mariotti, M.; Ridge, P.G.; Zhang, Y.; Lobanov, A.V.; Pringle, T.H.; Guigo, R.; Hatfield, D.L.; Gladyshev, V.N. Composition and Evolution of the Vertebrate and Mammalian Selenoproteomes. PLoS ONE 2012, 7, e33066. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular Pathways and Physiological Roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed]
- Allmang, C.; Wurth, L.; Krol, A. The Selenium to Selenoprotein Pathway in Eukaryotes: More Molecular Partners than Anticipated. Biochim. Biophys. Acta 2009, 1790, 1415–1423. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of Mammalian Selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef]
- Conrad, M.; Schweizer, U. Mouse Models That Target Individual Selenoproteins. In Selenium; Hatfield, D.L., Schweizer, U., Tsuji, P.A., Gladyshev, V.N., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 567–578. [Google Scholar] [CrossRef]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The Selenoprotein GPX4 Is Essential for Mouse Development and Protects from Radiation and Oxidative Damage Insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early Embryonic Lethality Caused by Targeted Disruption of the Mouse PHGPx Gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef]
- Jakupoglu, C.; Przemeck, G.K.H.; Schneider, M.; Moreno, S.G.; Mayr, N.; Hatzopoulos, A.K.; de Angelis, M.H.; Wurst, W.; Bornkamm, G.W.; Brielmeier, M.; et al. Cytoplasmic Thioredoxin Reductase Is Essential for Embryogenesis but Dispensable for Cardiac Development. Mol. Cell. Biol. 2005, 25, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Jakupoglu, C.; Moreno, S.G.; Lippl, S.; Banjac, A.; Schneider, M.; Beck, H.; Hatzopoulos, A.K.; Just, U.; Sinowatz, F.; et al. Essential Role for Mitochondrial Thioredoxin Reductase in Hematopoiesis, Heart Development, and Heart Function. Mol. Cell. Biol. 2004, 24, 9414–9423. [Google Scholar] [CrossRef] [PubMed]
- Boukhzar, L.; Hamieh, A.; Cartier, D.; Tanguy, Y.; Alsharif, I.; Castex, M.; Arabo, A.; El Hajji, S.; Bonnet, J.-J.; Errami, M.; et al. Selenoprotein T Exerts an Essential Oxidoreductase Activity That Protects Dopaminergic Neurons in Mouse Models of Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 557–574. [Google Scholar] [CrossRef]
- Avery, J.C.; Yamazaki, Y.; Hoffmann, F.W.; Folgelgren, B.; Hoffmann, P.R. Selenoprotein I Is Essential for Murine Embryogenesis. Arch. Biochem. Biophys. 2020, 689, 108444. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to Accessing Data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Schweizer, U.; Fradejas-Villar, N. Why 21? The Significance of Selenoproteins for Human Health Revealed by Inborn Errors of Metabolism. FASEB J. 2016, 30, 3669–3681. [Google Scholar] [CrossRef] [PubMed]
- Fradejas-Villar, N. Consequences of Mutations and Inborn Errors of Selenoprotein Biosynthesis and Functions. Free Radic. Biol. Med. 2018, 127, 206–214. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Chatterjee, K. Human Disorders Affecting the Selenocysteine Incorporation Pathway Cause Systemic Selenoprotein Deficiency. Antioxid. Redox Signal. 2020, 33, 481–497. [Google Scholar] [CrossRef]
- Schweizer, U.; Bohleber, S.; Zhao, W.; Fradejas-Villar, N. The Neurobiology of Selenium: Looking Back and to the Future. Front. Neurosci. 2021, 15, 652099. [Google Scholar] [CrossRef]
- Castets, P.; Lescure, A.; Guicheney, P.; Allamand, V. Selenoprotein N in Skeletal Muscle: From Diseases to Function. J. Mol. Med. 2012, 90, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Lescure, A.; Gautheret, D.; Carbon, P.; Krol, A. Novel Selenoproteins Identified in Silico and in Vivo by Using a Conserved RNA Structural Motif. J. Biol. Chem. 1999, 274, 38147–38154. [Google Scholar] [CrossRef]
- Moghadaszadeh, B.; Petit, N.; Jaillard, C.; Brockington, M.; Quijano Roy, S.; Merlini, L.; Romero, N.; Estournet, B.; Desguerre, I.; Chaigne, D.; et al. Mutations in SEPN1 Cause Congenital Muscular Dystrophy with Spinal Rigidity and Restrictive Respiratory Syndrome. Nat. Genet. 2001, 29, 17–18. [Google Scholar] [CrossRef]
- Jurynec, M.J.; Xia, R.; Mackrill, J.J.; Gunther, D.; Crawford, T.; Flanigan, K.M.; Abramson, J.J.; Howard, M.T.; Grunwald, D.J. Selenoprotein N Is Required for Ryanodine Receptor Calcium Release Channel Activity in Human and Zebrafish Muscle. Proc. Natl. Acad. Sci. USA 2008, 105, 12485–12490. [Google Scholar] [CrossRef] [PubMed]
- Petit, N.; Lescure, A.; Rederstorff, M.; Krol, A.; Moghadaszadeh, B.; Wewer, U.M.; Guicheney, P. Selenoprotein N: An Endoplasmic Reticulum Glycoprotein with an Early Developmental Expression Pattern. Hum. Mol. Genet. 2003, 12, 1045–1053. [Google Scholar] [CrossRef]
- Chernorudskiy, A.; Varone, E.; Colombo, S.F.; Fumagalli, S.; Cagnotto, A.; Cattaneo, A.; Briens, M.; Baltzinger, M.; Kuhn, L.; Bachi, A.; et al. Selenoprotein N Is an Endoplasmic Reticulum Calcium Sensor That Links Luminal Calcium Levels to a Redox Activity. Proc. Natl. Acad. Sci. USA 2020, 117, 21288–21298. [Google Scholar] [CrossRef]
- Rederstorff, M.; Castets, P.; Arbogast, S.; Lainé, J.; Vassilopoulos, S.; Beuvin, M.; Dubourg, O.; Vignaud, A.; Ferry, A.; Krol, A.; et al. Increased Muscle Stress-Sensitivity Induced by Selenoprotein N Inactivation in Mouse: A Mammalian Model for SEPN1-Related Myopathy. PLoS ONE 2011, 6, e23094. [Google Scholar] [CrossRef] [PubMed]
- Castets, P.; Bertrand, A.T.; Beuvin, M.; Ferry, A.; Le Grand, F.; Castets, M.; Chazot, G.; Rederstorff, M.; Krol, A.; Lescure, A.; et al. Satellite Cell Loss and Impaired Muscle Regeneration in Selenoprotein N Deficiency. Hum. Mol. Genet. 2011, 20, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Moghadaszadeh, B.; Desguerre, I.; Topaloglu, H.; Muntoni, F.; Pavek, S.; Sewry, C.; Mayer, M.; Fardeau, M.; Tomé, F.M.; Guicheney, P. Identification of a New Locus for a Peculiar Form of Congenital Muscular Dystrophy with Early Rigidity of the Spine, on Chromosome 1p35-36. Am. J. Hum. Genet. 1998, 62, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, A.; Quijano-Roy, S.; Pichereau, C.; Moghadaszadeh, B.; Goemans, N.; Bönnemann, C.; Jungbluth, H.; Straub, V.; Villanova, M.; Leroy, J.-P.; et al. Mutations of the Selenoprotein N Gene, Which Is Implicated in Rigid Spine Muscular Dystrophy, Cause the Classical Phenotype of Multiminicore Disease: Reassessing the Nosology of Early-Onset Myopathies. Am. J. Hum. Genet. 2002, 71, 739–749. [Google Scholar] [CrossRef]
- Clarke, N.F.; Kidson, W.; Quijano-Roy, S.; Estournet, B.; Ferreiro, A.; Guicheney, P.; Manson, J.I.; Kornberg, A.J.; Shield, L.K.; North, K.N. SEPN1: Associated with Congenital Fiber-Type Disproportion and Insulin Resistance. Ann. Neurol. 2006, 59, 546–552. [Google Scholar] [CrossRef]
- Ferreiro, A.; Ceuterick-de Groote, C.; Marks, J.J.; Goemans, N.; Schreiber, G.; Hanefeld, F.; Fardeau, M.; Martin, J.-J.; Goebel, H.H.; Richard, P.; et al. Desmin-Related Myopathy with Mallory Body-like Inclusions Is Caused by Mutations of the Selenoprotein, N. Gene. Ann. Neurol. 2004, 55, 676–686. [Google Scholar] [CrossRef]
- Villar-Quiles, R.N.; von der Hagen, M.; Métay, C.; Gonzalez, V.; Donkervoort, S.; Bertini, E.; Castiglioni, C.; Chaigne, D.; Colomer, J.; Cuadrado, M.L.; et al. The Clinical, Histologic, and Genotypic Spectrum of SEPN1-Related Myopathy: A Case Series. Neurology 2020, 95, e1512–e1527. [Google Scholar] [CrossRef]
- Allamand, V.; Richard, P.; Lescure, A.; Ledeuil, C.; Desjardin, D.; Petit, N.; Gartioux, C.; Ferreiro, A.; Krol, A.; Pellegrini, N.; et al. A Single Homozygous Point Mutation in a 3’untranslated Region Motif of Selenoprotein N MRNA Causes SEPN1-Related Myopathy. EMBO Rep. 2006, 7, 450–454. [Google Scholar] [CrossRef]
- Maiti, B.; Arbogast, S.; Allamand, V.; Moyle, M.W.; Anderson, C.B.; Richard, P.; Guicheney, P.; Ferreiro, A.; Flanigan, K.M.; Howard, M.T. A Mutation in the SEPN1 Selenocysteine Redefinition Element (SRE) Reduces Selenocysteine Incorporation and Leads to SEPN1-Related Myopathy. Hum. Mutat. 2009, 30, 411–416. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Brigelius-Flohé, R.; Aumann, K.D.; Roveri, A.; Schomburg, D.; Flohé, L. Diversity of Glutathione Peroxidases. Methods Enzymol. 1995, 252, 38–53. [Google Scholar] [CrossRef]
- Ursini, F.; Heim, S.; Kiess, M.; Maiorino, M.; Roveri, A.; Wissing, J.; Flohé, L. Dual Function of the Selenoprotein PHGPx during Sperm Maturation. Science 1999, 285, 1393–1396. [Google Scholar] [CrossRef] [PubMed]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of Ferroptosis Regulator Glutathione Peroxidase 4 in Forebrain Neurons Promotes Cognitive Impairment and Neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Sedaghatian, M.R. Congenital Lethal Metaphyseal Chondrodysplasia: A Newly Recognized Complex Autosomal Recessive Disorder. Am. J. Med. Genet. 1980, 6, 269–274. [Google Scholar] [CrossRef]
- Smith, A.C.; Mears, A.J.; Bunker, R.; Ahmed, A.; MacKenzie, M.; Schwartzentruber, J.A.; Beaulieu, C.L.; Ferretti, E.; FORGE Canada Consortium; Majewski, J.; et al. Mutations in the Enzyme Glutathione Peroxidase 4 Cause Sedaghatian-Type Spondylometaphyseal Dysplasia. J. Med. Genet. 2014, 51, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Aygun, C.; Celik, F.C.; Nural, M.S.; Azak, E.; Kucukoduk, S.; Ogur, G.; Incesu, L. Simplified Gyral Pattern with Cerebellar Hypoplasia in Sedaghatian Type Spondylometaphyseal Dysplasia: A Clinical Report and Review of the Literature. Am. J. Med. Genet. A 2012, 158A, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Sibbing, D.; Pfeufer, A.; Perisic, T.; Mannes, A.M.; Fritz-Wolf, K.; Unwin, S.; Sinner, M.F.; Gieger, C.; Gloeckner, C.J.; Wichmann, H.-E.; et al. Mutations in the Mitochondrial Thioredoxin Reductase Gene TXNRD2 Cause Dilated Cardiomyopathy. Eur. Heart J. 2011, 32, 1121–1133. [Google Scholar] [CrossRef]
- Prasad, R.; Chan, L.F.; Hughes, C.R.; Kaski, J.P.; Kowalczyk, J.C.; Savage, M.O.; Peters, C.J.; Nathwani, N.; Clark, A.J.L.; Storr, H.L.; et al. Thioredoxin Reductase 2 (TXNRD2) Mutation Associated with Familial Glucocorticoid Deficiency (FGD). J. Clin. Endocrinol. Metab. 2014, 99, E1556–E1563. [Google Scholar] [CrossRef]
- Kudin, A.P.; Baron, G.; Zsurka, G.; Hampel, K.G.; Elger, C.E.; Grote, A.; Weber, Y.; Lerche, H.; Thiele, H.; Nürnberg, P.; et al. Homozygous Mutation in TXNRD1 Is Associated with Genetic Generalized Epilepsy. Free Radic. Biol. Med. 2017, 106, 270–277. [Google Scholar] [CrossRef]
- Rojnueangnit, K.; Sirichongkolthong, B.; Wongwandee, R.; Khetkham, T.; Noojarern, S.; Khongkraparn, A.; Wattanasirichaigoon, D. Identification of Gene Mutations in Primary Pediatric Cardiomyopathy by Whole Exome Sequencing. Pediatr. Cardiol. 2020, 41, 165–174. [Google Scholar] [CrossRef]
- Ahmed, M.Y.; Al-Khayat, A.; Al-Murshedi, F.; Al-Futaisi, A.; Chioza, B.A.; Fernandez-Murray, J.P.; Self, J.E.; Salter, C.G.; Harlalka, G.V.; Rawlins, L.E.; et al. A Mutation of EPT1 (SELENOI) Underlies a New Disorder of Kennedy Pathway Phospholipid Biosynthesis. Brain 2017, 140, 547–554. [Google Scholar] [CrossRef]
- Horibata, Y.; Elpeleg, O.; Eran, A.; Hirabayashi, Y.; Savitzki, D.; Tal, G.; Mandel, H.; Sugimoto, H. EPT1 (Selenoprotein I) Is Critical for the Neural Development and Maintenance of Plasmalogen in Humans. J. Lipid Res. 2018, 59, 1015–1026. [Google Scholar] [CrossRef]
- Santesmasses, D.; Mariotti, M.; Gladyshev, V.N. Tolerance to Selenoprotein Loss Differs between Human and Mouse. Mol. Biol. Evol. 2020, 37, 341–354. [Google Scholar] [CrossRef]
- Turanov, A.A.; Xu, X.-M.; Carlson, B.A.; Yoo, M.-H.; Gladyshev, V.N.; Hatfield, D.L. Biosynthesis of Selenocysteine, the 21st Amino Acid in the Genetic Code, and a Novel Pathway for Cysteine Biosynthesis. Adv. Nutr. 2011, 2, 122–128. [Google Scholar] [CrossRef]
- Diamond, A.; Dudock, B.; Hatfield, D. Structure and Properties of a Bovine Liver UGA Suppressor Serine TRNA with a Tryptophan Anticodon. Cell 1981, 25, 497–506. [Google Scholar] [CrossRef]
- Bösl, M.R.; Takaku, K.; Oshima, M.; Nishimura, S.; Taketo, M.M. Early Embryonic Lethality Caused by Targeted Disruption of the Mouse Selenocysteine TRNA Gene (Trsp). Proc. Natl. Acad. Sci. USA 1997, 94, 5531–5534. [Google Scholar] [CrossRef]
- Carlson, B.A. Selenocysteine TRNA[Ser]Sec Mouse Models for Elucidating Roles of Selenoproteins in Health and Development. In Selenium; Springer International Publishing: Cham, Switzerland, 2016; pp. 555–566. [Google Scholar] [CrossRef]
- Itoh, Y.; Chiba, S.; Sekine, S.-I.; Yokoyama, S. Crystal Structure of Human Selenocysteine TRNA. Nucleic Acids Res. 2009, 37, 6259–6268. [Google Scholar] [CrossRef]
- Carlson, B.A.; Lee, B.J.; Tsuji, P.A.; Tobe, R.; Park, J.M.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L. Selenocysteine TRNA[Ser]Sec: From Nonsense Suppressor TRNA to the Quintessential Constituent in Selenoprotein Biosynthesis. In Selenium; Springer International Publishing: Cham, Switzerland, 2016; pp. 3–12. [Google Scholar] [CrossRef]
- Diamond, A.M.; Choi, I.S.; Crain, P.F.; Hashizume, T.; Pomerantz, S.C.; Cruz, R.; Steer, C.J.; Hill, K.E.; Burk, R.F.; McCloskey, J.A.; et al. Dietary Selenium Affects Methylation of the Wobble Nucleoside in the Anticodon of Selenocysteine TRNA([Ser]Sec). J. Biol. Chem. 1993, 268, 14215–14223. [Google Scholar] [CrossRef]
- Carlson, B.A.; Moustafa, M.E.; Sengupta, A.; Schweizer, U.; Shrimali, R.; Rao, M.; Zhong, N.; Wang, S.; Feigenbaum, L.; Lee, B.J.; et al. Selective Restoration of the Selenoprotein Population in a Mouse Hepatocyte Selenoproteinless Background with Different Mutant Selenocysteine TRNAs Lacking Um34. J. Biol. Chem. 2007, 282, 32591–32602. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Carlson, B.; Agostini, M.; Moran, C.; Rajanayagam, O.; Bochukova, E.; Tobe, R.; Peat, R.; Gevers, E.; Muntoni, F.; et al. Mutation in Human Selenocysteine Transfer RNA Selectively Disrupts Selenoprotein Synthesis. J. Clin. Investig. 2016, 126, 992–996. [Google Scholar] [CrossRef]
- Geslot, A.; Savagner, F.; Caron, P. Inherited Selenocysteine Transfer RNA Mutation: Clinical and Hormonal Evaluation of 2 Patients. Eur. Thyroid J. 2021, 1–6. [Google Scholar] [CrossRef]
- Palioura, S.; Sherrer, R.L.; Steitz, T.A.; Söll, D.; Simonovic, M. The Human SepSecS-TRNASec Complex Reveals the Mechanism of Selenocysteine Formation. Science 2009, 325, 321–325. [Google Scholar] [CrossRef]
- Copeland, P.R.; Fletcher, J.E.; Carlson, B.A.; Hatfield, D.L.; Driscoll, D.M. A Novel RNA Binding Protein, SBP2, Is Required for the Translation of Mammalian Selenoprotein MRNAs. EMBO J. 2000, 19, 306–314. [Google Scholar] [CrossRef]
- Seeher, S.; Atassi, T.; Mahdi, Y.; Carlson, B.A.; Braun, D.; Wirth, E.K.; Klein, M.O.; Reix, N.; Miniard, A.C.; Schomburg, L.; et al. Secisbp2 Is Essential for Embryonic Development and Enhances Selenoprotein Expression. Antioxid. Redox Signal. 2014, 21, 835–849. [Google Scholar] [CrossRef]
- Mehta, A.; Rebsch, C.M.; Kinzy, S.A.; Fletcher, J.E.; Copeland, P.R. Efficiency of Mammalian Selenocysteine Incorporation. J. Biol. Chem. 2004, 279, 37852–37859. [Google Scholar] [CrossRef] [PubMed]
- Copeland, P.R.; Stepanik, V.A.; Driscoll, D.M. Insight into Mammalian Selenocysteine Insertion: Domain Structure and Ribosome Binding Properties of Sec Insertion Sequence Binding Protein 2. Mol. Cell. Biol. 2001, 21, 1491–1498. [Google Scholar] [CrossRef]
- Allmang, C.; Carbon, P.; Krol, A. The SBP2 and 15.5 KD/Snu13p Proteins Share the Same RNA Binding Domain: Identification of SBP2 Amino Acids Important to SECIS RNA Binding. RNA 2002, 8, 1308–1318. [Google Scholar] [CrossRef][Green Version]
- Zhao, W.; Bohleber, S.; Schmidt, H.; Seeher, S.; Howard, M.T.; Braun, D.; Arndt, S.; Reuter, U.; Wende, H.; Birchmeier, C.; et al. Ribosome Profiling of Selenoproteins in Vivo Reveals Consequences of Pathogenic Secisbp2 Missense Mutations. J. Biol. Chem. 2019, jbc.RA119.009369. [Google Scholar] [CrossRef]
- Donovan, J.; Copeland, P.R. Selenocysteine Insertion Sequence Binding Protein 2L Is Implicated as a Novel Post-Transcriptional Regulator of Selenoprotein Expression. PLoS ONE 2012, 7, e35581. [Google Scholar] [CrossRef]
- Donovan, J.; Copeland, P.R. Evolutionary History of Selenocysteine Incorporation from the Perspective of SECIS Binding Proteins. BMC Evol. Biol. 2009, 9, 229. [Google Scholar] [CrossRef]
- Small-Howard, A.; Morozova, N.; Stoytcheva, Z.; Forry, E.P.; Mansell, J.B.; Harney, J.W.; Carlson, B.A.; Xu, X.-M.; Hatfield, D.L.; Berry, M.J. Supramolecular Complexes Mediate Selenocysteine Incorporation in Vivo. Mol. Cell. Biol. 2006, 26, 2337–2346. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Schoenmakers, N.; Chatterjee, K. Mutations in Humans That Adversely Affect the Selenoprotein Synthesis Pathway. In Selenium. Its Molecular Biology and Role in Human Health; Hatfield, D.L., Berry, M.J., Gladyshev, N.V., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 523–538. [Google Scholar] [CrossRef]
- Anttonen, A.-K.; Hilander, T.; Linnankivi, T.; Isohanni, P.; French, R.L.; Liu, Y.; Simonović, M.; Söll, D.; Somer, M.; Muth-Pawlak, D.; et al. Selenoprotein Biosynthesis Defect Causes Progressive Encephalopathy with Elevated Lactate. Neurology 2015, 85, 306–315. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Mentuccia, D.; Proietti-Pannunzi, L.; Tanner, K.; Bacci, V.; Pollin, T.I.; Poehlman, E.T.; Shuldiner, A.R.; Celi, F.S. Association between a Novel Variant of the Human Type 2 Deiodinase Gene Thr92Ala and Insulin Resistance: Evidence of Interaction with the Trp64Arg Variant of the Beta-3-Adrenergic Receptor. Diabetes 2002, 51, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Castagna, M.G.; Dentice, M.; Cantara, S.; Ambrosio, R.; Maino, F.; Porcelli, T.; Marzocchi, C.; Garbi, C.; Pacini, F.; Salvatore, D. DIO2 Thr92Ala Reduces Deiodinase-2 Activity and Serum-T3 Levels in Thyroid-Deficient Patients. J. Clin. Endocrinol. Metab. 2017, 102, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Jung, J.; Araki, O.; Tsunekawa, K.; Park, S.Y.; Kim, J.; Murakami, M.; Jeong, S.-Y.; Lee, S. Concurrent TSHR Mutations and DIO2 T92A Polymorphism Result in Abnormal Thyroid Hormone Metabolism. Sci. Rep. 2018, 8, 10090. [Google Scholar] [CrossRef]
- Meulenbelt, I.; Min, J.L.; Bos, S.; Riyazi, N.; Houwing-Duistermaat, J.J.; van der Wijk, H.-J.; Kroon, H.M.; Nakajima, M.; Ikegawa, S.; Uitterlinden, A.G.; et al. Identification of DIO2 as a New Susceptibility Locus for Symptomatic Osteoarthritis. Hum. Mol. Genet. 2008, 17, 1867–1875. [Google Scholar] [CrossRef]
- Mez, J.; Chung, J.; Jun, G.; Kriegel, J.; Bourlas, A.P.; Sherva, R.; Logue, M.W.; Barnes, L.L.; Bennett, D.A.; Buxbaum, J.D.; et al. Two Novel Loci, COBL and SLC10A2, for Alzheimer’s Disease in African Americans. Alzheimers. Dement. 2017, 13, 119–129. [Google Scholar] [CrossRef]
- Sreelatha, A.; Yee, S.S.; Lopez, V.A.; Park, B.C.; Kinch, L.N.; Pilch, S.; Servage, K.A.; Zhang, J.; Jiou, J.; Karasiewicz-Urbańska, M.; et al. Protein AMPylation by an Evolutionarily Conserved Pseudokinase. Cell 2018, 175, 809–821.e19. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.M.; Zhu, G.; Dy, V.; Heath, A.C.; Madden, P.A.F.; Kemp, J.P.; McMahon, G.; St Pourcain, B.; Timpson, N.J.; Golding, J.; et al. Genome-Wide Association Study Identifies Loci Affecting Blood Copper, Selenium and Zinc. Hum. Mol. Genet. 2013, 22, 3998–4006. [Google Scholar] [CrossRef]
- Gong, J.; Hsu, L.; Harrison, T.; King, I.B.; Stürup, S.; Song, X.; Duggan, D.; Liu, Y.; Hutter, C.; Chanock, S.J.; et al. Genome-Wide Association Study of Serum Selenium Concentrations. Nutrients 2013, 5, 1706–1718. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, M.C.; Fornage, M.; Foy, M.; Xun, P.; Gladyshev, V.N.; Morris, S.; Chasman, D.I.; Hu, F.B.; Rimm, E.B.; Kraft, P.; et al. Genome-Wide Association Study of Selenium Concentrations. Hum. Mol. Genet. 2015, 24, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Batai, K.; Trejo, M.J.; Chen, Y.; Kohler, L.N.; Lance, P.; Ellis, N.A.; Cornelis, M.C.; Chow, H.-H.S.; Hsu, C.-H.; Jacobs, E.T. Genome-Wide Association Study of Response to Selenium Supplementation and Circulating Selenium Concentrations in Adults of European Descent. J. Nutr. 2021, 151, 293–302. [Google Scholar] [CrossRef]
- Thomson, C.D.; Burton, C.E.; Robinson, M.F. On Supplementing the Selenium Intake of New Zealanders. 1. Short Experiments with Large Doses of Selenite or Selenomethionine. Br. J. Nutr. 1978, 39, 579–587. [Google Scholar] [CrossRef]
- Xia, Y.; Hill, K.E.; Byrne, D.W.; Xu, J.; Burk, R.F. Effectiveness of Selenium Supplements in a Low-Selenium Area of China. Am. J. Clin. Nutr. 2005, 81, 829–834. [Google Scholar] [CrossRef]
- White, L.; Romagné, F.; Müller, E.; Erlebach, E.; Weihmann, A.; Parra, G.; Andrés, A.M.; Castellano, S. Genetic Adaptation to Levels of Dietary Selenium in Recent Human History. Mol. Biol. Evol. 2015, 32, 1507–1518. [Google Scholar] [CrossRef]
- Canani, L.H.; Capp, C.; Dora, J.M.; Meyer, E.L.S.; Wagner, M.S.; Harney, J.W.; Larsen, P.R.; Gross, J.L.; Bianco, A.C.; Maia, A.L. The Type 2 Deiodinase A/G (Thr92Ala) Polymorphism Is Associated with Decreased Enzyme Velocity and Increased Insulin Resistance in Patients with Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2005, 90, 3472–3478. [Google Scholar] [CrossRef]
- Foster, C.B.; Aswath, K.; Chanock, S.J.; McKay, H.F.; Peters, U. Polymorphism Analysis of Six Selenoprotein Genes: Support for a Selective Sweep at the Glutathione Peroxidase 1 Locus (3p21) in Asian Populations. BMC Genet. 2006, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Engelken, J.; Espadas, G.; Mancuso, F.M.; Bonet, N.; Scherr, A.-L.; Jímenez-Álvarez, V.; Codina-Solà, M.; Medina-Stacey, D.; Spataro, N.; Stoneking, M.; et al. Signatures of Evolutionary Adaptation in Quantitative Trait Loci Influencing Trace Element Homeostasis in Liver. Mol. Biol. Evol. 2016, 33, 738–754. [Google Scholar] [CrossRef] [PubMed]
- Turanov, A.A.; Lobanov, A.V.; Hatfield, D.L.; Gladyshev, V.N. UGA Codon Position-Dependent Incorporation of Selenocysteine into Mammalian Selenoproteins. Nucleic Acids Res. 2013, 41, 6952–6959. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Weiss, S.L.; Sunde, R.A. UGA Codon Position Affects the Efficiency of Selenocysteine Incorporation into Glutathione Peroxidase-1. J. Biol. Chem. 1998, 273, 28533–28541. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santesmasses, D.; Gladyshev, V.N. Pathogenic Variants in Selenoproteins and Selenocysteine Biosynthesis Machinery. Int. J. Mol. Sci. 2021, 22, 11593. https://doi.org/10.3390/ijms222111593
Santesmasses D, Gladyshev VN. Pathogenic Variants in Selenoproteins and Selenocysteine Biosynthesis Machinery. International Journal of Molecular Sciences. 2021; 22(21):11593. https://doi.org/10.3390/ijms222111593
Chicago/Turabian StyleSantesmasses, Didac, and Vadim N. Gladyshev. 2021. "Pathogenic Variants in Selenoproteins and Selenocysteine Biosynthesis Machinery" International Journal of Molecular Sciences 22, no. 21: 11593. https://doi.org/10.3390/ijms222111593
APA StyleSantesmasses, D., & Gladyshev, V. N. (2021). Pathogenic Variants in Selenoproteins and Selenocysteine Biosynthesis Machinery. International Journal of Molecular Sciences, 22(21), 11593. https://doi.org/10.3390/ijms222111593