
Diabetes in Humans Activates Pancreatic Stellate Cells via RAGE in Pancreatic Ductal Adenocarcinoma

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Effects of High Fat Diet on the Activation of PSCs Isolated from Experimental Mice
2.2. AGE Stimulation Activated PSCs via RAGE
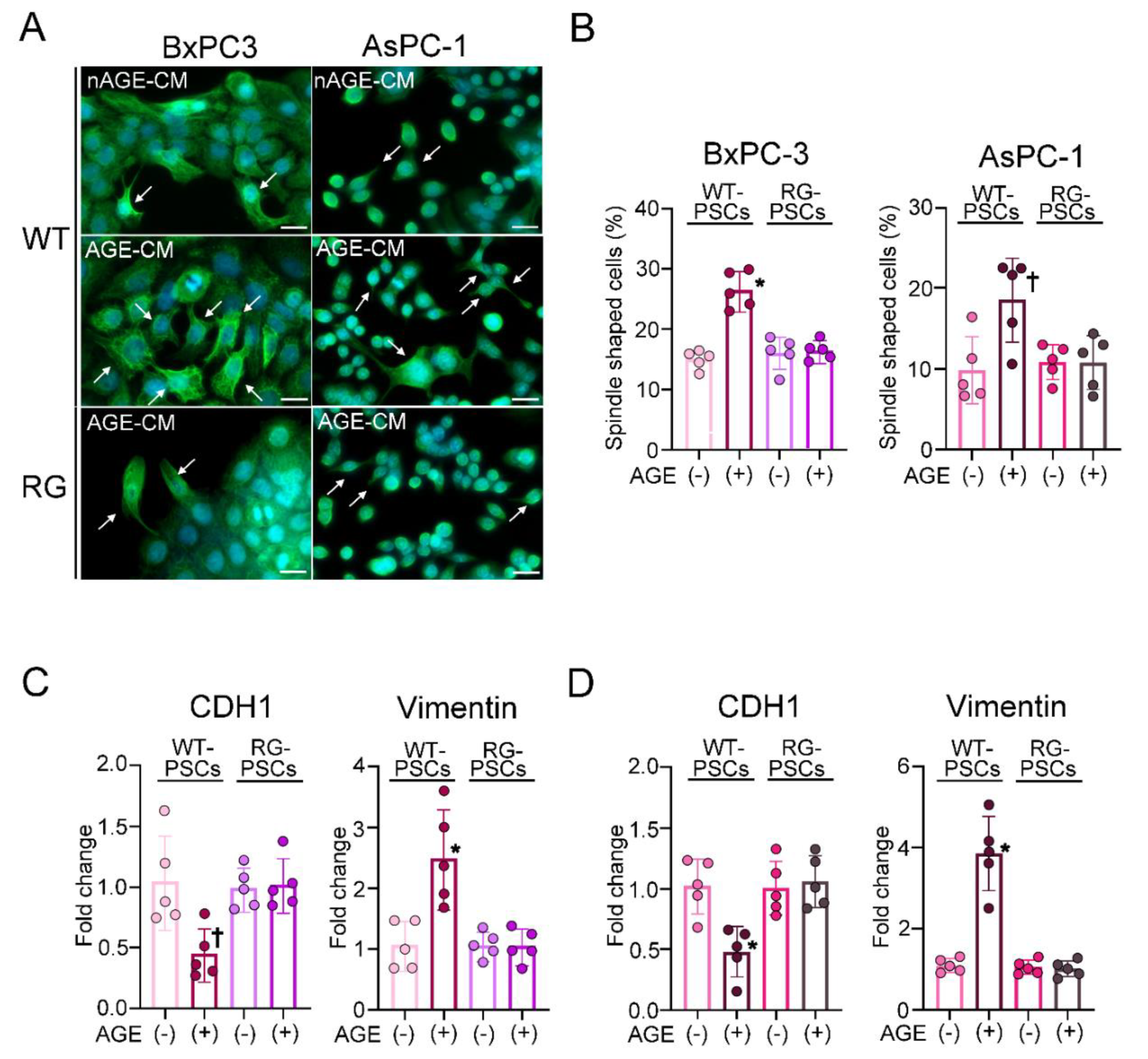
2.3. Humoral Factors Derived from PSCs Stimulated with AGEs Evoked EMT in PDAC
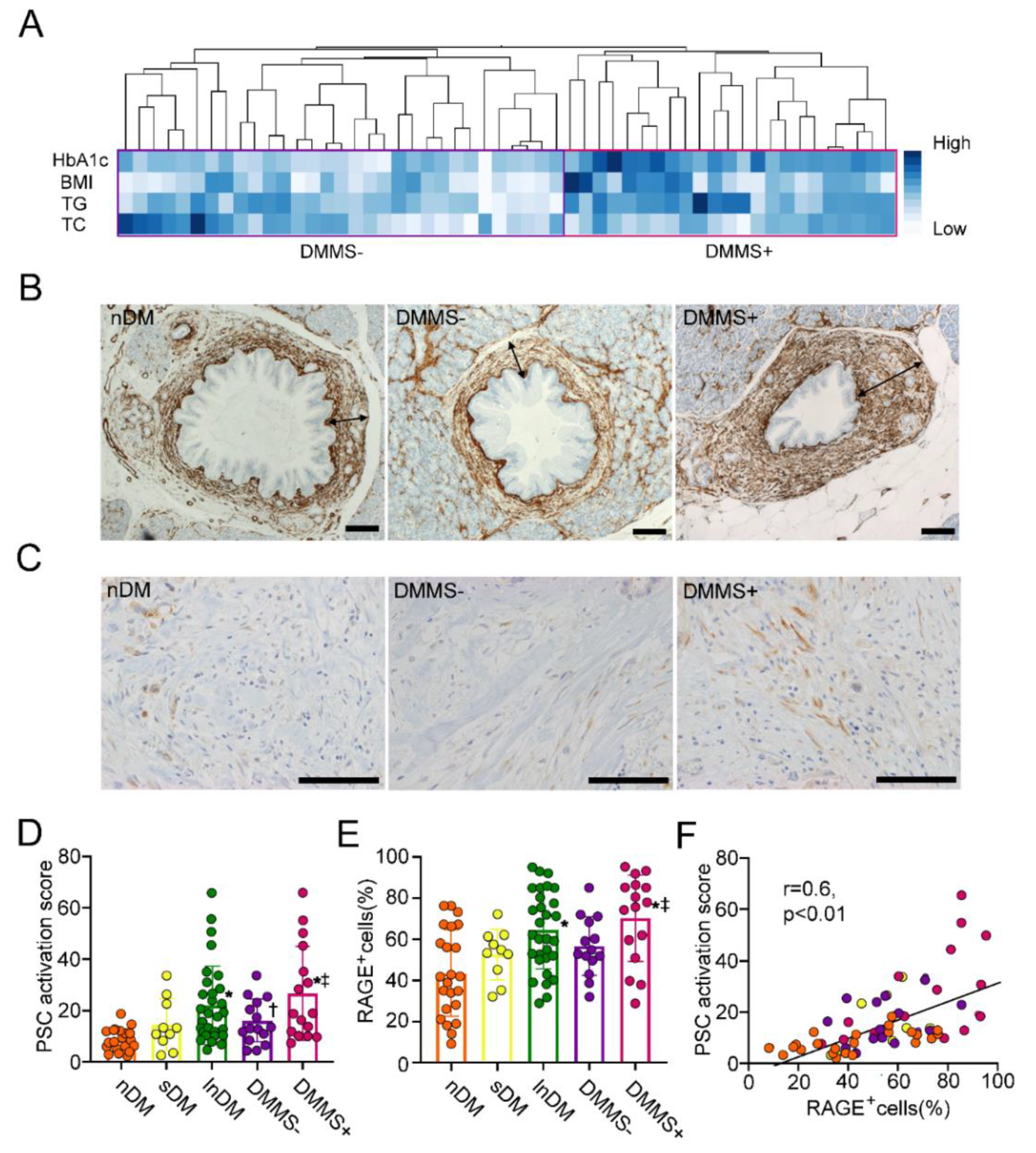
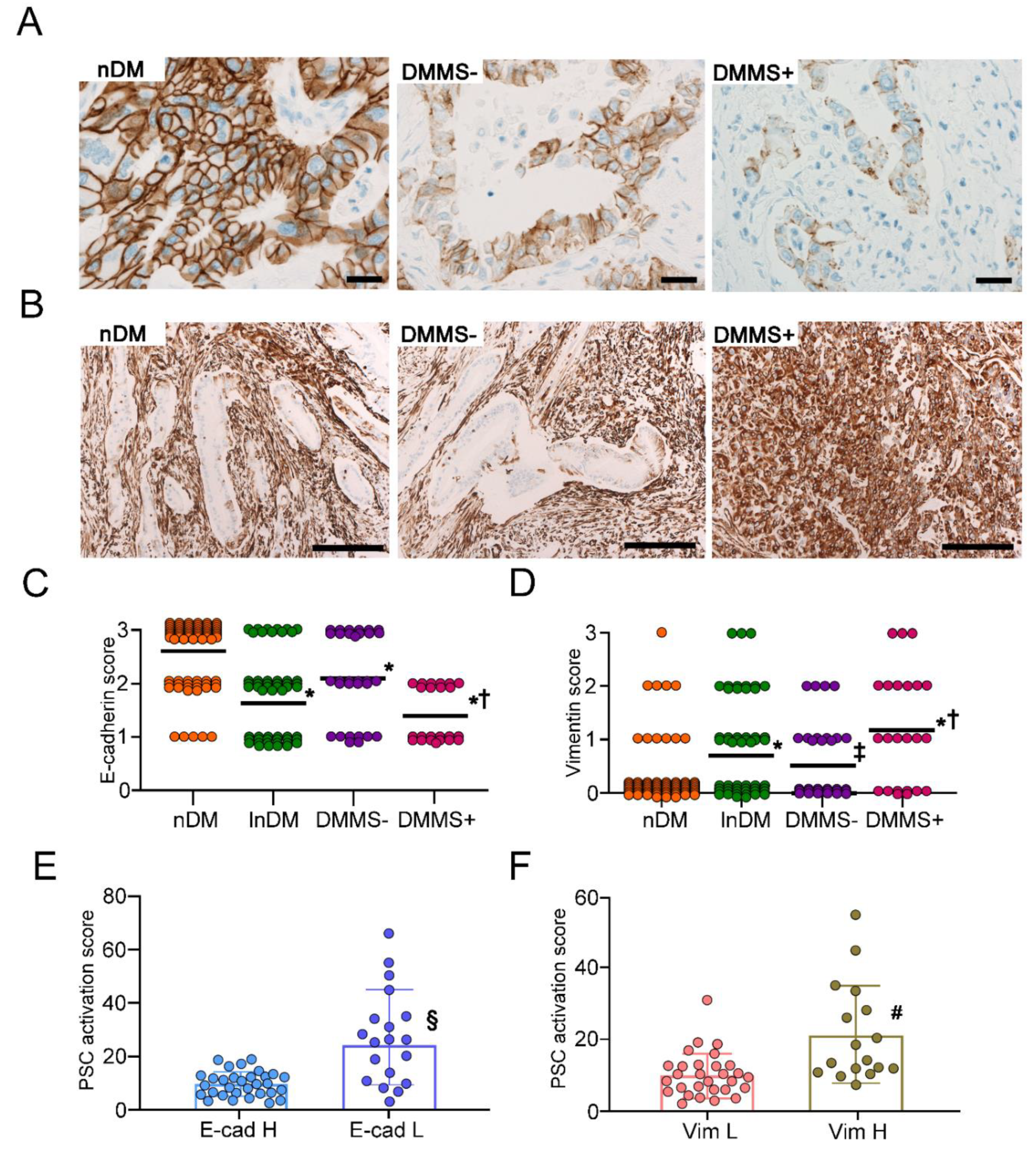
2.4. Increase in αSMA Expression Surrounding Pancreatic Intraepithelial Neoplasia in T2D Patients with Metabolic Syndrome
2.5. Expression of EMT Markers in PDAC Cells
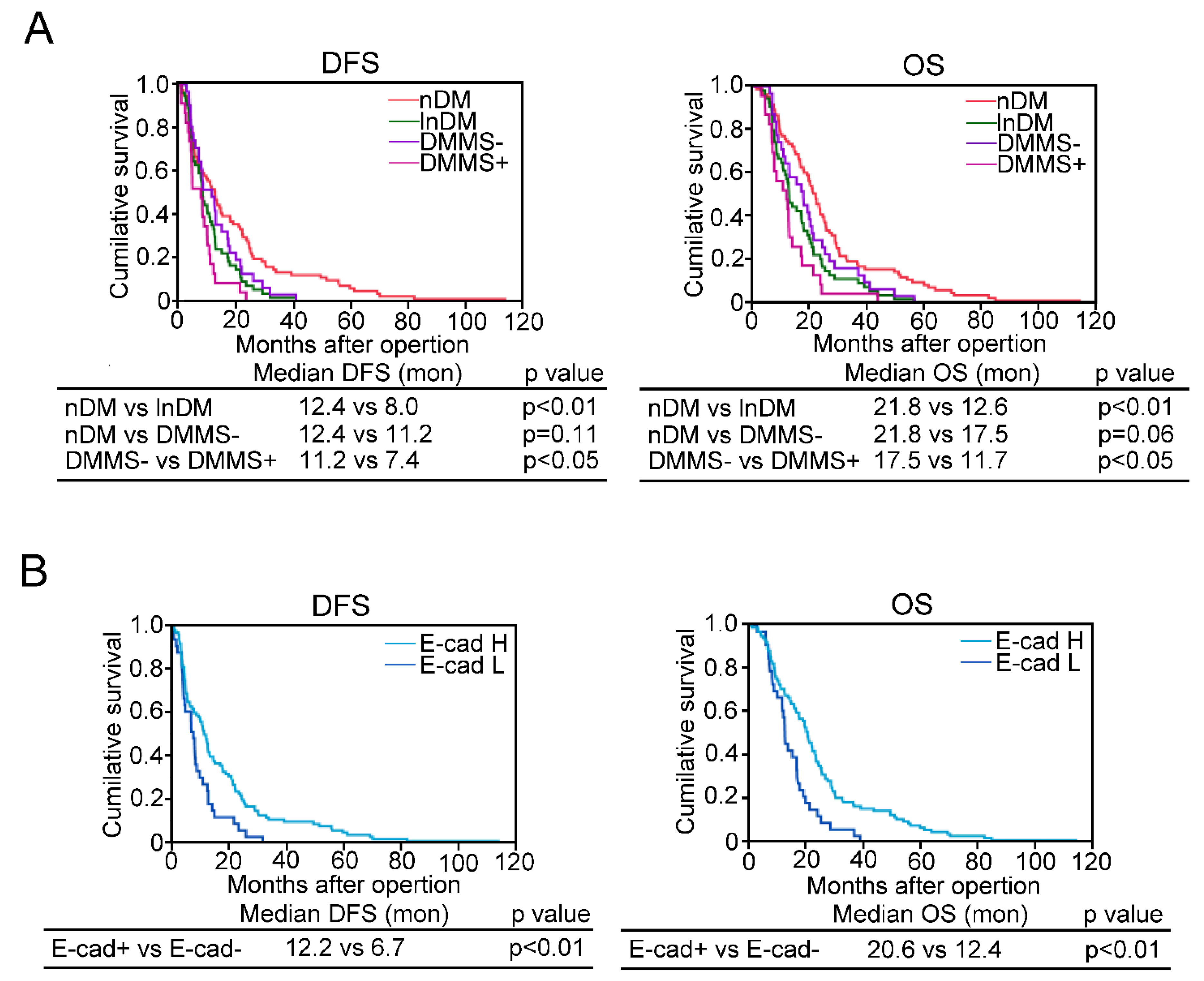
2.6. Diabetes Complicated with Metabolic Syndrome and EMT Are Associated with Short Disease Free Survival or Overall Survival in PDAC
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Animal Study
4.3. Measurement of Blood Glucose, Insulin, Total Cholesterol, and Triglycerides
4.4. Isolation and Culture of Mouse PSCs
4.5. Activation of mPSCs and Cytokine Secretion
4.6. Coculture Experiments with mPSCs and Human PDAC Cell Lines
4.7. Human PDAC Subjects
4.8. Histopathological Assessment
4.9. Immunohistochemical Analysis
4.10. PSC Activation Surrounding PanIN
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Mayor, S. Deaths from pancreatic cancer in Europe continue to increase while rates for other cancers fall. BMJ 2014, 348, g2914. [Google Scholar] [CrossRef] [PubMed]
- Huxley, R.; Ansary-Moghaddam, A.; Berrington de González, A.; Barzi, F.; Woodward, M. Type-II diabetes and pancreatic cancer: A meta-analysis of 36 studies. Br. J. Cancer 2005, 92, 2076–2083. [Google Scholar] [CrossRef] [Green Version]
- Kasuga, M.; Ueki, K.; Tajima, N.; Noda, M.; Ohashi, K.; Noto, H.; Goto, A.; Ogawa, W.; Sakai, R.; Tsugane, S.; et al. Report of the Japan Diabetes Society/Japanese Cancer Association Joint Committee on Diabetes and Cancer. Cancer Sci. 2013, 104, 965–976. [Google Scholar] [CrossRef]
- Saito, T.; Mizukami, H.; Umetsu, S.; Uchida, C.; Inaba, W.; Abe, M.; Takahashi, K.; Kudo, K.; Itabashi, C.; Yagihashi, S.; et al. Worsened outcome in patients with pancreatic ductal carcinoma on long-term diabetes: Association with E-cadherin1 (CDH1) promoter methylation. Sci Rep. 2017, 7, 18056. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schneider, E.; Gross, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grünert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [Green Version]
- Shields, M.A.; Dangi-Garimella, S.; Redig, A.J.; Munshi, H.G. Biochemical role of the collagen-rich tumor microenvironment in pancreatic cancer progression. Biochem. J. 2012, 441, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waghray, M.; Yalamanchili, M.; di Magliano, M.P.; Simeone, D.M. Deciphering the role of stroma in pancreatic cancer. Curr. Opin. Gastroenterol. 2013, 29, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.T.; Grahovac, J.; Kao, S.; et al. Obesity-Induced In-flammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016, 6, 852–869. [Google Scholar] [CrossRef] [Green Version]
- Hong, O.K.; Lee, S.H.; Rhee, M.; Ko, S.H.; Cho, J.H.; Choi, Y.H.; Song, K.H.; Son, H.Y.; Yoon, K.H. Hyperglycemia and hyperinsulinemia have additive effects on activation and proliferation of pancreatic stellate cells: Possible explanation of islet-specific fibrosis in type 2 diabetes mellitus. J. Cell Biochem. 2007, 101, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, Y.; Tashiro, M.; Yamaguchi, T.; Watanabe, S.; Taguchi, M.; Asaumi, H.; Nakamura, H.; Makoto, O. High glucose activates rat pancreatic stellate cells through protein kinase C and p38 mitogen-activated protein kinase pathway. Pancreas 2007, 34, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.H.; Hong, O.K.; Kim, J.W.; Ahn, Y.B.; Song, K.H.; Cha, B.Y.; Son, H.Y.; Kim, M.J.; Jeong, I.K.; Yoon, K.H. High glucose increases extracellular matrix production in pancreatic stellate cells by activating the renin-angiotensin system. J. Cell Biochem. 2006, 98, 343–355. [Google Scholar] [CrossRef]
- Ryu, G.R.; Lee, E.; Chun, H.J.; Yoon, K.H.; Ko, S.H.; Ahn, Y.B.; Songl, K.H. Oxidative stress plays a role in high glucose-induced ac-tivation of pancreatic stellate cells. Biochem. Biophys. Res. Commun. 2013, 439, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Kiss, K.; Baghy, K.; Spisák, S.; Szanyi, S.; Tulassay, Z.; Zalatnai, A.; Löhr, J.M.; Jesenofsky, R.; Kovalszky, I.; Firneisz, G. Chronic hyperglycemia induces trans-differentiation of human pancreatic stellate cells and enhances the malignant molecular communication with human pancreatic cancer cells. PLoS ONE 2015, 10, e0128059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Waldron, R.T.; Su, H.Y.; Moro, A.; Chang, H.H.; Eibl, G.; Ferreri, K.; Kandeel, F.R.; Lugea, A.; Li, L.; et al. Insulin promotes proliferation and fibrosing responses in activated pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G675–G687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Zhang, J.; Yuan, Z.; Zhang, H.; Chong, Y.; Huang, Y.; Wang, J.; Xiong, Q.; Wang, S.; Wu, Q.; et al. PSC-derived Galectin-1 inducing epithelial-mesenchymal transition of pancreatic ductal adenocarcinoma cells by activating the NF-κB pathway. Oncotarget 2017, 8, 86488–86502. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, H.; Kihara, Y.; Taguchi, M.; Yamaguchi, T.; Nakamura, H.; Otsuki, M. Role of TGF-beta1 in the development of pancreatic fibrosis in Otsuka Long-Evans Tokushima Fatty rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G549–G558. [Google Scholar] [CrossRef]
- Mizukami, H.; Inaba, W.; Takahashi, K.; Kamata, K.; Tsuboi, K.; Yagihashi, S. The effects of dipeptidyl-peptidase-IV inhibitor, vildagliptin, on the exocrine pancreas in spontaneously diabetic Goto-Kakizaki rats. Pancreas 2013, 42, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, N.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manigrasso, M.B.; Juranek, J.; Ramasamy, R.; Schmidt, A.M. Unlocking the biology of RAGE in diabetic microvascular complica-tions. Trends Endocrinol. Metab. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehrenbach, H.; Weiskirchen, R.; Kasper, M.; Gressner, A.M. Up-regulated expression of the receptor for advanced glycation end products in cultured rat hepatic stellate cells during transdifferentiation to myofibroblasts. Hepatology 2001, 34, 943–952. [Google Scholar] [CrossRef]
- Miller-Ocuin, J.L.; Liang, X.; Boone, B.A.; Doerfler, W.R.; Singhi, A.D.; Tang, D.; Kang, R.; Lotze, M.T.; Zeh, H.J., 3rd. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology 2019, 8, e1605822. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schürmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.I.; Mittendorfer, B.; Klein, S. Metabolically healthy obesity: Facts and fantasies. J. Clin. Investig. 2019, 129, 3978–3989. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Wadden, T.; Sugerman, H.J. AGA technical review on obesity. Gastroenterology 2002, 123, 882–932. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, H.; Su, S.; Harshfield, G.; Sullivan, J.; Webb, C.; Blumenthal, J.A.; Wang, X.; Huang, Y.; Treiber, F.A.; et al. High-Mobility Group Box-1 Is Associated with Obesity, Inflammation, and Subclinical Cardiovascular Risk Among Young Adults: A Longitudinal Cohort Study. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2776–2784. [Google Scholar] [CrossRef]
- Wang, H.; Qu, H.; Deng, H. Plasma HMGB-1 Levels in Subjects with Obesity and Type 2 Diabetes: A Cross-Sectional Study in China. PLoS ONE 2015, 10, e0136564. [Google Scholar] [CrossRef] [Green Version]
- Arner, P.; Petrus, P.; Esteve, D.; Boulomié, A.; Näslund, E.; Thorell, A.; Gao, H.; Dahlman, I.; Rydén, M. Screening of potential adipokines identifies S100A4 as a marker of pernicious adipose tissue and insulin resistance. Int. J. Obes. 2018, 42, 2047–2056. [Google Scholar] [CrossRef]
- Gaens, K.H.J.; Goossens, G.H.; Niessen, P.M.; van Greevenbroek, M.M.; van der Kallen, C.J.H.; Niessen, H.W.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.M.; Blaak, E.E.; et al. N ε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1199–1208. [Google Scholar] [CrossRef] [Green Version]
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pan-creatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 17, 380–384. [Google Scholar] [CrossRef]
- Hwang, H.J.; Oh, M.S.; Lee, D.W.; Kuh, H.J. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix re-modeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J. Exp. Clin. Cancer Res. 2019, 38, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Kim, S.K.; Khawar, I.A.; Jeong, S.Y.; Chung, S.; Kuh, H.J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Ohuchida, K.; Fei, S.; Zheng, B.; Guan, W.; Feng, H.; Kibe, S.; Ando, Y.; Koikawa, K.; Abe, T.; et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 221. [Google Scholar] [CrossRef]
- Wu, Y.S.; Chung, I.; Wong, W.F.; Masamune, A.; Sim, M.S.; Looi, C.Y. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 296–306. [Google Scholar] [CrossRef]
- Mizuuchi, Y.; Aishima, S.; Ohuchida, K.; Shindo, K.; Fujino, M.; Hattori, M.; Miyazaki, T.; Mizumoto, K.; Tanaka, M.; Oda, Y. An-terior gradient 2 downregulation in a subset of pancreatic ductal adenocarcinoma is a prognostic factor indicative of epitheli-al-mesenchymal transition. Lab. Investig. 2015, 95, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Basso, D.; Brigato, L.; Veronesi, A.; Panozzo, M.P.; Amadori, A.; Plebani, M. The pancreatic cancer cell line MIA PaCa2 produces one or more factors able to induce hyperglycemia in SCID mice. Anticancer Res. 1995, 15, 2585–2588. [Google Scholar] [PubMed]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL-1-induced JAK/ STAT signaling is antago-nized by TGF-beta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2018, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Hara, A.; Kobayashi, H.; Asai, N.; Saito, S.; Higuchi, T.; Kato, K.; Okumura, T.; Bando, Y.K.; Takefuji, M.; Mizutani, Y.; et al. Roles of the mesenchymal stromal/stem cell marker Meflin in cardiac tissue repair and the development of diastolic dysfunction. Circ. Res. 2019, 125, 414–430. [Google Scholar] [CrossRef]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Bento, C.F.; Pereira, P. Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia 2011, 54, 1946–1956. [Google Scholar] [CrossRef] [PubMed]
- van Geenen, E.J.; Smits, M.M.; Schreuder, T.C.; van der Peet, D.L.; Bloemena, E.; Mulder, C.J. Nonalcoholic fatty liver disease is related to nonalcoholic fatty pancreas disease. Pancreas 2010, 39, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Azuma, K.; Nonaka, Y.; Kamada, Y.; Tomoeda, M.; Kishida, M.; Tanemura, M.; Miyoshi, E. Pancreatic fatty degeneration and fibrosis as predisposing factors for the development of pancreatic ductal adenocarcinoma. Pancreas 2014, 43, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Strathdee, G. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin. Cancer Biol. 2002, 12, 373–379. [Google Scholar] [CrossRef]
- Myint, K.M.; Yamamoto, Y.; Doi, T.; Kato, I.; Harashima, A.; Yonekura, H.; Watanabe, T.; Shinohara, H.; Takeuchi, M.; Tsuneyama, K.; et al. RAGE control of diabetic nephropathy in a mouse model: Effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes 2006, 55, 2510–2522. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Mizukami, H.; Osonoi, S.; Takahashi, K.; Ogasawara, S.; Kudo, K.; Sasaki, T.; Yagihashi, S. Beneficial effects of combination therapy of canagliflozin and teneligliptin on diabetic polyneuropathy and β-cell volume density in spontaneously type 2 dia-betic Goto-Kakizaki rats. Metabolism 2020, 107, 154232. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Mizukami, H.; Kamata, K.; Inaba, W.; Kato, N.; Hibi, C.; Yagihashi, S. Amelioration of acute kidney injury in lip-opolysaccharide-induced systemic inflammatory response syndrome by an aldose reductase inhibitor, fidarestat. PLoS ONE 2012, 7, e30134. [Google Scholar] [CrossRef]
- Takahashi, K.; Mizukami, H.; Osonoi, S.; Ogasawara, S.; Hara, Y.; Kudoh, K.; Takeuchi, Y.; Sasaki, T.; Daimon, M.; Yagihashi, S. Inhibitory effects of xanthine oxidase inhibitor, topiroxostat, on development of neuropathy in db/db mice. Neurobiol. Dis. 2021, 155, 105392. [Google Scholar] [CrossRef]
- Seino, Y.; Nanjo, K.; Tajima, N.; Kadowaki, T.; Kashiwagi, A.; Araki, E.; Ito, C.; Inagaki, N.; Iwamoto, Y.; Kasuga, M.; et al. Report of the committee on the classification and diagnostic criteria of diabetes mellitus. J. Diabetes Investig. 2010, 1, 212–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO Classification of Tumors Editional Boards. WHO Classification of Tumours of the Digestive System, 5th ed.; IARC WHO Classification of Tumors No. 10; WHO: Geneva, Switzerland, 2019; pp. 322–940. [Google Scholar]
- Zhang, Y.J.; Bai, D.N.; Du, J.X.; Jin, L.; Ma, J.; Yang, J.L.; Cai, W.B.; Feng, Y.; Xing, C.Y.; Yuan, L.J.; et al. Ultrasound-guided imaging of junctional adhesion molecule-A-targeted microbubbles identifies vulnerable plaque in rabbits. Biomaterials 2016, 94, 20–30. [Google Scholar] [CrossRef]
- Willett, P. Recent trends in hierarchic document clustering: A critical review. Inf. Process Manag. 1988, 24, 577–597. [Google Scholar] [CrossRef]
- Mizukami, H.; Takahashi, K.; Inaba, W.; Tsuboi, K.; Osonoi, S.; Yoshida, T.; Yagihashi, S. Involvement of oxidative stress-induced DNA damage, endoplasmic reticulum stress, and autophagy deficits in the decline of β-cell mass in Japanese type 2 diabetic patients. Diabetes Care 2014, 37, 1966–1974. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | WTHFD | RG | RGHFD | |
|---|---|---|---|---|
| Body weight (g): | ||||
| Start (0 week) | 21.3 ± 0.9 (n = 8) | 21.2± 0.7 (n = 8) | 21.4 ± 1.5 (n = 7) | 21.3 ± 0.8 (n = 8) |
| End (8 weeks) | 24.7 ± 1.4 (n = 7) | 30.6 ± 2.8 * (n = 8) | 25.8 ± 3.8 (n = 7) | 30.3 ± 1.5 † (n = 8) |
| Fasting blood glucose (mmol/L) (8 weeks) | 4.8 ± 0.5 (n = 4) | 5.7 ± 1.0 * (n = 4) | 4.4 ± 0.6 (n = 4) | 5.7 ± 0.6 † (n = 4) |
| Fed blood glucose (mmol/L) (8 weeks) | 10.7 ± 0.8 (n = 4) | 12.9 ± 1.6 ‡ (n = 4) | 10.9 ± 0.7 (n = 7) | 13.5 ± 2.2 † (n = 8) |
| Triglycerides (mmol/L)(8 weeks) | 1.4 ± 0.1 (n = 7) | 3.1 ± 0.3 * (n = 8) | 1.4 ± 0.1 (n = 7) | 3.0 ± 0.2 † (n = 8) |
| Total cholesterol (mmol/L)(8 weeks) | 0.3 ± 0.1 (n = 7) | 0.5 ± 0.1 * (n = 8) | 0.3 ± 0.1 (n = 7) | 0.5 ± 0.1 † (n = 8) |
| Plasma insulin (ng/mL)(8 weeks) | 1.8 ± 0.9 (n = 7) | 2.9 ± 1.4 ‡ (n = 7) | 2.0 ± 1.1 (n = 7) | 2.7 ± 1.7 § (n = 7) |
| nDM | sDM | lnDM | |
|---|---|---|---|
| Number (Male/Female) | 83 (34/49) | 18 (10/8) | 54 (27/27) |
| Age (years) | 66.3 ± 8.1 | 66.6 ± 8.9 | 68.6 ± 8.2 |
| Body mass index | 22.5 ± 3.7 | 21.9 ± 3.0 | 22.7 ± 3.4 |
| Diabetes duration (years) | 1.8 ± 0.9 | 11.3 ± 7.0 * | |
| HbA1c (NGSP, %) | 5.7 ± 0.6 | 7.7 ± 1.5 † | 7.9 ± 1.6 † |
| Triglycerides (mmol/L) | 1.7 ± 1.0 | 1.3 ± 0.5 | 1.5± 1.0 |
| Total cholesterol (mmoL/L) | 5.2 ± 1.5 | 4.9 ± 1.5 | 4.6 ± 1.2 ‡ |
| Tumor size (mm) | 37.9 ± 16.7 | 39.4 ± 16.5 | 36.9 ± 15.5 |
| ly−factor | 2.1 ± 0.8 | 2.1 ± 0.8 | 2.3 ± 0.6 |
| v−factor | 2.0 ± 0.9 | 1.9 ± 0.9 | 2.4 ± 0.7 *† |
| Histological grade: | |||
| wel−mod | 81.9% (68/83) | 88.9% (16/2) | 44.4% (23/54) |
| por | 18.1% (15/83) | 11.1% (2/16) | 55.6% (30/54) *† |
| T stage (UICC 8th): | |||
| 1/2 | 67.5% (56/83) | 66.7% (12/18) | 50.0% (26/32) |
| 3/4 | 32.5% (27/83) | 33.3% (6/18) | 50.0% (26/54) ठ|
| N stage: | |||
| 0 | 28.9% (24/83) | 38.9% (7/18) | 33.3% (18/54) |
| 1/2 | 71.1% (59/83) | 61.1% (11/18) | 66.7% (36/54) |
| DMMS− | DMMS+ | p Value | |
|---|---|---|---|
| Number (male/female) | 31(15/16) | 23 (12/11) | |
| Age (years) | 69.3 ± 8.0 | 68.4 ± 8.5 | 0.714 |
| Body mass index | 21.1 ± 2.8 | 25.1 ± 2.8 | <0.001 |
| Diabetes duration (years) | 11.4± 7.1 | 11.0 ± 7.1 | 0.949 |
| HbA1c (NGSP, %) | 8.3 ± 1.8 | 7.7 ± 0.9 | 0.138 |
| Triglycerides (mmol/L) | 1.0 ± 0.3 | 2.3 ± 1.2 | <0.001 |
| Total cholesterol (mmol/L) | 4.1 ± 0.9 | 5.5 ± 1.1 | <0.001 |
| Tumor size (mm) | 38.0 ± 18.2 | 35.4 ± 10.8 | 0.549 |
| Histological grade: | |||
| wel-mod | 51.6% (16/31) | 34.8% (8/23) | 0.022 |
| por | 48.4% (15/31) | 65.2% (15/23) | |
| T stage (UICC 8th): | |||
| T1−T2 | 58.1% (18/31) | 41.9% (13/23) | <0.01 |
| T3−T4 | 39.1% (9/31) | 60.9% (14/23) | |
| N stage: | |||
| 0 | 38.7% (12/31) | 26.1% (6/23) | 0.049 |
| 1/2 | 61.3% (19/31) | 73.9% (17/23) |
| Variables | Median DFS (Month) | p Value |
|---|---|---|
| BMI: <24 vs. ≧24 | 9.8 vs. 8.0 | 0.472 |
| History of T2D: (−) vs. (+) | 12.4 vs. 8.0 | 0.002 |
| HbA1c (%): <7.0 vs. ≧7.0 | 12.3 vs. 7.7 | 0.008 |
| DMMS−: (−) vs. (+) | 9.8 vs. 11.2 | 0.402 |
| DMMS+: (−) vs. (+) | 12.2 vs. 7.4 | <0.001 |
| TG (mmol/L): <1.7 vs. ≧1.7 | 11.2 vs. 15.8 | 0.458 |
| TC (mmol/L): <5.7 vs. ≧5.7 | 10.7 vs. 7.6 | 0.499 |
| E−cadherin expression: high vs. low | 12.2 vs. 6.7 | <0.001 |
| Vimentin expression: low vs. high | 13.0 vs. 7.2 | 0.011 |
| Variables | Median OS (Month) | p Value |
|---|---|---|
| BMI: <24 vs. ≧24 | 18.9 vs. 16.8 | 0.826 |
| History of T2D: (−) vs. (+) | 21.8 vs. 12.6 | <0.001 |
| HbA1c (%): <7.0 vs. ≧7.0 | 20.5 vs. 12.4 | 0.001 |
| DMMS−: (−) vs. (+) | 19.15 vs. 17.5 | 0.292 |
| DMMS+: (−) vs. (+) | 20.0 vs. 11.7 | <0.001 |
| TG: <1.7 vs. ≧1.7 | 19.5 vs. 12.5 | 0.234 |
| TC: <5.7 vs. ≧5.7 | 19.4 vs. 11.7 | 0.068 |
| E−cadherin expression: high vs. low | 20.6 vs. 12.4 | <0.001 |
| Vimentin expression: low vs. high | 20.0 vs. 11.2 | <0.001 |
| Variables | Hazard Ratio | 95% CI | p Value |
|---|---|---|---|
| History of T2D: (−) vs. (+) | 1.149 | 0.573–2.146 | 0.681 |
| HbA1c (%): <7.0 vs. ≧7.0 | 0.980 | 0.540–1.883 | 0.949 |
| DMMS+ | 1.528 | 0.840–2.725 | 0.163 |
| E−cadherin low | 1.351 | 0.787–2.298 | 0.029 |
| Vimentin high | 1.721 | 1.060–2.742 | 0.273 |
| Variables | Hazard Ratio | 95% CI | p Value |
|---|---|---|---|
| History of T2D: (−) vs. (+) | 1.008 | 0.497–1.898 | 0.979 |
| HbA1c (%): <7.0 vs. ≧7.0 | 1.337 | 0.689–2.733 | 0.398 |
| DMMS+ | 1.609 | 0.900–2.839 | 0.107 |
| E−cadherin low | 1.184 | 1.246–3.089 | 0.004 |
| Vimentin high | 1.984 | 0.677–2.059 | 0.552 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uchida, C.; Mizukami, H.; Hara, Y.; Saito, T.; Umetsu, S.; Igawa, A.; Osonoi, S.; Kudoh, K.; Yamamoto, Y.; Yamamoto, H.; et al. Diabetes in Humans Activates Pancreatic Stellate Cells via RAGE in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 11716. https://doi.org/10.3390/ijms222111716
Uchida C, Mizukami H, Hara Y, Saito T, Umetsu S, Igawa A, Osonoi S, Kudoh K, Yamamoto Y, Yamamoto H, et al. Diabetes in Humans Activates Pancreatic Stellate Cells via RAGE in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2021; 22(21):11716. https://doi.org/10.3390/ijms222111716
Chicago/Turabian StyleUchida, Chiaki, Hiroki Mizukami, Yutaro Hara, Takeshi Saito, Satoko Umetsu, Akiko Igawa, Sho Osonoi, Kazuhiro Kudoh, Yasuhiko Yamamoto, Hiroshi Yamamoto, and et al. 2021. "Diabetes in Humans Activates Pancreatic Stellate Cells via RAGE in Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 22, no. 21: 11716. https://doi.org/10.3390/ijms222111716