Abstract
Background. Scleroderma renal crisis (SRC) is a life-threatening complication of systemic sclerosis (SSc). Autoantibodies (Abs) against endothelial cell antigens have been implicated in SSc and SRC. However, their detailed roles remain poorly defined. Pro-inflammatory cytokine interleukin-6 (IL-6) has been found to be increased in SSc, but its role in SRC is unclear. Here, we aimed to determine how the autoantibodies from patients with SSc and SRC affect IL-6 secretion by micro-vascular endothelial cells (HMECs). Methods. Serum IgG fractions were isolated from either SSc patients with SRC (n = 4) or healthy individuals (n = 4) and then each experiment with HMECs was performed with SSc-IgG from a separate patient or separate healthy control. IL-6 expression and release by HMECs was assessed by quantitative reverse transcription and quantitative PCR (RT-qPCR) and immunoassays, respectively. The mechanisms underlying the production of IL-6 were analyzed by transient HMEC transfections with IL-6 promoter constructs, electrophoretic mobility shift assays, Western blots and flow cytometry. Results. Exposure of HMECs to IgG from SSc patients, but not from healthy controls, resulted in a time- and dose-dependent increase in IL-6 secretion, which was associated with increased AKT, p70S6K, and ERK1/2 signalling, as well as increased c-FOS/AP-1 transcriptional activity. All these effects could be reduced by the blockade of the endothelial PAR-1 receptor and/or c-FOS/AP-1silencing. Conclusions. Autoantibodies against PAR-1 found in patients with SSc and SRC induce IL-6 production by endothelial cells through signalling pathways controlled by the AP-1 transcription factor. These observations offer a greater understanding of adverse endothelial cell responses to autoantibodies present in patients with SRC.
1. Introduction
Scleroderma renal crisis (SRC) is a rare, but potentially life-threatening complication of systemic sclerosis (SSc) [1]. It may occur in up to 13% of all SSc patients [2,3,4,5] with those with early (<5 years) or rapidly progessing diffuse cutaneous SSc (dcSSc) being predominantly affected [6]. Typically, clinical symptoms of SRC are associated with a rise in blood pressure and serum creatinine [6]. In addition, there may be symptoms of microangiopathic hemolysis or thrombosis. As a result, SRC was until recently the leading cause of death in SSc [7]. However, the introduction of angiotensin-converting enzyme inhibitors (ACEi) markedly improved the prognosis in these patients [7]. Nevertheless, renal involvement is still a severe, but nowadays often more silent complication of the disease, and as such determines the patient survival [8]. So far, studies on the mechanisms of SRC are scarce.
The formation of autoantibodies is a cardinal feature of systemic autoimmune diseases [9]. The presence of SSc autoantibodies is associated with kidney fibrosis and vasculopathy during SRC [10]. Some of these antibodies target endothelial antigens and therefore, the vascular endothelium forms a critical interphase between parenchymal cells and the immune system. Indeed, antibody-induced endothelial dysfunction leads to the induction of pro-inflammatory cytokines, perivascular infiltration of immune cells, and vascular remodeling, contributing to SRC [11]. As serum levels of IL-6 are increased in SSc [12], it has been hypothesized that IL-6 may serve as a biomarker to stratify patients for SRC risk and response to therapy [13]. However, for this purpose, it would be essential to elucidate the mechanisms involved in the regulation of IL-6 secretion in SRC.
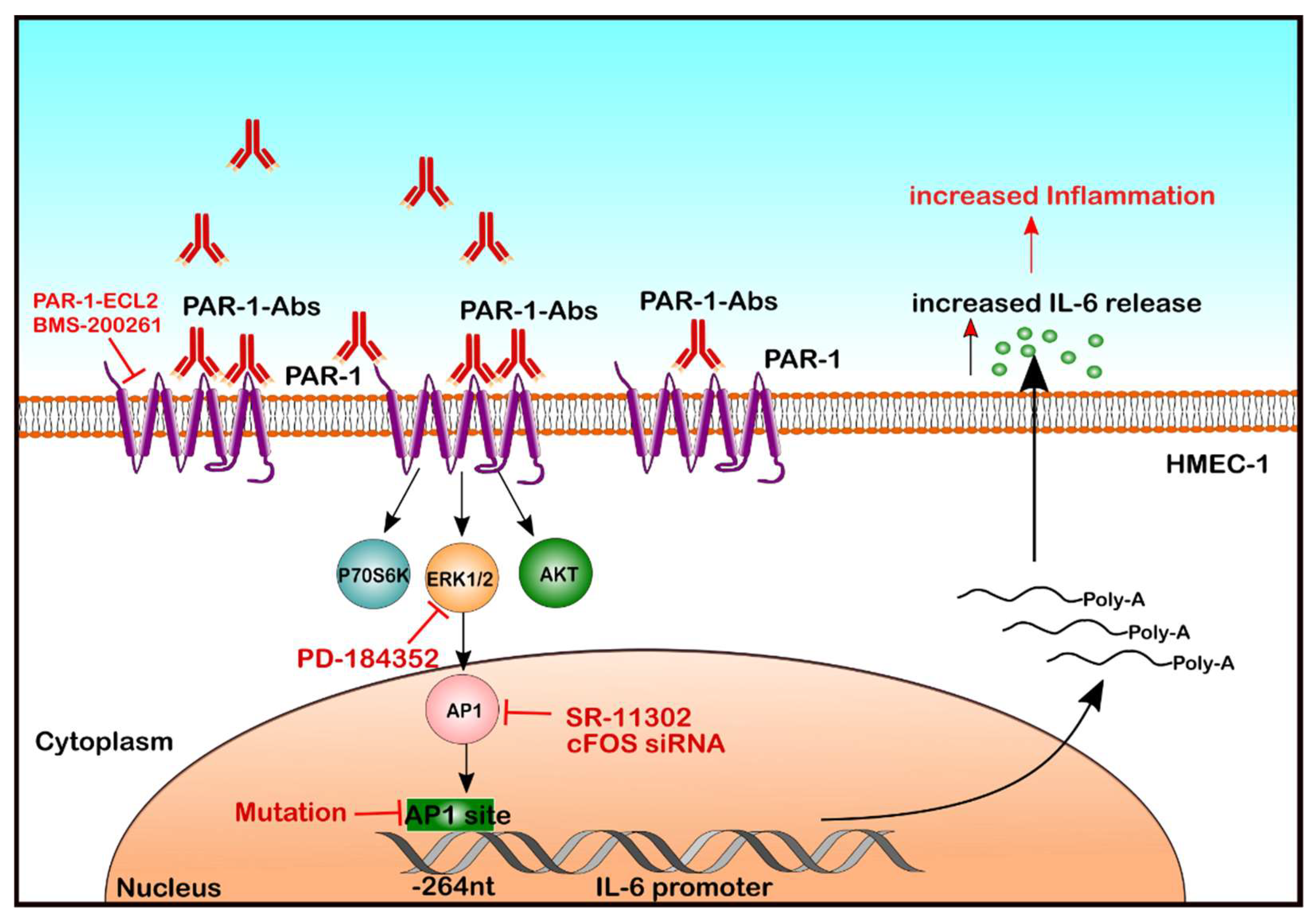
In recent years, a new group of autoantibodies that react with cell surface receptors like G-Protein-coupled receptors (GPCRs), and contribute to the pathological activation of intracellular signalling has been identified [14]. Anti-GPCR autoantibodies against endothelial angiotensin II type 1 receptor (AT1-R) and endothelin-1 type A receptor (ETA-R) are commonly detected in SSc [15], suggesting their involvement in the SSc pathogenesis [11]. In contrast, the role of autoantibodies against Protease-activated receptor-1 (PAR-1), another GPCR, is less clear. It is known that thrombin-activated PAR-1 can contribute to inflammation [16,17,18], which may include IL-6 production [19]. Thus, the objective of the present study was to analyze the mechanisms by which the IgG fraction found in SSc patients modulates signalling through PAR-1 and affects IL-6 secretion by human endothelial cells (ECs) (Figure 1).
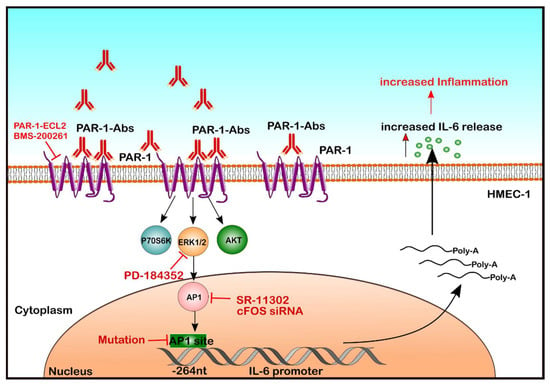
Figure 1.
Molecular mechanisms underlying endothelial IL-6 release in response to autoantibodies from patients with SRC. Here, we demonstrate that autoantibodies present in SRC patients can activate GPCRs on EC and engage a signalling cascade involving PAR-1, PI3K/mTOR/ERK1/2 and AP-1/c-FOS to activate the IL-6 promoter (Position @-264 nucleotides from the IL-6 gene), which leads to IL-6 mRNA expression and IL-6 protein release.
2. Results
2.1. Effect of SSc-IgG on IL-6 Release in HMECs
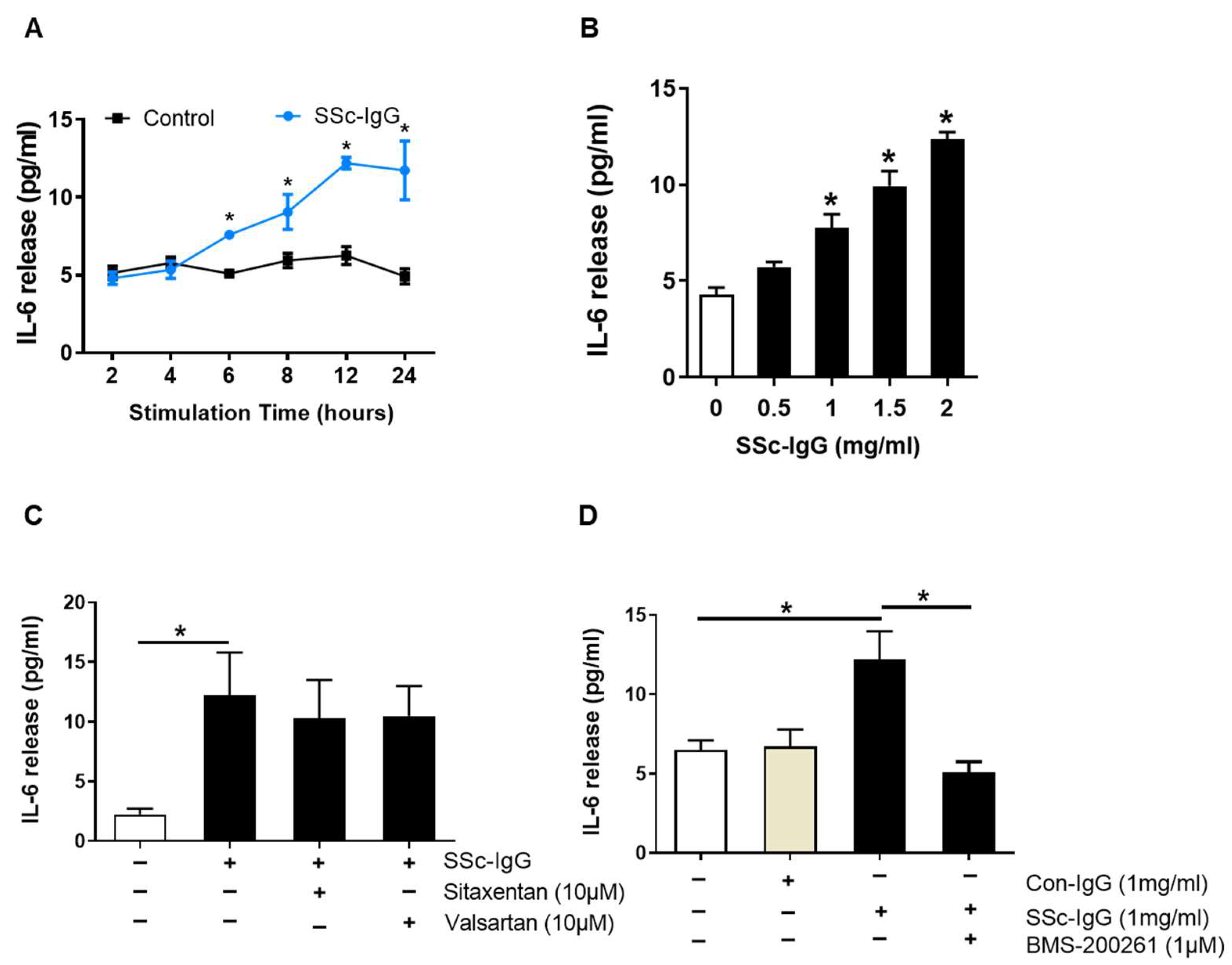
Demograhic data, clinical manifestation, histological findings and antibo6dy levels for each SSc patient with SRC included are shown in Table S1. Exposure of HMECs to the IgG fraction of serum from SSc patients (SSc-IgG) resulted in a time- and dose-dependent increase in IL-6 release. The effect of SSc-IgG at 1 mg/mL became apparent after 6 h of stimulation, peaked at 12 h and plateaued at 24 h (Figure 2A). Such an effect was also dose-dependent and reached maximum at the highest dose of SSc-IgG tested (2 mg/mL; Figure 2B). In contrast, the IgG fraction of serum from patients with rheumatoid arthritis (RA), a chronic systemic disease with immunologically mediated inflammation [20] had no effect on IL-6 production by HMECs (Figure S1).
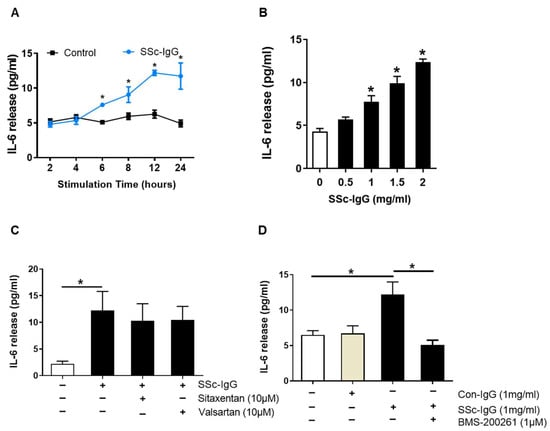
Figure 2.
Effect of SSc-IgG on IL-6 production by human microvascular endothelial cells. HMECs were cultured in the presence or absence of SSc-IgG and assessed for (A) time- and (B) dose-dependent IL-6 secretion (n = 4). The exposure time in (B) was 24 h, while the dose of SSc-IgG in (A) was 1.0 mg/mL. In separate experiments, HMECs were pretreated for 1 h with (C) either valsartan (AT1-R inhibitor) or sitaxentan (ETA-R inhibitor) or (D) BMS-200261 (PAR-1 inhibitor) at doses indicated and then stimulated with SSc-IgG (1 mg/mL) for 24 h (n = 5). To check for specificity of the effect of SSc-IgG, sister HMECs in D were treated with IgG from healthy controls (Con-IgG) at the same dose. For comparison, separate HMECs in D were treated with thrombin. ANOVA mean +/− SEM with * p < 0.05.
Since antibodies against AT1-R and ETA-R have been linked to the pathogenesis of SSc [11], we analyzed whether signalling through AT1-R and ETA-R mediated the effect of SSc-IgG on IL-6 release. To this end, HMECs were preincubated with either AT1-R or ETA-R inhibitors (valsartan or sitaxentan, respectively) at pharmacologically relevant doses [21]. The use of these inhibitors had no effect on IL-6 secretion induced by SSc-IgG (Figure 2C). In contrast, pretreatment of HMECs with BMS-200261, a PAR-1 inhibitor, abolished the stimulating effect of SSc-IgG on IL-6 release (Figure 2D). BMS-200261 was selected after preliminary experiments, which showed that BMS-200261 had alone no effect on IL-6 release (Figure S2). PAR-1 functionality was confirmed by stimulating HMECs with thrombin, which is known to signal through PAR-1 [18]. These experiments showed that thrombin induced IL-6 release by HMECs and that this effect was reduced when HMECs were pretreated with BMS-200261 (Figure S3).
2.2. SSc-lgG-Induced PAR-1 Activation
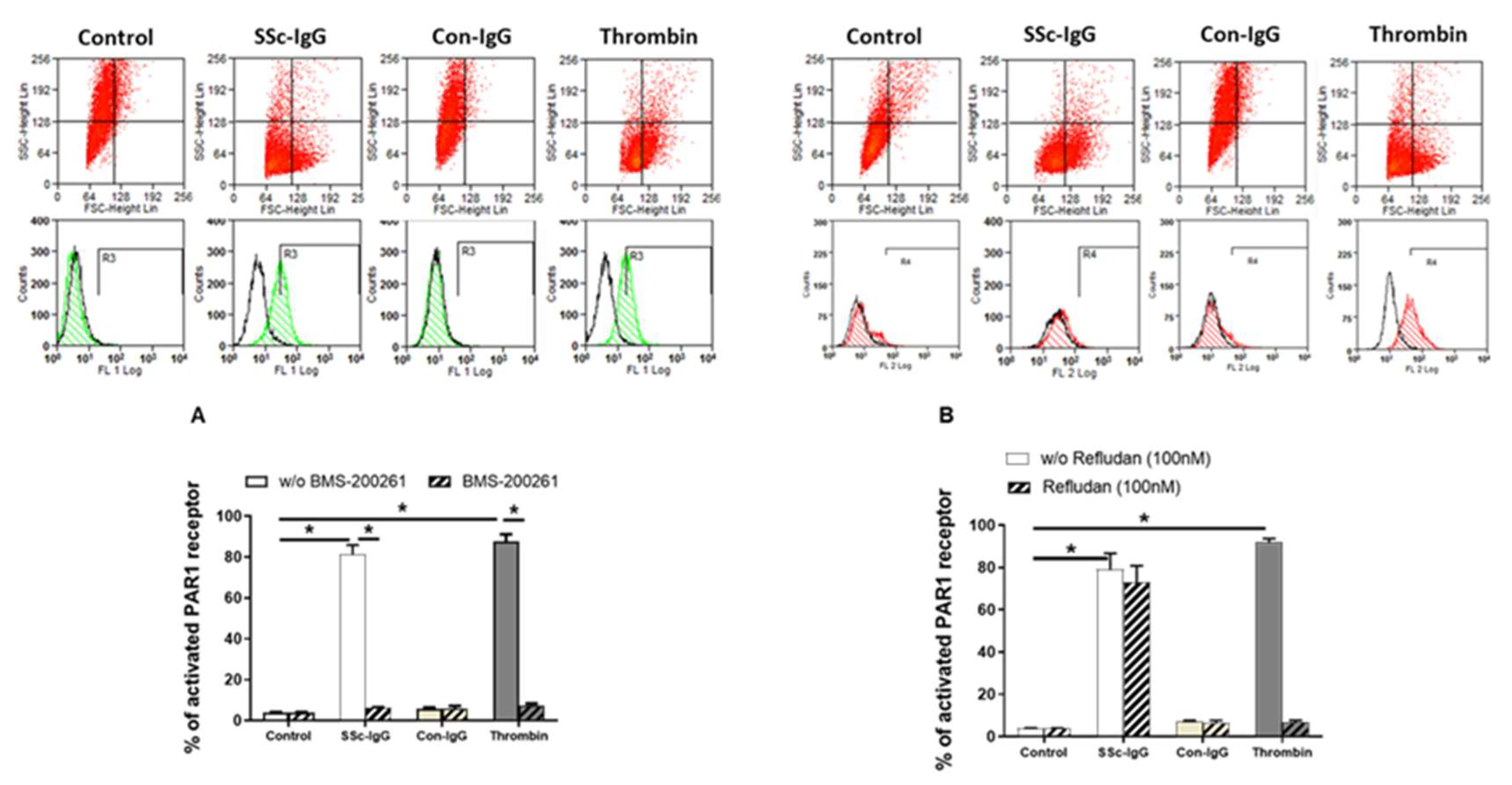
Next, we assessed the expression of PAR-1 on the surface of HMECs. Expression of activated PAR-1 on unstimulated HMECs or on HMECs stimulated with IgG from healthy controls (Con-IgG) was less than 10% of total PAR-1 expression (Figure 3A). In contrast, expression of activated PAR-1 on HMECs treated with SSc-IgG (1 mg/mL) was >80%, which was similar to PAR-1 activation exerted by thrombin (Figure 3A). The effect of SSc-IgG and thrombin was almost completely eliminated by the PAR-1 Inhibitor BMS-200261 (Figure 3A). In contrast, PAR-1 activation by SSc-IgG was not reduced by Refludan, a thrombin inhibitor (Figure 3B), which however abolished PAR-1 activation by thrombin (Figure S2B). These effects suggested that SSc-IgG activates PAR-1 through a mechanism different from that induced by thrombin.
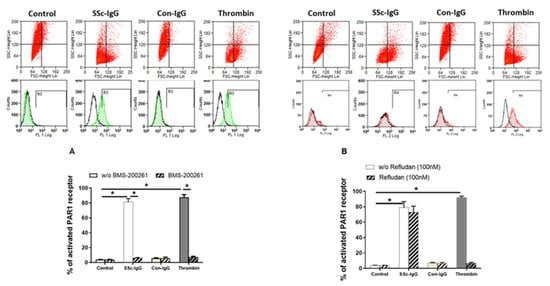
Figure 3.
Effect of SSc-IgG on PAR-1 activation. HMECs were stimulated with SSc-IgG, Con-IgG or thrombin (0.1 U/mL) for 60 min and PAR-1 activation was then assessed by measuring the percentage of cleaved PAR-1 on the cell surface. HMECs were pretreated with either (A) BMS-200261 (PAR-1 inhibitor) or (B) refludan (thrombin inhibitor) for 24 h prior to stumulation. Representative histograms are from flow cytometry (FACS) with HMECs labelled with or without SPAN12 antibody, as described in Methods. The data on bar graphs are means ± SEM from 4 experiments with * p < 0.05.
2.3. PAR-1 Blockade Impairs SSc-IgG-Induced Intracellular Signalling in HMECs
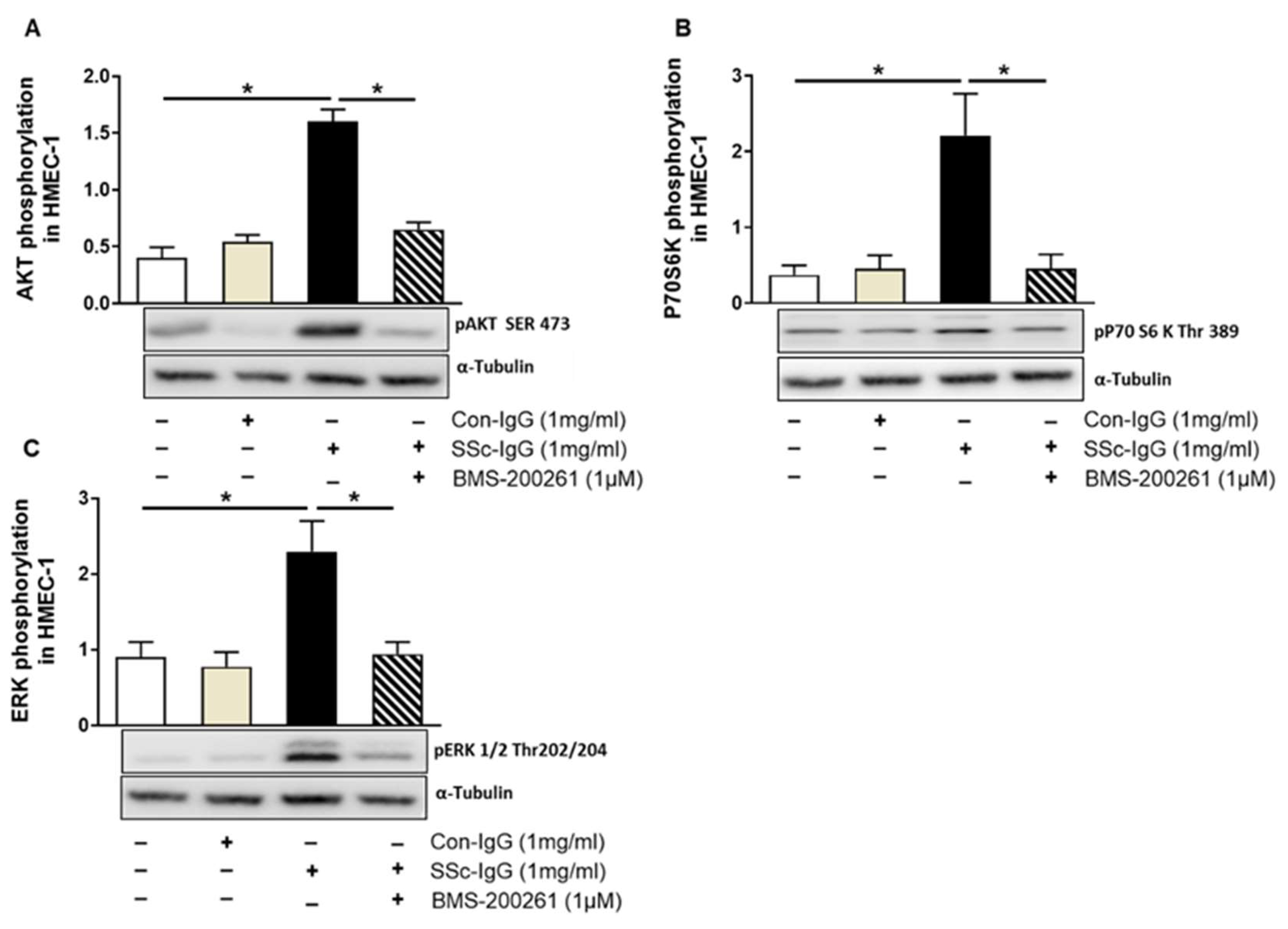
Since thrombin signalling through PAR-1 has previously been shown to involve phosphatidylinositol 3-kinase (PI3K), as mammalian target of rapamycin (mTOR) and the extracellular signal-regulated kinases 1/2 (ERK1/2) [22], we examined whether SSc-IgG could initiate similar signalling pathways in HMECs. Indeed, exposure of HMECs to SSc-IgG at a dose of 1 mg/mL resulted in an approximately 2-fold increase in the expression of phosphorylated pAKT (a central effector of the PI3K pathway upstream of mTORC1), p70S6K (a substrate for the mTOR pathway) and pERK1/2 (Figure 4). Importantly, such effects were not detected in HMECs stimulated with IgG from healthy donors and were almost entirely abolished by preincubation of HMECs with the PAR-1 inhibitor, BMS-200261 (Figure 4).
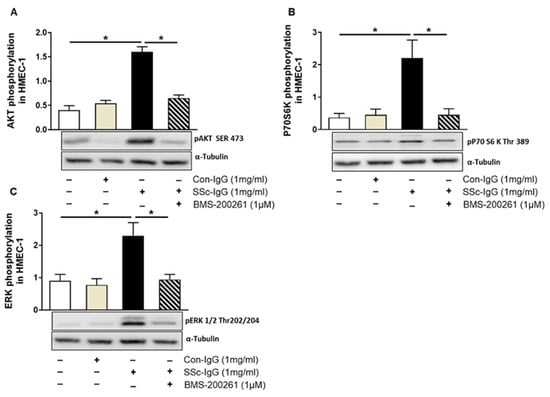
Figure 4.
Effect of SSc-IgG on AKT, 70S6K and ERK1/2 phosphorylation. HMECs were pretreated with or without the PAR-1 inhibitor BMS-200261 for 1 h, followed by stimulation with either SSc-IgG or Con-IgG (both at 1 mg/mL) for 15 min and assessed for the presence of phosphorylated (A) AKT, (B) p70S6K or (C) ERK1/2 (n = 4). The data are expressed as the ratio of target protein expression to α-tubulin expression. ANOVA mean +/− SEM with * p < 0.05.
2.4. SSc-IgG Induces ERK1/2-Mediated IL-6 Release by HMECs
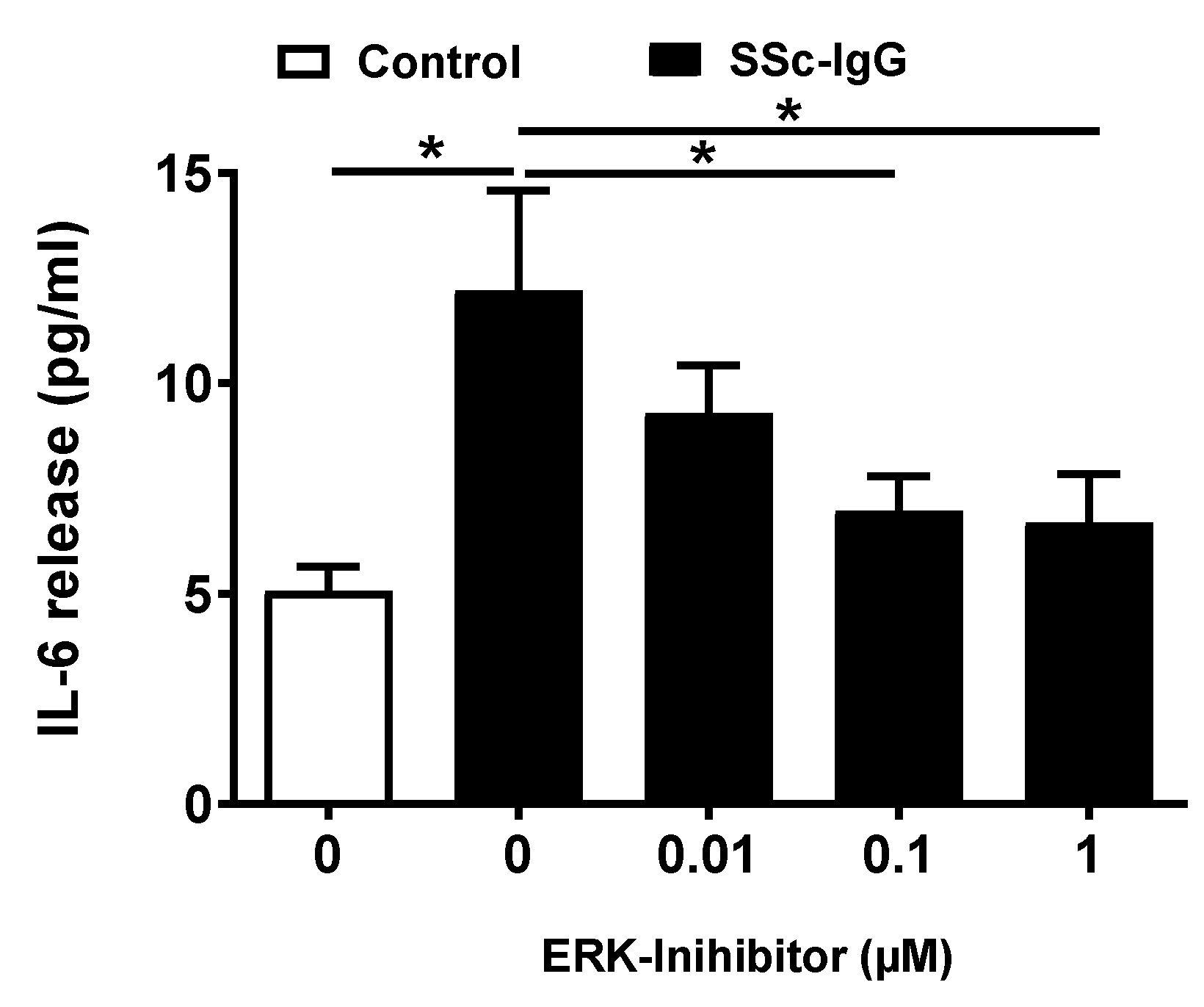
We next analyzed whether activation of ERK1/2 affects the release of IL-6 by HMECs. Pretreatment of HMECs with PD-184352, a specific ERK1/2 inhibitor, resulted in a dose-dependent decrease in IL-6 release, with the significant effect observed at concentrations ≥ 0.1 μM (Figure 5).
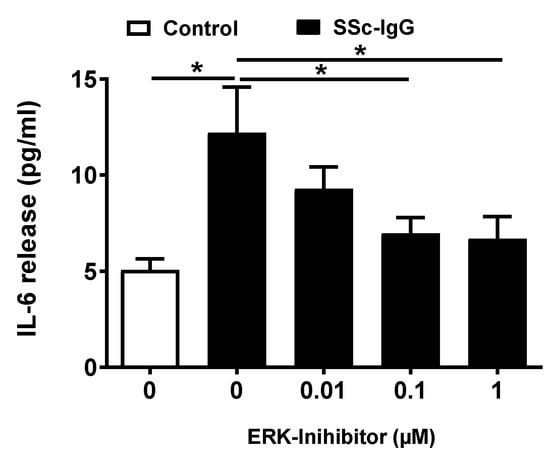
Figure 5.
Effect of ERK1/2 signalling on SSc-IgG-induced IL-6 release by HMECs. Cells were pretreated with or without the specific ERK 1/2 inhibitor PD-184352 for 1 h, then stimulated with SSc-IgG (1 mg/mL) for 24 h after which the level of IL-6 released was measured (n = 8). ANOVA mean +/− SEM with * p < 0.05.
2.5. SSc-IgG Activated the IL-6 Promoter in Microvascular Endothelial Cells
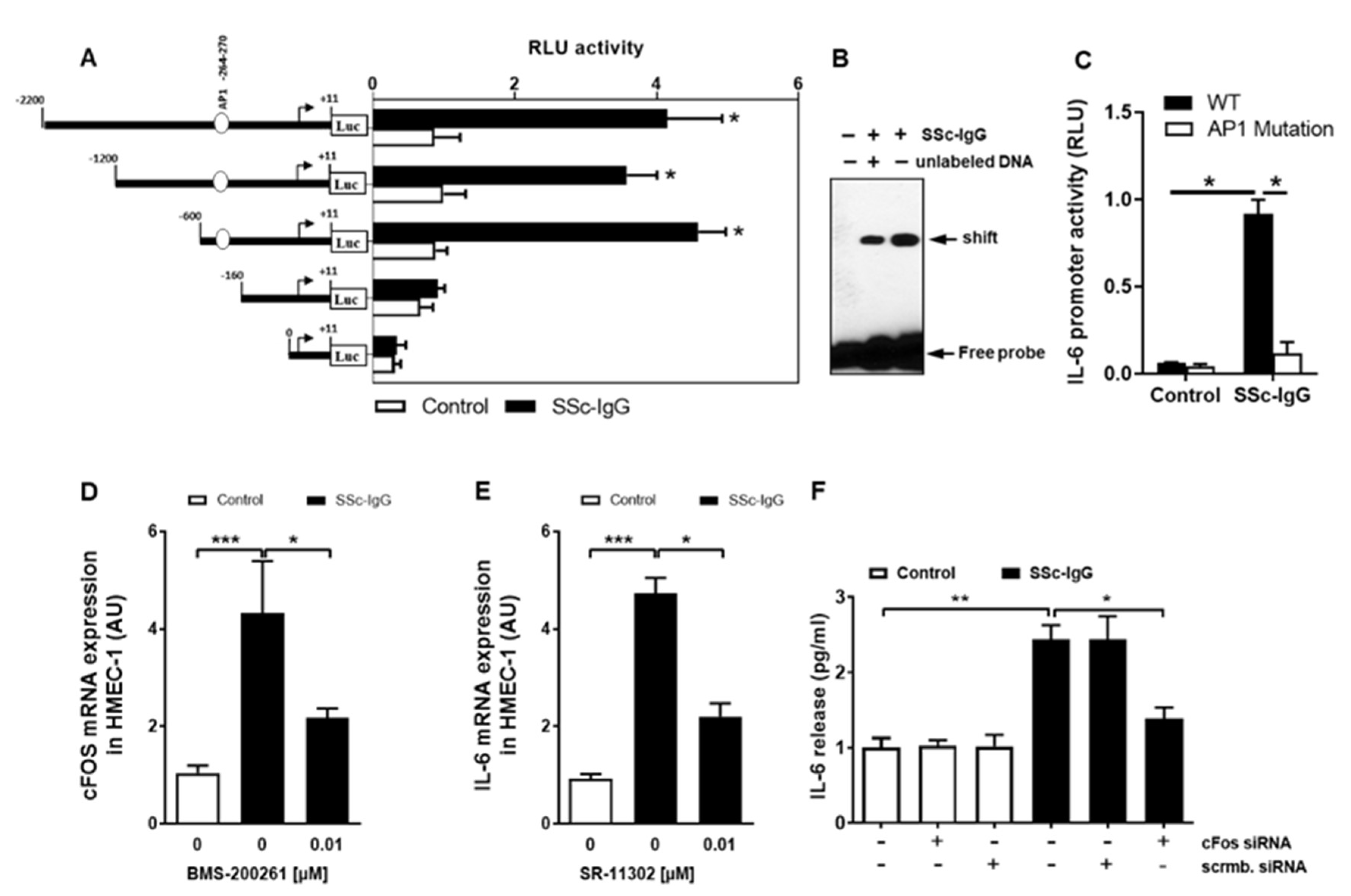
To investigate how SSc-IgG affects the activity of the IL-6 gene promoter, we employed a set of established assays [23,24]. First, HMECs were transiently transfected with IL-6 luciferase reporter gene constructs and stimulated with SSc-IgG (1 mg/mL). Exposure of HMECs to SSc-IgG led to a significant increase in the full-length IL-6 promoter activity (Figure 6A). To identify IL-6 promoter regions responsive to SSc-IgG, progressive deletions of the IL-6 promoter were performed. Truncation of the promoter region spanning positions −160 to +11 abolished the ability of the IL-6 promoter to respond to SSc-IgG (Figure 6A), suggesting that the region contained regulatory elements essential for the IL-6 promoter activity. The in silico analysis pointed to the presence of high-affinity binding sites for c-FOS. To determine whether c-FOS mediated the effect of SSc-IgG on the IL-6 promoter, an electrophoretic mobility shift assay (EMSA) was performed using a biotin-labeled consensus oligonucleotide for c-FOS binding that corresponded to positions −278 to −257 of the IL-6 promoter (Figure 6B). This experiment demonstrated that nuclear extracts from cells stimulated with SSc-IgG formed a DNA–protein complex with the specific oligonucleotide and that the effect was less in the presence of competing unlabeled DNA.
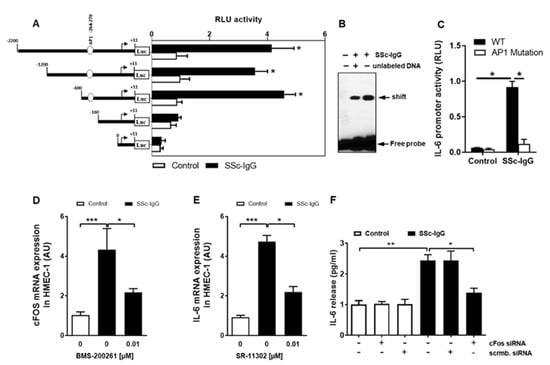
Figure 6.
Characterization of the effect of SSc-IgG on the activity of the IL-6 promoter. HMECs were transiently transfected with IL-6 promoter constructs, stimulated with SSc-IgG (1 mg/mL) for 6 h and analyzed for luciferase activity. (A) Effect of progressive 5′ deletions of the IL-6 promoter on its activity upon stimulation with SSc-IgG (n = 8). (B) EMSA identifying the role of c-FOS in activation of the IL-6 promoter by SSc-IgG (n = 4). Nuclear extracts were obtained from HMECs treated with or without SSc-IgG (1 mg/mL) for 6 h, and EMSA was performed using c-FOS consensus oligonucleotide probes. (C) Effect of mutation in the AP-1 (c-FOS complex with cJUN) binding site within the IL-6 promoter on its activity after stimulation with SSc-IgG. HMECs were transfected with a normal or a mutated construct and stimulated with SSc-IgG (1 mg/mL) for 6 h (n = 4). (D) c-FOS mRNA expression by HMECs stimulated with SSc-IgG (1 mg/mL) for 1 h. Cells were pretreated with or without BMS-200261 for 1 h prior to stimulation (n = 4). (E) IL-6 mRNA expression by HMECs stimulated with SSc-IgG (1 mg/mL) for 3 h. Cells were pretreated with or without SR-11302 (AP-1 inhibitor) for 1 h prior to stimulation (n = 4). (F) IL-6 release by HMECs transfected with either c-FOS siRNA or control scrambled siRNA and stimulated with SSc-IgG (1 mg/mL) for 24 h (n = 4). ANOVA mean +/− SEM with * p < 0.05, ** p < 0.01, and *** p < 0.001.
We then examined the binding specificity of AP-1/c-FOS by analyzing the effect of the mutation in the AP-1 binding site on the activity of the IL-6 promoter. In contrast to cells transfected with a normal construct, transfection with a construct mutated within the AP-1 binding site abolished IL-6 promoter activation after stimulation with SSc-IgG (Figure 6C) indicating that functional binding of active AP-1 plays a crucial role for IL-6 promoter activation and subsequent IL-6 release.
Since c-FOS is part of the AP-1 transcription complex, we also assessed the effect of SSc-IgG on c-FOS mRNA expression. Exposure of HMECs to SSc-IgG resulted in an approximately 5-fold increase in c-FOS mRNA expression and this effect was significantly diminished by pretreatment of the cells with the PAR-1 blocker, BMS-200261 (Figure 6D). The contribution of AP-1/c-FOS signalling to the SSc-IgG-driven IL-6 release was confirmed by the observation that the effect of SSc-IgG was significantly reduced by SR-11302, a specific AP-1 inhibitor (Figure 6E) and by silencing c-FOS with specific siRNA (Figure 6F).
3. Discussion
The main observation of the present study is that antibodies from SSc patients with SRC, but not from healthy individuals, are capable of inducing IL-6 production in endothelial cells, acting through the PAR-1. Intriguingly, this effect of SSc-IgG on PAR-1 seems to resemble that of thrombin [17] and engages downstream signalling through the PI3K/mTOR/ERK1/2 pathway and AP-1/c-FOS transcription factor.
The exact role of IL-6 in SSc and SRC is not well understood. Pro-inflammatory functions of IL-6 in SSc have been recognized in several studies [12,25,26,27]. Surprisingly, however, no obvious signs of inflammation were found in kidney biopsies from SRC patients [28]. To the best of our knowledge, there is only one clinical case report that suggests an association between SRC and IL-6. It documents improvement in creatinine clearance following therapy with tocilizumab, an anti-IL-6 receptor antibody, in a patient with renal failure due to SRC [29]. The lack of apparent inflammatory lesions in the kidneys during SRC may be related to the fact that pro-inflammatory effects of IL-6 are seen only during a specific stage of SSc [30] and serum IL-6 levels decrease gradually with disease duration [31]. Nevertheless, SRC is present in patients with early and rapidly progressing disease with an inflammatory phenotype. Our data indicate a potential role of IL-6-targeted therapies also in SRC. In addition, addressing signalling of SSc-IgG, targeting the AP-1/c-FOS transcription factor complex could be another therapeutic option.
Functional antibodies against AT1-R and ETA-R are detected commonly in SSc with higher levels associated with a more severe course of disease and increased mortality [15]. They were found to induce interleukin-8 (IL-8) and vascular cell adhesion molecule-1 (VCAM-1) expression in HMECs in vitro and in naive C57BL/6J mice in vivo [11]. Importantly, ACE-inhibitors are known to improve endothelial cell function and their introduction into clinical practice led to a significant decrease in SRC-related mortality [28]. Moreover, the blockade of ETA-R signalling was shown to reduce IL-6 expression in a rat model of chronic kidney disease [32]. However, our findings do not support the idea that an increase in IL-6 secretion by HMECs stimulated with SSc-IgG is mediated by functional anti-AT1-R and/or ETA-R Abs, since the use of specific inhibitors of AT1-R and ETA-R didn’t reduce the secretion of IL-6 in vitro. In contrast, we have demonstrated that this effect is mediated rather through PAR-1 signalling (Figure 2). These data show that in addition to AT1-R/ETA-R Abs, PAR-1 Abs contribute to the pathogenesis of SSc, particularly to that of SRC. Thus, PAR-1 inhibition could be a specific therapeutic option in the treatment of SRC.
The role of PAR-1 as a key receptor for thrombin is recognized [33,34]. Here, we demonstrate that SSc-IgG acts in a thrombin-like fashion to activate PAR-1. First, the effect of SSc-IgG on PAR-1 activation can be blocked by the thrombin inhibitor refludan. Second, the signalling pathway evoked by SSc-IgG resembles closely that induced by thrombin on endothelial VEGF expression [35].
Studies on the function of autoantibodies against PAR-1 are rare, with only one report [36] of clinical relevance published. That analysis of 197 women with primary epithelial ovarian cancer revealed a negative correlation between the levels of anti-PAR-1 antibodies and histological grading of the tumor.
This finding is in line with our observation that AKTSer473, which is an effector of the PI3K-pathway involved in endothelial proliferation, survival and angiogenesis [37], can be affected by anti-PAR-1 Abs in SSc-sera. A stimulatory effect of SSc-IgG on AKTSer473 was also found in vascular smooth muscle cells (VSMCs) [38]. In that study antibodies against the platelet-derived growth factor receptor (PDGFR) were found to transactivate the epidermal growth factor receptor (EGFR) leading to phosphorylation of AKT at Ser473 and ERK1/2 activation, resulting in increased protein synthesis, as well as increased expression of pro-fibrotic genes. Another study [39] demonstrated that anti-fibroblast antibodies found in patients with SSc stimulated fibroblasts to produce pro-fibrotic and pro-angiogenic chemokines through a mechanism controlled by ERK1/2. Involvement of ERK1/2 signalling has also been implicated in renal fibrosis associated with systemic lupus erythematosus [40]. In addition, ERK1/2 signalling was demonstrated to underlie IL-6 production in human primary mesangial and proximal tubular cells [41]. Sustained activation of ERK1/2 was also found to be involved in renal endothelial inflammation [42], myofibroblast differentiation, extracellular matrix production [43] and fibrosis [44] in the course of SSc. Interplay between PAR-1 and p70S6K was also demonstrated in animal models of neuronal ischemia [45] and endometriosis [46]. Corroborating our results, phosphorylation of p70S6K was also detected in the renal endothelium of patients with SRC [47].
Organs affected by autoimmune disorders are infiltrated by immune cells that crosstalk with resident stromal cells, e.g., fibroblasts or endothelial cells to augment and perpetuate the inflammatory response and tissue remodeling [48]. In this respect, by connecting metabolic cues with inflammatory cytokines, mTOR may be another promising therapeutic target in autoimmune disorders [49]. Though, in the present manuscript, we have identified a role of IL-6 in SRC independent of inflammation.
As a member of the FOS multigene family, c-Fos heterodimerizes with other components to form the AP-1 transcription factor [50]. Being a crucial integrator of a myriad of extracellular signals, AP-1 switches on/off different transcriptional programs depending on the nature of the signal, the cellular context and/or the combination of the dimer components involved [50]. A prominent role of AP-1 in SSc-associated tissue fibrosis has been elucidated before. In this respect, pathological activation of fibroblasts resulting in the accumulation of extracellular matrix (ECM) was demonstrated to involve the AP-1 components: fos-related antigen-2 (Fra-2) [51] from the Fos multigene family [50], and JunD from the Jun multigene family [52]. Moreover, the AP-1 constituents c-Jun and c-Fos were implicated in generating a pro-fibrotic tissue milieu by inducting a pro-fibrotic and pro-inflammatory secretory phenotype in monocytes [53]. Additionally, our data are in line with animal studies showing severe diffuse SSc in mice transgenic for Fra-2 (Fra-2 tg) [54]. With extensive changes in ECM blood vessels, Fra-2 tg mice are considered a promising preclinical model to study the complex interaction between vasculopathy and fibrosis in SSc [55]. These studies revealed increased perivascular inflammation in the skin of Fra-2 tg mice [55] and increased systemic concentration of IL-6 in Fra-2 tg mice exposed to pulmonary infection. In accordance, the osteoblast-specific overexpression of Fra-2 resulted in increased systemic levels of IL-6 [56].
Among other effects, c-FOS has also been implicated in pro-inflammatory cytokine-induced IL-6 expression in vascular cells [23]. Additionally, more recently, c-FOS has been shown to mediate hyper-inflammatory endothelial IL-6 trans-signalling in response to the SARS-CoV-2 spike protein [57]. Other in vitro studies showed that the natural PAR-1 ligand, thrombin, provoked a time- and concentration-dependent IL-6 release in endothelial cells [58]. The application of hirudin, a natural thrombin inhibitor, resulted in a reduced expression of both PAR-1 and IL-6, as well as decreased accumulation of ECM in the renal interstitium [59].
Although the advent of ACEi reduced mortality in SRC patients, 1-year-outcomes remain poor [28]. This may indicate that novel treatments are required to improve the prognosis in SRC patients [28]. These may be helped by detailed characterization of interactions between SSc-associated autoantibodies and vascular endothelial cells. We here show, that PAR-1 Abs contribute to pathological endothelial processes in SSc, especially in SRC by signalling through PI3K/mTOR/ERK1/2 and AP-1/c-FOS resulting in an IL-6 release, for the first time. Thus, these results are of more decisive clinical significance. Hirudin-analogs [60] and Voraxapar, an orally applicable PAR-1 inhibitor, are already in clinical use [61]. PD-184352, the ERK1/2 inhibitor, was successfully applied in phase 1 studies as orally available compound Cl-1040 [62]. For inhibition of mTOR, Everolimus is an orally administered, accessible approved drug [63]. Tocilizumab, an anti Il-6 receptor antibody, appears to demonstrate beneficial effects in a phase 3 study of systemic sclerosis-associated interstitial lung disease (SSc-ILD) [64]. Small molecule inhibitors of AP-1 have also been proposed as an attractive therapeutic strategy [65,66]. For example, T-5224, a small-molecule inhibitor suppressing the DNA-binding activity of the dimer c-Jun/c-Fos revealed an excellent safety profile in a preclinical animal study of arthritis [67]. Therefore, based on our preclinical results, a therapy applying one or more of the aforementioned accessible therapeutic agents appears reasonable to treat SRC in SSc patients.
4. Materials and Methods
4.1. Materials
Unless stated otherwise, all chemicals were from Sigma-Aldrich (St Louis, MO, USA) and all culture plastics were Falcon from Becton Dickinson (Franklin Lakes, NJ, USA). Cell culture media and buffers were from Thermo Fisher Scientific (Waltham, MA, USA) and fetal calf serum (FCS) was from Invitrogen (Darmstadt, Germany). The source and characteristics of antibodies used is given in Supplemental Material. Thrombin used was α-thrombin from human plasma with concentrations reported in NIH units of activity.
4.2. Endothelial Cell Culture
Human dermal microvascular endothelial cells (HMECs, catalogue no. CRL-3243) were purchased from ATCC® (Manassas, VA, USA) and cultured as described previously [35].
4.3. Patient Samples
SSc patients were those who were treated for SRC at the Charité University Hospital Campus Mitte in Berlin from 1 January 2010, through 31 July 2014. Samples of serum were obtained from all patients and screened for agonistic antibodies targeting AT1-R, ETA-R and PAR-1, as described previously [15]. Control samples were obtained from healthy subjects. Written informed consent to use serum samples for research purposes was obtained from each participant. The study was approved by the institutional review board of the Charité Universitätsmedizin Berlin (AZ/Nr.: EA2/068/07).
4.4. IgG Isolation
IgG was isolated by protein-G sepharose chromatography in 20 mM phosphate buffer pH 7.0 and eluted with 0.1 M glycine/HCl, pH 2.7; then the pH was neutralized with 1 M Tris/HCl, pH 9.0 and eluted IgG was dialyzed against HMEC medium MCDB131. Absorbance of the solution obtained was measured at 280 nm (Emax, Molecular Devices, San Jose, CA, USA) [11].
4.5. IL-6 Protein Measurement
IL-6 protein concentration was measured by immunoassay using a human IL-6 Antibody Pair Kit (Thermo Fisher Scientific, Waltham, MA, USA), as previously described [68]. The limit of detection was 2 pg/mL as indicated in the product specification.
4.6. Gene Expression Analysis
Expression of the IL-6 gene as well as β2M as a housekeeping gene was assessed with reverse transcription and quantitative PCR (RT-qPCR) as detailed elsewhere [23].
4.7. DNA Construct Transfection and Luciferase Assays
Genomic DNA from HMECs was isolated with the Isol-RNA Lysis Reagent (5Prime, VWR, Radnor, PA, USA) and used to generate progressive IL-6 5′-deletion luciferase plasmid constructs (pLuc 2211, pLuc 1211, pLuc 611 and pLuc 171) via PCR amplification using appropriate primer pairs. The Infusion Cloning Kit (Clontech; Takara Bio USA, Mojntain View, CA, USA) was used with the pGL4.10 vector backbone to create the luciferase reporter constructs as described elsewhere [69]. The length of the promoter segments was checked for correctness by restriction digestion and by sequencing (LC Genomics, Houston, TX, USA).
4.8. Transient Transfection and Luciferase Assays
For transient transfection studies cells were seeded into 6-well culture plates at a density that allowed them to reach 70–80% confluence within 24 h. Transfections were then performed in the absence of serum using TurboFect™ transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA) at a ratio of 1 mL/0.33 mg according to the manufacturer’s instructions and as detailed previously [69]. HMECs were transfected with the IL-6 reporter plasmid (0.2 lg/well) and co-transfected with the reference pRL-TK Renilla plasmid (0.02 lg/well). Luciferase activity was assessed with the dual-luciferase reporter assay system (Promega, Mannheim, Germany) with a microplate luminometer (Fluostar Optima; BMG Labtech, Ortenberg, Germany) and normalized to background levels of Renilla luciferase activity from the cotransfected control vectors. Transfection of HMECs with either siRNA for c-FOS (sc-29221) or scrambled siRNA control (sc-37007) was performed with the siRNA Transfection Reagent (all from Santa Cruz Biotechnology, Dallas, TX, USA). The region -599 to -161 of the human IL-6 promoter (GenBank NC_000007.14) was analyzed with Transcription Element Search Software (http://www.cbil.upenn.edu/cgi-bin/tess/tess, accessed on 11 April 2021) to predict and locate potential transcription factor binding sites.
4.9. Nuclear Extracts and Electrophoretic Mobility Shift Assay
Nuclear extracts were prepared using NE-PER Nuclear and Cytoplasmic Extraction Kit and oligonucleotide probes labeled with Biotin 3′-End DNA Labeling Kit (both from ThermoFisher Scientific, Waltham, MA, USA). For electrophoretic mobility shift assay (EMSA) [68], the following AP-1 probe was used (the corresponding region of the IL-6 promoter is given in parenthesis): 5′-CAAAGTGCTGAGTCACTAATAA-3′ (−278 to −257). Each binding mixture (20 µL) contained 5 µg of nuclear extract, 20 fmol labeled double-stranded probe, 1 µg poly-dI/dC and 2 µL 10× reaction buffer and was incubated at room temperature for 30 min. In supershift experiments nuclear extracts were incubated with SSc-IgG for 20 min at room temperature before addition of the biotin-labeled probe. Protein–DNA complexes then were analyzed by electrophoresis in 6% non-denaturing polyacrylamide gels and visualized using the LightShift Chemiluminescent EMSA Kit (ThermoFisher Scientific, Waltham, MA, USA).
4.10. Western Blotting
Preparation of cell extracts was conducted as described before [23], electrophoresed on sodium dodecyl sulfate-polyacrylamide gels and Western blotted using antibodies against pAKT, p70S6K, pERK1/2 and α-Tubulin (Cell Signaling Technology, Frankfurt, Germany) as well as appropriate secondary peroxidase-conjugated IgG (Dianova, Hamburg, Germany). The bands obtained were visualized and analyzed using Enhanced Chemiluminescence Detection System (ThermoFisher Scientific, Waltham, MA, USA) and Image J 1.43 software (National Institutes of Health, Bethesda, MD, USA).
4.11. PAR-1 Expression and Activation
PAR-1 receptor expression and cleavage was assessed by flow cytometry (FACS Aria; Becton Dickinson, Franklin Lakes, NJ, USA). Activation of PAR-1 was monitored using SPAN12, a monoclonal antibody detecting an epitope of PAR-1 that exists only in the uncleaved, i.e., non-active PAR-1. Thus, loss of SPAN12 staining points to PAR-1 receptor activation [70]. To prevent unspecific PAR-1 activation and internalization all experimental steps were performed at 4 °C. HMECs were diluted (3:7) in PBS and incubated with either SPAN12 or a buffer for 10 min, and then fixed with 1% paraformaldehyde and analyzed immediately by flow cytometry.
4.12. Statistics
Statistical analysis was performed using GraphPad Prism 6.05 software (GraphPad Software, San Diego, CA, USA). The data were analyzed by t-test or repeated measures analysis of variance, as appropriate. Results were expressed as means ± SEM. Differences with a p-value <0.05 were considered significant. Asterisks represent p values as follows: * for p < 0.05, ** for p < 0.01, and *** for p < 0.001.
Supplementary Materials
The following data are available online at https://www.mdpi.com/article/10.3390/ijms222111793/s1.
Author Contributions
Conceptualization, R.C., M.S., G.R. and J.W.; Data curation, R.C., M.S., H.Z., D.W., C.L.,; Formal analysis, R.C., M.S., C.L., H.Z., D.W.; Funding acquisition, R.C.; Methodology, R.C., M.S., I.H., H.Z., D.W., C.L.; Project administration, R.C.; Software, R.C. and C.L., M.S.; Supervision, R.C.; Validation, R.C.; Visualization, R.C., M.S., C.L. and J.W.; Writing—original draft, M.S., R.C., J.W., H.H., G.R., K.B.; Writing—review & editing, M.S., R.C., J.W., H.H., G.R., K.B. All authors have read and agreed to the published version of the manuscript.
Funding
The study was further supported by DFG through the CRC 1365 (Nephroprotection;project number 394046635, subproject A03 to R.C.).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Ethics Committee of Charité Universitätsmedizin Berlin (AZ/Nr.: EA2/068/07).
Informed Consent Statement
Informed consent was obtained from all patients involved in the Study.
Data Availability Statement
Original data are available upon Request.
Acknowledgments
We would like to thank Marc Eigen for excellent technical assistance. We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité—Universitätsmedizin Berlin.
Conflicts of Interest
None of the authors have any conflicts of interest related to this manuscript.
References
- Woodworth, T.G.; Suliman, Y.A.; Li, W.; Furst, D.E.; Clements, P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat. Rev. Nephrol. 2016, 12, 678–691. [Google Scholar] [CrossRef]
- Teixeira, L.; Mouthon, L.; Mahr, A.; Berezne, A.; Agard, C.; Mehrenberger, M.; Noel, L.-H.; Trolliet, P.; Frances, C.; Cabane, J.; et al. Mortality and risk factors of scleroderma renal crisis: A French retrospective study of 50 patients. Ann. Rheum. Dis. 2008, 67, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Muangchan, C.; Research, G.C.S.; Baron, M.; Pope, J. The 15% rule in scleroderma: The frequency of severe organ complications in systemic sclerosis—A systematic review. J. Rheumatol. 2013, 40, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Penn, H.; Howie, A.J.; Kingdon, E.J.; Bunn, C.C.; Stratton, R.J.; Black, C.M.; Burns, A.; Denton, C.P. Scleroderma renal crisis: Patient characteristics and long-term outcomes. QJM 2007, 100, 485–494. [Google Scholar] [CrossRef]
- DeMarco, P.J.; Weisman, M.H.; Seibold, J.R.; Furst, D.E.; Wong, W.K.; Hurwitz, E.L.; Mayes, M.; White, B.; Wigley, F.; Barr, W.; et al. Predictors and outcomes of scleroderma renal crisis: The high-dose versus low-dose D-penicillamine in early diffuse systemic sclerosis trial. Arthritis Rheumatol. 2002, 46, 2983–2989. [Google Scholar] [CrossRef]
- Steen, V.D.; Medsger, T.A., Jr.; Osial, T.A., Jr.; Ziegler, G.L.; Shapiro, A.P.; Rodnan, G.P. Factors predicting development of renal involvement in progressive systemic sclerosis. Am. J. Med. 1984, 76, 779–786. [Google Scholar] [CrossRef]
- Steen, V.D.; Medsger, T.A. Changes in causes of death in systemic sclerosis, 1972–2002. Ann. Rheum. Dis. 2007, 66, 940–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyndall, A.J.; Bannert, B.; Vonk, M.; Airo, P.; Cozzi, F.; Carreira, P.E.; Farge Bancel, D.; Allanore, Y.; Mueller-Ladner, U.; Distler, O.; et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis. 2010, 69, 1809–1815. [Google Scholar] [CrossRef] [Green Version]
- Fujii, T. Direct and indirect pathogenic roles of autoantibodies in systemic autoimmune diseases. Allergol. Int. 2014, 63, 515–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrielli, A.; Avvedimento, E.V.; Krieg, T. Scleroderma. N. Engl. J. Med. 2009, 360, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Kill, A.; Tabeling, C.; Undeutsch, R.; Kuhl, A.A.; Gunther, J.; Radic, M.; Becker, M.O.; Heidecke, H.; Worm, M.; Witzenrath, M.; et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res. Ther. 2014, 16, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needleman, B.W.; Wigley, F.M.; Stair, R.W. Interleukin-1, interleukin-2, interleukin-4, interleukin-6, tumor necrosis factor alpha, and interferon-gamma levels in sera from patients with scleroderma. Arthritis Rheum. 1992, 35, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Ong, V.H. Interleukin-6 and related proteins as biomarkers in systemic sclerosis. J. Scleroderma Relat. Disord. 2017, 2 (Suppl. S2), S13–S19. [Google Scholar] [CrossRef] [Green Version]
- Moroncini, G.; Baroni, S.S.; Gabrielli, A. Agonistic antibodies in systemic sclerosis. Immunol. Lett. 2018, 195, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Riemekasten, G.; Philippe, A.; Nather, M.; Slowinski, T.; Muller, D.N.; Heidecke, H.; Matucci-Cerinic, M.; Czirjak, L.; Lukitsch, I.; Becker, M.; et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 530–536. [Google Scholar] [CrossRef]
- Ma, L.; Dorling, A. The roles of thrombin and protease-activated receptors in inflammation. Semin. Immunopathol. 2012, 34, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Coughlin, S.R. How the protease thrombin talks to cells. Proc. Natl. Acad. Sci. USA 1999, 96, 11023–11027. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.C.; Fong, Y.C.; Lai, C.H.; Hung, C.H.; Hsu, H.C.; Lee, T.S.; Yang, R.-S.; Fu, W.-M.; Tang, C.-H. Thrombin-induced IL-6 production in human synovial fibroblasts is mediated by PAR1, phospholipase C, protein kinase C α, c-Src, NF-kappaB and p300 pathway. Mol. Immunol. 2008, 45, 1587–1599. [Google Scholar] [CrossRef]
- Kay, J.; Upchurch, K.S. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatology 2012, 51 (Suppl. S6), vi5–vi9. [Google Scholar] [CrossRef] [Green Version]
- Dragun, D.; Muller, D.N.; Brasen, J.H.; Fritsche, L.; Nieminen-Kelha, M.; Dechend, R.; Kintscher, U.; Rudolph, B.; Hoebeke, J.; Eckert, D.; et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N. Engl. J. Med. 2005, 352, 558–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.H.; Chen, C.H.; Huang, D.Y.; Pai, H.C.; Pan, S.L.; Teng, C.M. Thrombin induces expression of twist and cell motility via the hypoxia-inducible factor-1alpha translational pathway in colorectal cancer cells. J. Cell. Physiol. 2011, 226, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Zickler, D.; Luecht, C.; Willy, K.; Chen, L.; Witowski, J.; Girndt, M.; Fiedler, R.; Storr, M.; Kamhieh-Milz, J.; Schoon, J.; et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol. Dial. Transpl. 2018, 33, 574–585. [Google Scholar] [CrossRef]
- Catar, R.; Witowski, J.; Wagner, P.; Schramm, I.A.; Kawka, E.; Philippe, A.; Dragun, D.; Jorres, A. The proto-oncogene c-Fos transcriptionally regulates VEGF production during peritoneal inflammation. Kidney Int. 2013, 84, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, A.E.; Kronfeld-Harrington, L.B.; Szekanecz, Z.; Cho, M.M.; Haines, G.K.; Harlow, L.A.; Strieter, R.M.; Kunkel, S.L.; Massa, M.C.; Barr, W.G.; et al. In situ expression of cytokines and cellular adhesion molecules in the skin of patients with systemic sclerosis: Their role in early and late disease. Pathobiology 2013, 61, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Sato, S.; Fujimoto, M.; Ihn, H.; Kikuchi, K.; Takehara, K. Serum levels of interleukin 6 (IL-6), oncostatin M, soluble IL-6 receptor, and soluble gp130 in patients with systemic sclerosis. J. Rheumatol. 1998, 25, 308–313. [Google Scholar] [PubMed]
- Scala, E.; Pallotta, S.; Frezzolini, A.; Abeni, D.; Barbieri, C.; Sampogna, F.; De Pità, O.; Puddu, P.; Paganelli, R.; Russo, G. Cytokine and chemokine levels in systemic sclerosis: Relationship with cutaneous and internal organ involvement. Clin. Exp. Immunol. 2004, 138, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.; Ghossein, C.; Steen, V. Scleroderma renal crisis. Presse Med. 2021, 50, 104063. [Google Scholar] [CrossRef]
- Shima, Y.; Kuwahara, Y.; Murota, H.; Kitaba, S.; Kawai, M.; Hirano, T.; Arimitsu, J.; Narazaki, M.; Hagihara, K.; Ogata, A.; et al. The skin of patients with systemic sclerosis softened during the treatment with anti-IL-6 receptor antibody tocilizumab. Rheumatology 2010, 49, 2408–2412. [Google Scholar] [CrossRef] [Green Version]
- Shima, Y. Cytokines Involved in the Pathogenesis of SSc and Problems in the Development of Anti-Cytokine Therapy. Cells 2021, 10, 1104. [Google Scholar] [CrossRef]
- Matsushita, T.; Hasegawa, M.; Hamaguchi, Y.; Takehara, K.; Sato, S. Longitudinal analysis of serum cytokine concentrations in systemic sclerosis: Association of interleukin 12 elevation with spontaneous regression of skin sclerosis. J. Rheumatol. 2006, 33, 275–284. [Google Scholar]
- Lariviere, R.; Gauthier-Bastien, A.; Ung, R.V.; St-Hilaire, J.; Mac-Way, F.; Richard, D.E.; Agharazii, M. Endothelin type A receptor blockade reduces vascular calcification and inflammation in rats with chronic kidney disease. J. Hypertens. 2017, 35, 376–384. [Google Scholar] [CrossRef]
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Vu, T.K.; Wheaton, V.I.; Hung, D.T.; Charo, I.; Coughlin, S.R. Domains specifying thrombin-receptor interaction. Nature 1991, 353, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Catar, R.; Moll, G.; Hosp, I.; Simon, M.; Luecht, C.; Zhao, H.; Wu, D.; Chen, L.; Kamhieh-Milz, J.; Korybalska, K.; et al. Transcriptional Regulation of Thrombin-Induced Endothelial VEGF Induction and Proangiogenic Response. Cells 2021, 10, 910. [Google Scholar] [CrossRef]
- Kreienbring, K.; Franz, A.; Richter, R.; Dragun, D.; Heidecke, H.; Mueller, D.; Mentze, M.; Dechend, R.; Sehouli, J.; Breicu, E.I. The Role of PAR1 Autoantibodies in Patients with Primary Epithelial Ovarian Cancer. Anticancer Res. 2018, 38, 3619–3625. [Google Scholar] [CrossRef]
- Cohen, M.M., Jr. The AKT genes and their roles in various disorders. Am. J. Med. Genet. A 2013, 161, 2931–2937. [Google Scholar] [CrossRef] [PubMed]
- Arts, M.R.; Baron, M.; Chokr, N.; Fritzler, M.J.; Canadian Scleroderma Research Group; Servant, M.J. Systemic sclerosis immunoglobulin induces growth and a pro-fibrotic state in vascular smooth muscle cells through the epidermal growth factor receptor. PLoS ONE 2014, 9, e100035. [Google Scholar] [CrossRef] [Green Version]
- Fineschi, S.; Goffin, L.; Rezzonico, R.; Cozzi, F.; Dayer, J.M.; Meroni, P.L.; Chizzolini, C. Antifibroblast antibodies in systemic sclerosis induce fibroblasts to produce profibrotic chemokines, with partial exploitation of toll-like receptor 4. Arthritis Rheum. 2008, 58, 3913–3923. [Google Scholar] [CrossRef]
- Zhang, C.; Chan, C.Y.; Cheung, K.F.; Chau, M.K.M.; Yap, D.Y.H.; Ma, M.K.M.; Chan, K.W.; Yung, S.; Chan, T.M. Effect of mycophenolate and rapamycin on renal fibrosis in lupus nephritis. Clin. Sci. 2019, 133, 1721–1744. [Google Scholar] [CrossRef]
- Leonard, M.; Ryan, M.P.; Watson, A.J.; Schramek, H.; Healy, E. Role of MAP kinase pathways in mediating IL-6 production in human primary mesangial and proximal tubular cells. Kidney Int. 1999, 56, 1366–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, X.; Yang, J.; Yang, X.; Wei, Y.; Zhu, L.; Gao, D.; Li, M. IL-17A induces endothelial inflammation in systemic sclerosis via the ERK signaling pathway. PLoS ONE 2013, 8, e85032. [Google Scholar]
- Akhmetshina, A.; Dees, C.; Pileckyte, M.; Szucs, G.; Spriewald, B.M.; Zwerina, J.; Distler, O.; Schett, G.; Distler, J.H.W. Rho-associated kinases are crucial for myofibroblast differentiation and production of extracellular matrix in scleroderma fibroblasts. Arthritis Rheum. 2008, 58, 2553–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.; Misra, D.P.; Agarwal, V. Interleukin-17 pathways in systemic sclerosis-associated fibrosis. Rheumatol. Int. 2019, 39, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Yamashita, S.; Hua, Y.; Keep, R.F.; Liu, W.; Xi, G. Thrombin-induced neuronal protection: Role of the mitogen activated protein kinase/ribosomal protein S6 kinase pathway. Brain Res. 2010, 1361, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Takashima, H.; Fumoto, K.; Kajihara, T.; Uchino, S.; Ishihara, O.; Yoshie, M.; Kusama, K.; Tachikawa, E. Possible Role of alpha1-Antitrypsin in Endometriosis-Like Grafts from a Mouse Model of Endometriosis. Reprod. Sci. 2015, 22, 1088–1097. [Google Scholar] [CrossRef]
- Salituri, J.; Patey, N.; Takano, T.; Fiset, P.; Del Rincon, S.; Berkson, L.; Baron, M.; Hudson, M. Mammalian target of rapamycin is activated in the kidneys of patients with scleroderma renal crisis. J. Scleroderma Relat. Dis. 2020, 5, 152–158. [Google Scholar] [CrossRef]
- Buckley, C.D.; Barone, F.; Nayar, S.; Benezech, C.; Caamano, J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu. Rev. Immunol. 2015, 33, 715–745. [Google Scholar] [CrossRef] [PubMed]
- Suto, T.; Karonitsch, T. The immunobiology of mTOR in autoimmunity. J. Autoimmun. 2020, 110, 102373. [Google Scholar] [CrossRef] [PubMed]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 11–23. [Google Scholar] [CrossRef]
- Reich, N.; Maurer, B.; Akhmetshina, A.; Venalis, P.; Dees, C.; Zerr, P.; Palumbo, K.; Zwerina, J.; Nevskaya, T.; Gay, S.; et al. The transcription factor Fra-2 regulates the production of extracellular matrix in systemic sclerosis. Arthritis Rheum. 2010, 62, 280–290. [Google Scholar] [CrossRef]
- Palumbo, K.; Zerr, P.; Tomcik, M.; Vollath, S.; Dees, C.; Akhmetshina, A.; Avouac, J.; Yaniv, M.; Distler, O.; Schett, G.; et al. The transcription factor JunD mediates transforming growth factor {beta}-induced fibroblast activation and fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 1320–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avouac, J.; Palumbo, K.; Tomcik, M.; Zerr, P.; Dees, C.; Horn, A.; Maurer, B.; Akhmetshina, A.; Beyer, C.; Sadowski, A.; et al. Inhibition of activator protein 1 signaling abrogates transforming growth factor beta-mediated activation of fibroblasts and prevents experimental fibrosis. Arthritis Rheum. 2012, 64, 1642–1652. [Google Scholar] [CrossRef]
- Maurer, B.; Reich, N.; Juengel, A.; Kriegsmann, J.; Gay, R.E.; Schett, G.; Michel, B.A.; Gay, S.; Distler, J.H.; Distler, O. Fra-2 transgenic mice as a novel model of pulmonary hypertension associated with systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1382–1387. [Google Scholar] [CrossRef] [Green Version]
- Maurer, B.; Busch, N.; Jungel, A.; Pileckyte, M.; Gay, R.E.; Michel, B.A.; Schett, G.; Gay, S.; Distler, J.; Distler, O. Transcription factor fos-related antigen-2 induces progressive peripheral vasculopathy in mice closely resembling human systemic sclerosis. Circulation 2009, 120, 2367–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Grotsch, B.; Hannemann, N.; Jimenez, M.; Ipseiz, N.; Uluckan, O.; Lin, N.; Schett, G.; Wagner, E.F.; Bozec, A. Fra-2 Expression in Osteoblasts Regulates Systemic Inflammation and Lung Injury through Osteopontin. Mol. Cell. Biol. 2018, 38, e00022-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, T.; Meyer, K.; Geerling, L.; Isbell, T.S.; Hoft, D.F.; Brien, J.; Pinto, A.K.; Ray, R.B.; Ray, R. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020, 16, e1009128. [Google Scholar] [CrossRef]
- Chi, L.; Li, Y.; Stehno-Bittel, L.; Gao, J.; Morrison, D.C.; Stechschulte, D.J.; Dileepan, K.N. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J. Interferon Cytokine Res. 2001, 21, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Fan, B.; Zhao, Q.; Ji, Y.; Liu, P.; Gao, S.; Ren, T.; Dou, Y.; Pei, M.; Yang, H. Hirudin Ameliorates Renal Interstitial Fibrosis via Regulating TGF-beta1/Smad and NF-kappaB Signaling in UUO Rat Model. Evid. Based Complement. Altern. Med. 2020, 2020, 7291075. [Google Scholar] [CrossRef]
- Nowak, G. Pharmacology of recombinant hirudin. Semin. Thromb. Hemost. 2002, 28, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.A.; Braunwald, E.; Bonaca, M.P.; Ameriso, S.F.; Dalby, A.J.; Fish, M.P.; Fox, K.A.; Lipka, L.J.; Liu, X.; Nicolau, J.C.; et al. Vorapaxar in the secondary prevention of atherothrombotic events. N. Engl. J. Med. 2012, 366, 1404–1413. [Google Scholar] [CrossRef]
- Allen, L.F.; Sebolt-Leopold, J.; Meyer, M.B. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin. Oncol. 2003, 30, 105–116. [Google Scholar] [CrossRef]
- Hasskarl, J. Everolimus. Recent Results Cancer Res. 2018, 211, 101–123. [Google Scholar]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y.; et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef]
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. 2018, 128, 366–375. [Google Scholar] [CrossRef]
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small molecule inhibitors targeting activator protein 1 (AP-1). J. Med. Chem. 2014, 57, 6930–6948. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, Y.; Morimoto, K.; Yamamoto, T.; Chaki, H.; Hashiramoto, A.; Narita, H.; Hirono, S.; Shiozawa, S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol. 2008, 26, 817–823. [Google Scholar] [CrossRef]
- Catar, R.; Witowski, J.; Zhu, N.; Lucht, C.; Soria, A.; Fernandez, J.U.; Chen, L.; Jones, S.A.; Fielding, C.A.; Rudolf, A.; et al. IL-6 Trans-Signaling Links Inflammation with Angiogenesis in the Peritoneal Membrane. J. Am. Soc. Nephrol. 2017, 28, 1188–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Schuff-Werner, P.; Steiner, M. Thrombin/thrombin receptor (PAR-1)-mediated induction of IL-8 and VEGF expression in prostate cancer cells. Biochem. Biophys. Res. Commun. 2006, 343, 183–189. [Google Scholar] [CrossRef]
- Brass, L.F.; Pizarro, S.; Ahuja, M.; Belmonte, E.; Blanchard, N.; Stadel, J.M.; Hoxie, J.A. Changes in the structure and function of the human thrombin receptor during receptor activation, internalization, and recycling. J. Biol. Chem. 1994, 269, 2943–2952. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).