
Contribution of Dysregulated DNA Methylation to Autoimmunity

 and
and
Abstract
:1. Introduction
1.1. Overview of DNA Methylation in Immune Cells
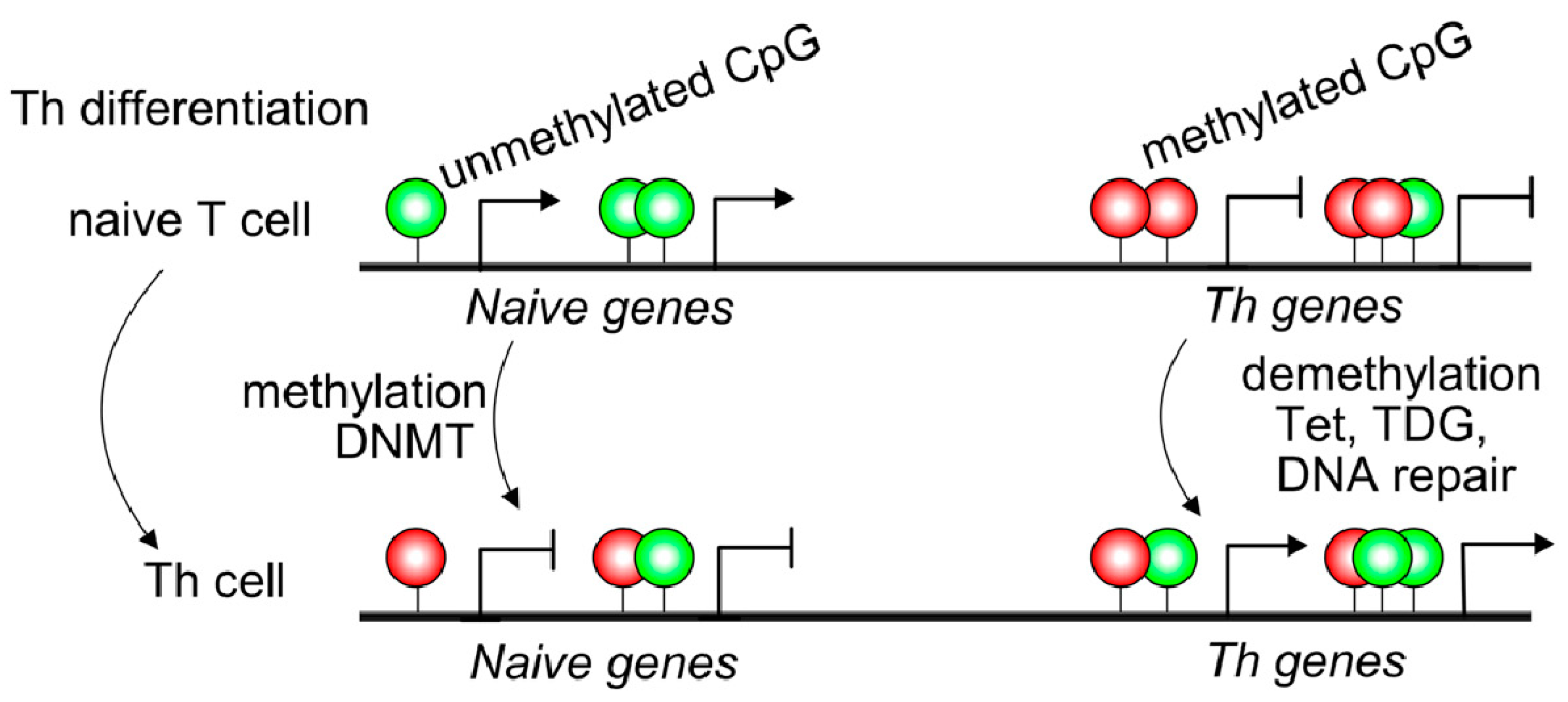
1.2. Contribution of Demethylases to T and B Cell Differentiation
2. Dysregulated Epigenetic Modifications in Autoimmune Diseases
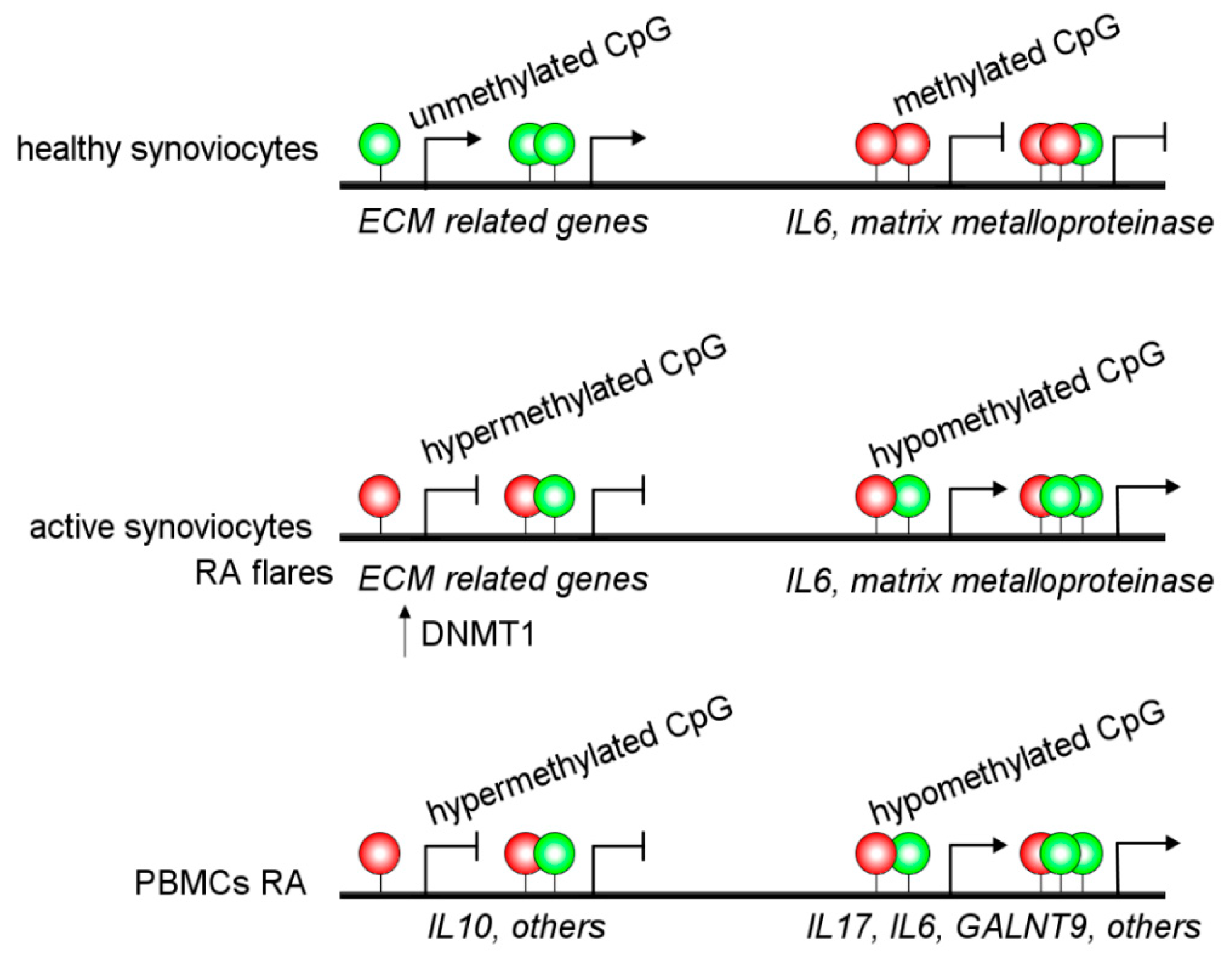
2.1. Rheumatoid Arthritis (RA)
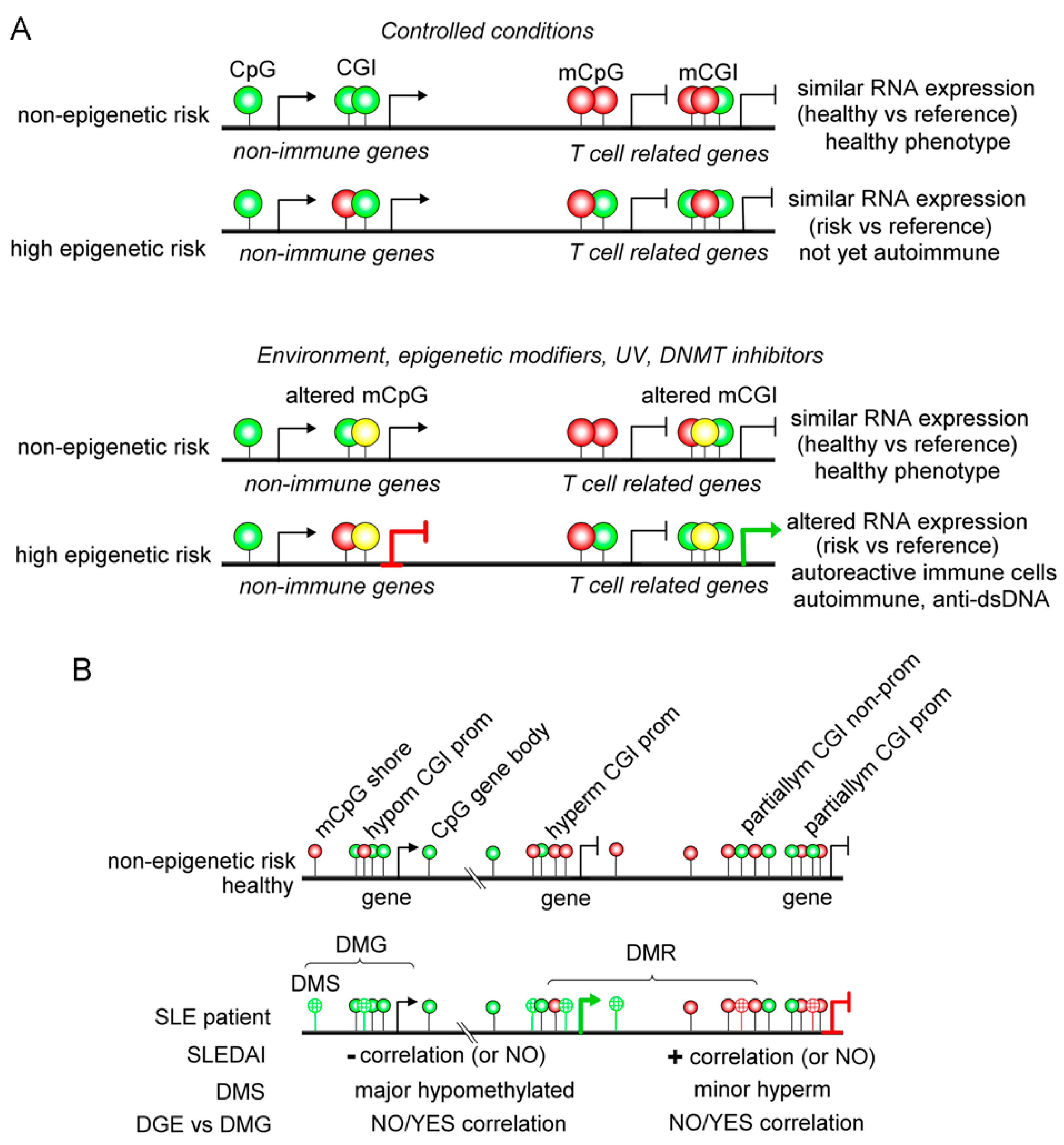
2.2. Systemic Lupus Erythematosus (SLE)
2.3. Sjogren’s Syndrome (SS)
2.4. T Cell-Mediated Diseases: Multiple Sclerosis and Psoriasis
2.4.1. Multiple Sclerosis (MS)
2.4.2. DNA Methylation in Psoriasis
3. Female and X-Linked DNA Methylation
4. Advantages and Disadvantages of Epigenetic Therapy in Autoimmunity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddington, C.H. The epigenotype. Endeavour 1942, 1, 18–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Chen, Y.; Zhu, H.; Zhao, M.; Lu, Q. The pathogenic role of dysregulated epigenetic modifications in autoimmune diseases. Front. Immunol. 2019, 10, 2305. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Lu, Q. Genetic and epigenetic influences on the loss of tolerance in autoimmunity. Cell. Mol. Immunol. 2018, 15, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Floreani, A.; Leung, P.S.C.; Gershwin, M.E. Environmental Basis of Autoimmunity. Clin. Rev. Allergy Immunol. 2016, 50, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E. Epigenetics lessons from twins: Prospects for autoimmune disease. Clin. Rev. Allergy Immunol. 2010, 39, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Liberman, N.; Wang, S.Y.; Greer, E.L. Transgenerational epigenetic inheritance: From phenomena to molecular mechanisms. Curr. Opin. Neurobiol. 2019, 59, 189–206. [Google Scholar] [CrossRef]
- Zouali, M. DNA methylation signatures of autoimmune diseases in human B lymphocytes. Clin. Immunol. 2020, 222, 108622. [Google Scholar] [CrossRef]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Mamrut, S.; Avidan, N.; Staun-Ram, E.; Ginzburg, E.; Truffault, F.; Berrih-Aknin, S.; Miller, A. Integrative analysis of methylome and transcriptome in human blood identifies extensive sex- and immune cell-specific differentially methylated regions. Epigenetics 2015, 10, 943–957. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, B.; Zhang, Q.; Cao, X. The function and regulation of TET2 in innate immunity and inflammation. Protein Cell 2021, 12, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300. [Google Scholar] [CrossRef] [Green Version]
- Schoeler, K.; Aufschnaiter, A.; Messner, S.; Derudder, E.; Herzog, S.; Villunger, A.; Rajewsky, K.; Labi, V. TET enzymes control antibody production and shape the mutational landscape in germinal centre B cells. FEBS J. 2019, 286, 3566–3581. [Google Scholar] [CrossRef] [PubMed]
- Wittkopp, P.J.; Kalay, G. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2011, 13, 59–69. [Google Scholar] [CrossRef]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, L.; Sun, X.; Deng, T.; Huang, G.; Li, X.; Xie, Z.; Zhou, Z. Role of Tet2 in Regulating Adaptive and Innate Immunity. Front. Cell Dev. Biol. 2021, 9, 665897. [Google Scholar] [CrossRef] [PubMed]
- Minichiello, E.; Semerano, L.; Boissier, M.-C. Time trends in the incidence, prevalence, and severity of rheumatoid arthritis: A systematic literature review. Jt. Bone Spine 2016, 83, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Bang, S.-Y.; Lee, H.-S.; Bae, S.-C. Update on the genetic architecture of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Aho, K.; Koskenvuo, M.; Tuominen, J.; Kaprio, J. Occurrence of rheumatoid arthritis in a nationwide series of twins. J. Rheumatol. 1986, 13, 899–902. [Google Scholar] [PubMed]
- Silman, A.; MacGregor, A.; Thomson, W.; Holligan, S.; Carthy, D.; Farhan, A.; Ollier, W. Twin concordance rates for rheumatoid arthritis: Results from a nationwide study. Rheumatology 1993, 32, 903–907. [Google Scholar] [CrossRef]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Karouzakis, E.; Gay, R.E.; Michel, B.A.; Gay, S.; Neidhart, M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2009, 60, 3613–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Whitaker, J.W.; Boyle, D.L.; Wang, W.; Firestein, G.S. DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 110–117. [Google Scholar] [CrossRef]
- Whitaker, J.W.; Shoemaker, R.; Boyle, D.L.; Hillman, J.; Anderson, D.; Wang, W.; Firestein, G.S. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med. 2013, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- de la Rica, L.; Urquiza, J.M.; Gómez-Cabrero, D.; Islam, A.B.M.M.K.; López-Bigas, N.; Tegnér, J.; Toes, R.E.M.; Ballestar, E. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J. Autoimmun. 2013, 41, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Aryee, M.J.; Padyukov, L.; Fallin, M.D.; Hesselberg, E.; Runarsson, A.; Reinius, L.; Acevedo, N.; Taub, M.; Ronninger, M.; et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013, 31, 142–147. [Google Scholar] [CrossRef]
- Ai, R.; Laragione, T.; Hammaker, D.; Boyle, D.L.; Wildberg, A.; Maeshima, K.; Palescandolo, E.; Krishna, V.; Pocalyko, D.; Whitaker, J.W. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat. Commun. 2018, 9, 1921. [Google Scholar] [CrossRef]
- Coit, P.; Dozmorov, M.G.; Merrill, J.T.; McCune, W.J.; Maksimowicz-McKinnon, K.; Wren, J.D.; Sawalha, A.H. Epigenetic Reprogramming in Naive CD4+ T Cells Favoring T Cell Activation and Non-Th1 Effector T Cell Immune Response as an Early Event in Lupus Flares. Arthritis Rheumatol. 2016, 68, 2200–2209. [Google Scholar] [CrossRef] [PubMed]
- Coit, P.; Ortiz-Fernandez, L.; Lewis, E.E.; McCune, W.J.; Maksimowicz-McKinnon, K.; Sawalha, A.H. A longitudinal and transancestral analysis of DNA methylation patterns and disease activity in lupus patients. JCI Insight 2020, 5, e143654. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Pu, W.; Guo, S.; Jin, L.; He, D.; Wang, J. Genome-Wide DNA Methylation Profiles Reveal Common Epigenetic Patterns of Interferon-Related Genes in Multiple Autoimmune Diseases. Front. Genet. 2019, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Almlöf, J.C.; Leonard, D.; Sjöwall, C.; Syvänen, A.-C.; Rönnblom, L.; Sandling, J.K.; Nordmark, G. Shared and Unique Patterns of DNA Methylation in Systemic Lupus Erythematosus and Primary Sjögren’s Syndrome. Front. Immunol. 2019, 10, 1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altorok, N.; Coit, P.; Hughes, T.; Koelsch, K.A.; Stone, D.U.; Rasmussen, A.; Radfar, L.; Scofield, R.H.; Sivils, K.L.; Farris, A.D. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjögren’s syndrome. Arthritis Rheumatol. 2014, 66, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhrmann, S.; Ewing, E.; Piket, E.; Kular, L.; Cetrulo Lorenzi, J.C.; Fernandes, S.J.; Morikawa, H.; Aeinehband, S.; Sayols-Baixeras, S.; Aslibekyan, S. Hypermethylation of MIR21 in CD4+ T cells from patients with relapsing-remitting multiple sclerosis associates with lower miRNA-21 levels and concomitant up-regulation of its target genes. Mult. Scler. J. 2018, 24, 1288–1300. [Google Scholar] [CrossRef] [Green Version]
- Ong, L.T.; Schibeci, S.D.; Fewings, N.L.; Booth, D.R.; Parnell, G.P. Age-dependent VDR peak DNA methylation as a mechanism for latitude-dependent multiple sclerosis risk. Epigenetics Chromatin 2021, 14, 9. [Google Scholar] [CrossRef]
- Gervin, K.; Vigeland, M.D.; Mattingsdal, M.; Hammerø, M.; Nygård, H.; Olsen, A.O.; Brandt, I.; Harris, J.R.; Undlien, D.E.; Lyle, R. DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: Identification of epigenetically dysregulated genes. PLoS Genet. 2012, 8, e1002454. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Yao, T.; Fang, M.; Zheng, X.; Chen, G.; Li, M.; Wang, D.; Li, X.; Ma, H.; Wang, X.; et al. Genomic DNA methylation in HLA-Cw*0602 carriers and non-carriers of psoriasis. J. Dermatol. Sci. 2020, 99, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Fang, T.-J.; Ou, T.-T.; Wu, C.-C.; Li, R.-N.; Lin, Y.-C.; Lin, C.-H.; Tsai, W.-C.; Liu, H.-W.; Yen, J.-H. Global DNA methylation, DNMT1, and MBD2 in patients with rheumatoid arthritis. Immunol. Lett. 2011, 135, 96–99. [Google Scholar] [CrossRef]
- Fu, L.-H.; Ma, C.-L.; Cong, B.; Li, S.-J.; Chen, H.-Y.; Zhang, J.-G. Hypomethylation of proximal CpG motif of interleukin-10 promoter regulates its expression in human rheumatoid arthritis. Acta Pharmacol. Sin. 2011, 32, 1373–1380. [Google Scholar] [CrossRef] [Green Version]
- Ishida, K.; Kobayashi, T.; Ito, S.; Komatsu, Y.; Yokoyama, T.; Okada, M.; Abe, A.; Murasawa, A.; Yoshie, H. Interleukin-6 gene promoter methylation in rheumatoid arthritis and chronic periodontitis. J. Periodontol. 2012, 83, 917–925. [Google Scholar] [CrossRef]
- Liu, H.-W.; Lin, H.-L.; Yen, J.-H.; Tsai, W.-C.; Chiou, S.-S.; Chang, J.-G.; Ou, T.-T.; Wu, C.-C.; Chao, N.-C. Demethylation within the proximal promoter region of human estrogen receptor alpha gene correlates with its enhanced expression: Implications for female bias in lupus. Mol. Immunol. 2014, 61, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Nile, C.J.; Read, R.C.; Akil, M.; Duff, G.W.; Wilson, A.G. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 2008, 58, 2686–2693. [Google Scholar] [CrossRef] [PubMed]
- Glossop, J.R.; Emes, R.D.; Nixon, N.B.; Haworth, K.E.; Packham, J.C. Genome-wide DNA methylation profiling in rheumatoid arthritis identifies disease-associated methylation changes that are distinct to individual T-and B-lymphocyte populations. Epigenetics 2014, 9, 1228–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niimoto, T.; Nakasa, T.; Ishikawa, M.; Okuhara, A.; Izumi, B.; Deie, M.; Suzuki, O.; Adachi, N.; Ochi, M. MicroRNA-146a expresses in interleukin-17 producing T cells in rheumatoid arthritis patients. BMC Musculoskelet. Disord. 2010, 11, 209. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Haupt, S.; Kreuzer, J.T.; Hammitzsch, A.; Proft, F.; Neumann, C.; Leipe, J.; Witt, M.; Schulze-Koops, H.; Skapenko, A. Decreased expression of miR-146a and miR-155 contributes to an abnormal Treg phenotype in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1265–1274. [Google Scholar] [CrossRef]
- Yang, G.; Wu, D.; Zeng, G.; Jiang, O.; Yuan, P.; Huang, S.; Zhu, J.; Tian, J.; Weng, Y.; Rao, Z. Correlation between miR-126 expression and DNA hypomethylation of CD4+ T cells in rheumatoid arthritis patients. Int. J. Clin. Exp. Pathol. 2015, 8, 8929. [Google Scholar]
- Sokka, T.; Toloza, S.; Cutolo, M.; Kautiainen, H.; Makinen, H.; Gogus, F.; Skakic, V.; Badsha, H.; Peets, T.; Baranauskaite, A. Women, men, and rheumatoid arthritis: Analyses of disease activity, disease characteristics, and treatments in the QUEST-RA study. Arthritis Res. Ther. 2009, 11, R7. [Google Scholar]
- Feng, X.; Hao, X.; Shi, R.; Xia, Z.; Huang, L.; Yu, Q.; Zhou, F. Detection and comparative analysis of methylomic biomarkers of rheumatoid arthritis. Front. Genet. 2020, 11, 238. [Google Scholar] [CrossRef]
- Toussirot, E.; Abbas, W.; Khan, K.A.; Tissot, M.; Jeudy, A.; Baud, L.; Bertolini, E.; Wendling, D.; Herbein, G. Imbalance between HAT and HDAC activities in the PBMCs of patients with ankylosing spondylitis or rheumatoid arthritis and influence of HDAC inhibitors on TNF alpha production. PLoS ONE 2013, 8, e70939. [Google Scholar] [CrossRef]
- Iancu-Rubin, C.; Gajzer, D.; Mosoyan, G.; Feller, F.; Mascarenhas, J.; Hoffman, R. Panobinostat (LBH589)-induced acetylation of tubulin impairs megakaryocyte maturation and platelet formation. Exp. Hematol. 2012, 40, 564–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göschl, L.; Preglej, T.; Boucheron, N.; Saferding, V.; Müller, L.; Platzer, A.; Hirahara, K.; Shih, H.-Y.; Backlund, J.; Matthias, P. Histone deacetylase 1 (HDAC1): A key player of T cell-mediated arthritis. J. Autoimmun. 2020, 108, 102379. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.C.; Brock, M.; Hemmatazad, H.; Giger, O.T.; Moritz, F.; Trenkmann, M.; Distler, J.H.; Gay, R.E.; Kolling, C.; Moch, H. Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. 2007, 56, 1087–1093. [Google Scholar] [CrossRef]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; Van Baarsen, I.M.; Ferguson, B.S.; García, S.; Fernandez, B.M.; McKinsey, T.A.; Tak, P.P.; Fossati, G. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum. Dis. 2017, 76, 277–285. [Google Scholar] [CrossRef]
- Park, J.K.; Jang, Y.J.; Oh, B.R.; Shin, J.; Bae, D.; Ha, N.; il Choi, Y.; Youn, G.S.; Park, J.; Lee, E.Y. Therapeutic potential of CKD-506, a novel selective histone deacetylase 6 inhibitor, in a murine model of rheumatoid arthritis. Arthritis Res. Ther. 2020, 22, 176. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Leoni, F.; Meghji, S.; Mascagni, P. Inhibition of HDAC activity by ITF2357 ameliorates joint inflammation and prevents cartilage and bone destruction in experimental arthritis. Mol. Med. 2011, 17, 391–396. [Google Scholar] [CrossRef]
- Nafie, M.S.; Arafa, K.; Sedky, N.K.; Alakhdar, A.A.; Arafa, R.K. Triaryl dicationic DNA minor-groove binders with antioxidant activity display cytotoxicity and induce apoptosis in breast cancer. Chem.-Biol. Interact. 2020, 324, 109087. [Google Scholar] [CrossRef]
- Vojinovic, J.; Damjanov, N.; D’Urzo, C.; Furlan, A.; Susic, G.; Pasic, S.; Iagaru, N.; Stefan, M.; Dinarello, C.A. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011, 63, 1452–1458. [Google Scholar] [CrossRef]
- Herrada, A.A.; Llanos, C.; Mackern-Oberti, J.P.; Carreño, L.J.; Henriquez, C.; Gómez, R.S.; Gutierrez, M.A.; Anegon, I.; Jacobelli, S.H.; Kalergis, A.M. Haem oxygenase 1 expression is altered in monocytes from patients with systemic lupus erythematosus. Immunology 2012, 136, 414–424. [Google Scholar] [CrossRef] [Green Version]
- Mackern-Oberti, J.P.; Llanos, C.; Carreño, L.J.; Riquelme, S.A.; Jacobelli, S.H.; Anegon, I.; Kalergis, A.M. Carbon monoxide exposure improves immune function in lupus-prone mice. Immunology 2013, 140, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Funes, S.C.; Ríos, M.; Gómez-Santander, F.; Fernández-Fierro, A.; Altamirano-Lagos, M.J.; Rivera-Perez, D.; Pulgar-Sepúlveda, R.; Jara, E.L.; Rebolledo-Zelada, D.; Villarroel, A.; et al. Tolerogenic dendritic cell transfer ameliorates systemic lupus erythematosus in mice. Immunology 2019, 158, 322–339. [Google Scholar] [CrossRef] [PubMed]
- Mackern-Oberti, J.P.; Obreque, J.; Méndez, G.P.; Llanos, C.; Kalergis, A.M. Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 182, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulton, V.R.; Tsokos, G.C. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J. Clin. Investig. 2015, 125, 2220–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffries, M.; Dozmorov, M.; Tang, Y.; Merrill, J.T.; Wren, J.D.; Sawalha, A.H. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics 2011, 6, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Quddus, J.; Johnson, K.J.; Gavalchin, J.; Amento, E.P.; Chrisp, C.E.; Yung, R.L.; Richardson, B.C. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J. Clin. Investig. 1993, 92, 38–53. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Tsokos, M.G.; Bickerton, S.; Sharabi, A.; Li, Y.; Moulton, V.R.; Kong, P.; Fahmy, T.M.; Tsokos, G.C. Precision DNA demethylation ameliorates disease in lupus-prone mice. JCI Insight 2018, 3, e120880. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Ise, W.; Inoue, T.; Ito, A.; Ono, C.; Shima, Y.; Sakakibara, S.; Nakayama, M.; Fujii, K.; Miura, I.; et al. Tet2 and Tet3 in B cells are required to repress CD86 and prevent autoimmunity. Nat. Immunol. 2020, 21, 950–961. [Google Scholar] [CrossRef]
- Furukawa, H.; Oka, S.; Matsui, T.; Hashimoto, A.; Arinuma, Y.; Komiya, A.; Fukui, N.; Tsuchiya, N.; Tohma, S. Genome, epigenome and transcriptome analyses of a pair of monozygotic twins discordant for systemic lupus erythematosus. Hum. Immunol. 2013, 74, 170–175. [Google Scholar] [CrossRef]
- Yung, R.; Powers, D.; Johnson, K.; Amento, E.; Carr, D.; Laing, T.; Yang, J.; Chang, S.; Hemati, N.; Richardson, B. Mechanisms of drug-induced lupus. II. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J. Clin. Investig. 1996, 97, 2866–2871. [Google Scholar] [CrossRef]
- Somers, E.C.; Richardson, B.C. Environmental exposures, epigenetic changes and the risk of lupus. Lupus 2014, 23, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Bian, S.; Chen, H.; Wang, L.; Zhao, L.; Zhang, X.; Zhao, Y.; Zeng, X.; Zhang, F. Clinical characteristics and risk factors for overlapping rheumatoid arthritis and Sjögren’s syndrome. Sci. Rep. 2018, 8, 6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didier, K.; Bolko, L.; Giusti, D.; Toquet, S.; Robbins, A.; Antonicelli, F.; Servettaz, A. Autoantibodies associated with connective tissue diseases: What meaning for clinicians? Front. Immunol. 2018, 9, 541. [Google Scholar] [CrossRef] [Green Version]
- Thabet, Y.; Le Dantec, C.; Ghedira, I.; Devauchelle, V.; Cornec, D.; Pers, J.-O.; Renaudineau, Y. Epigenetic dysregulation in salivary glands from patients with primary Sjögren’s syndrome may be ascribed to infiltrating B cells. J. Autoimmun. 2013, 41, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Cabellos, J.S.; Seco-Cervera, M.; Osca-Verdegal, R.; Pallardó, F.V.; García-Giménez, J.L. Epigenetic regulation in the pathogenesis of Sjögren Syndrome and Rheumatoid Arthritis. Front. Genet. 2019, 10, 1104. [Google Scholar] [CrossRef] [PubMed]
- Jamebozorgi, K.; Rostami, D.; Pormasoumi, H.; Taghizadeh, E.; Barreto, G.E.; Sahebkar, A. Epigenetic aspects of multiple sclerosis and future therapeutic options. Int. J. Neurosci. 2021, 131, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Diniz, S.N.; da Silva, C.F.; de Almeida, I.T.; da Silva Costa, F.E.; de Oliveira, E.M.L. INFβ treatment affects global DNA methylation in monocytes of patients with multiple sclerosis. J. Neuroimmunol. 2021, 355, 577563. [Google Scholar] [CrossRef]
- Elder, J.T.; Bruce, A.T.; Gudjonsson, J.E.; Johnston, A.; Stuart, P.E.; Tejasvi, T.; Voorhees, J.J.; Abecasis, G.R.; Nair, R.P. Molecular dissection of psoriasis: Integrating genetics and biology. J. Investig. Dermatol. 2010, 130, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christophers, E. Psoriasis--epidemiology and clinical spectrum. Clin. Exp. Dermatol. 2001, 26, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef]
- Cheuk, S.; Wiken, M.; Blomqvist, L.; Nylen, S.; Talme, T.; Stahle, M.; Eidsmo, L. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J. Immunol. 2014, 192, 3111–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sosa, T.; Sánchez, M.B.; Pietrobon, E.O.; Fernandez-Muñoz, J.M.; Zoppino, F.C.M.; Neira, F.J.; Germanó, M.J.; Cargnelutti, D.E.; Innocenti, A.C.; Jahn, G.A.; et al. Desmoglein-4 Deficiency Exacerbates Psoriasiform Dermatitis in Rats While Psoriasis Patients Displayed a Decreased Gene Expression of DSG4. Front. Immunol. 2021, 12, 708. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, X.; Liu, N.; Chen, H. Prediction of crucial epigenetically-associated, differentially expressed genes by integrated bioinformatics analysis and the identification of S100A9 as a novel biomarker in psoriasis. Int. J. Mol. Med. 2020, 45, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostensen, M.; Andreoli, L.; Brucato, A.; Cetin, I.; Chambers, C.; Clowse, M.E.; Costedoat-Chalumeau, N.; Cutolo, M.; Dolhain, R.; Fenstad, M.H.; et al. State of the art: Reproduction and pregnancy in rheumatic diseases. Autoimmun. Rev. 2015, 14, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Golden, L.C.; Itoh, Y.; Itoh, N.; Iyengar, S.; Coit, P.; Salama, Y.; Arnold, A.P.; Sawalha, A.H.; Voskuhl, R.R. Parent-of-origin differences in DNA methylation of X chromosome genes in T lymphocytes. Proc. Natl. Acad. Sci. USA 2019, 116, 26779–26787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, P.C.; Ponnampalam, A.P.; Steiner, M.; Mitchell, M.D. Effect of cyclic AMP and estrogen/progesterone on the transcription of DNA methyltransferases during the decidualization of human endometrial stromal cells. Mol. Hum. Reprod. 2013, 19, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bynote, K.K.; Hackenberg, J.M.; Korach, K.S.; Lubahn, D.B.; Lane, P.H.; Gould, K.A. Estrogen receptor-alpha deficiency attenuates autoimmune disease in (NZB x NZW)F1 mice. Genes Immun. 2008, 9, 137–152. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; McMurray, R.W. Effects of estrogen receptor subtype-selective agonists on autoimmune disease in lupus-prone NZB/NZW F1 mouse model. Clin. Immunol. 2007, 123, 219–226. [Google Scholar] [CrossRef]
- Svenson, J.L.; EuDaly, J.; Ruiz, P.; Korach, K.S.; Gilkeson, G.S. Impact of estrogen receptor deficiency on disease expression in the NZM2410 lupus prone mouse. Clin. Immunol. 2008, 128, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, M.A.; Wirth, J.R.; Naga, O.; Eudaly, J.; Gilkeson, G.S. Estrogen Receptor Alpha Binding to ERE is Required for Full Tlr7- and Tlr9-Induced Inflammation. SOJ Immunol. 2014, 2, 7. [Google Scholar] [CrossRef]
- Maselli, A.; Conti, F.; Alessandri, C.; Colasanti, T.; Barbati, C.; Vomero, M.; Ciarlo, L.; Patrizio, M.; Spinelli, F.R.; Ortona, E.; et al. Low expression of estrogen receptor beta in T lymphocytes and high serum levels of anti-estrogen receptor alpha antibodies impact disease activity in female patients with systemic lupus erythematosus. Biol. Sex Differ. 2016, 7, 016–0057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierdominici, M.; Maselli, A.; Varano, B.; Barbati, C.; Cesaro, P.; Spada, C.; Zullo, A.; Lorenzetti, R.; Rosati, M.; Rainaldi, G.; et al. Linking estrogen receptor beta expression with inflammatory bowel disease activity. Oncotarget 2015, 6, 40443–40451. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Gilkeson, G. Mouse models of lupus: What they tell us and what they don’t. Lupus Sci. Med. 2018, 5, e000199. [Google Scholar] [CrossRef] [PubMed]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.-C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3, eaap8855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawalha, A.H.; Wang, L.; Nadig, A.; Somers, E.C.; McCune, W.J.; Hughes, T.; Merrill, J.T.; Scofield, R.H.; Strickland, F.M.; Richardson, B. Sex-specific differences in the relationship between genetic susceptibility, T cell DNA demethylation and lupus flare severity. J. Autoimmun. 2012, 38, J216–J222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronson, J.K.; Ferner, R.E. Biomarkers—A general review. Curr. Protoc. Pharmacol. 2017, 76, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liao, J.; Li, Q.; Yang, M.; Zhao, M.; Lu, Q. Epigenetics as biomarkers in autoimmune diseases. Clin. Immunol. 2018, 196, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Mok, A.; Rhead, B.; Holingue, C.; Shao, X.; Quach, H.L.; Quach, D.; Sinclair, E.; Graf, J.; Imboden, J.; Link, T. Hypomethylation of CYP 2E1 and DUSP 22 Promoters Associated With Disease Activity and Erosive Disease Among Rheumatoid Arthritis Patients. Arthritis Rheumatol. 2018, 70, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhou, Y.; Zhu, B.; Wan, M.; Jiang, T.; Tan, Q.; Liu, Y.; Jiang, J.; Luo, S.; Tan, Y. IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann. Rheum. Dis. 2016, 75, 1998–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coit, P.; Jeffries, M.; Altorok, N.; Dozmorov, M.G.; Koelsch, K.A.; Wren, J.D.; Merrill, J.T.; McCune, W.J.; Sawalha, A.H. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J. Autoimmun. 2013, 43, 78–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coit, P.; Renauer, P.; Jeffries, M.A.; Merrill, J.T.; McCune, W.J.; Maksimowicz-McKinnon, K.; Sawalha, A.H. Renal involvement in lupus is characterized by unique DNA methylation changes in naïve CD4+ T cells. J. Autoimmun. 2015, 61, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liggett, T.; Melnikov, A.; Tilwalli, S.; Yi, Q.; Chen, H.; Replogle, C.; Feng, X.; Reder, A.; Stefoski, D.; Balabanov, R.; et al. Methylation patterns of cell-free plasma DNA in relapsing–remitting multiple sclerosis. J. Neurol. Sci. 2010, 290, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewes, D.; Tatomir, A.; Kruszewski, A.M.; Rao, G.; Tegla, C.A.; Ciriello, J.; Nguyen, V.; Royal, W.; Bever, C.; Rus, V.; et al. SIRT1 as a potential biomarker of response to treatment with glatiramer acetate in multiple sclerosis. Exp. Mol. Pathol. 2017, 102, 191–197. [Google Scholar] [CrossRef]
- Murata, K.; Furu, M.; Yoshitomi, H.; Ishikawa, M.; Shibuya, H.; Hashimoto, M.; Imura, Y.; Fujii, T.; Ito, H.; Mimori, T. Comprehensive microRNA analysis identifies miR-24 and miR-125a-5p as plasma biomarkers for rheumatoid arthritis. PLoS ONE 2013, 8, e69118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assmann, T.S.; Recamonde-Mendoza, M.; Puñales, M.; Tschiedel, B.; Canani, L.H.; Crispim, D. MicroRNA expression profile in plasma from type 1 diabetic patients: Case-control study and bioinformatic analysis. Diabetes Res. Clin. Pract. 2018, 141, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Liang, Y.; Zhao, Y.; Chen, L.; Wang, X.; Zhang, C. Meta-analysis of association of microRNAs genetic variants with susceptibility to rheumatoid arthritis and systemic lupus erythematosus. Medicine 2021, 100, e25689. [Google Scholar] [CrossRef]
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Amjadi, P.; Green, P.; Read, J.E.; Brennan, F.; Gregory, B.; Williams, R.O. Methotrexate restores regulatory T cell function through demethylation of the FoxP3 upstream enhancer in patients with rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 1182–1192. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Y.; Wang, X.; Kong, L.; Johnston, L.J.; Lu, L.; Ma, X. Dietary nutrients shape gut microbes and intestinal mucosa via epigenetic modifications. Crit. Rev. Food Sci. Nutr. 2020, 1–15. [Google Scholar] [CrossRef]
- Tsaprouni, L.G.; Yang, T.-P.; Bell, J.; Dick, K.J.; Kanoni, S.; Nisbet, J.; Viñuela, A.; Grundberg, E.; Nelson, C.P.; Meduri, E. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 2014, 9, 1382–1396. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Methylation Modification (hypo/hyper) | Methods | Tissue/Cells | Disease Activity | Model/Population | Reference |
|---|---|---|---|---|---|---|
| Global genomic hypomethylation. Fewer 5-methylcytosine and methylated CG sites upstream of an L1 open-reading frame | Immunohistochemistry for global 5-methylcytosine (5-MeC) determination and L1 promoter bisulfite sequencing | synovial fibroblasts from synovial tissue | Associated with activated phenotype in synovial fibroblasts | RA patients | [25] | |
| Hypomethylated loci in key genes (CHI3L1, CASP1, STAT3, MAP3K5, MEFV and WISP3). Hypermethylation in (TGFBR2 and FOXO1) | Infinium HumanMethylation450 BeadChip. Methylation confirmed by pyrosequencing and gene expression by qPCR | fibroblast-like synoviocytes from synovial tissues | not mentioned | female RA patients | [26] | |
| 1091 hypomethylated CpG sites (in 575 genes) and 1479 hypermethylated CpG sites (in 714 genes) | Integrated analysis of the DNA methylation, miRNA expression and mRNA expression data | fibroblast-like synoviocytes from synovial tissues | not mentioned | RA patients | [28] | |
| Two clusters within MHC regions with differential methylation potentially mediating genetic risk for RA | Illumina Human Hap300 v1.0 chip, Hap370CNVduo chip or Hap550duo chip | peripheral blood cells and monocyte cell fraction | not mentioned | RA patients with citrullinated protein antibodies, Swedish population | [29] | |
| No DNA methylation patterns identified but Huntingtin interacting protein-1 regulates FLS invasion into matrix | Histone modifications, WGBS, ATAC-seq and RNA-seq | synovial fibroblasts from synovial tissue | not mentioned | RA patients | [30] | |
| SLE | 4,839 hypomethylated and 1,568 hypermethylated CpG sites correlated | bisulfite genome-wide methylation assesment on Illumina platform. mRNA expression data | CD4+T cells | correlated negatively and positively with active disease | SLE patients, American | [31] |
| 487 hypomethylated and 420 hypermethylated CpG sites; SNX18, GALNT18, IFN signature genes | bisulfite genome-wide methylation assessment; Single nucleotide polymorphisms; Illumina platform | Neutrophils | correlated with Lupus nephritis | SLE patiens, African American and European American | [32] | |
| 7889 hypomethylated and 7400 hypermethylated CpG sites; IFI44L | bisulfite genome-wide methylation assessment | CD4+ T cells | not mentioned | SLE, GD, RA and SSc | [33] | |
| SS | 509 Differentially methylated CpG sites, 5 unique for SS | EWAS with Illumina Human Methylation 450k Array | peripheral blood cells | Correlated with active disease | primary SS patients | [34] |
| 553 hypomethylated and 200 hypermethylated CpG sites | Genome wide DNA methylation with Illumina Human Methylation 450k Array | Naive CD4+ T cells | Correlated with changes in the pathogenesis of SS and with active disease | primary SS patients | [35] | |
| MS | 11 Hypermethylated CpG sites; VMP1, MIR21 | Illumina Human Methylation 450k Array | CD4+ T cells | Correlated with Relapsing remitting MS | Relapsing remitting and secondary progressive form of MS patients | [36] |
| 502 Differentially methylated CpG sites | Bisulfite genome wide methylation assessment; Illumina platform; RADmeth software | CD14+ cells from haematopoietic progenitor cells | Correlation to incidence of MS and others autoimmune diseases | Adult and pediatric population | [37] | |
| Psoriasis | IL13, TNFSF11, others | bisulfite genome-wide methylation assessment; Illumina platform | CD4+ and CD8+ T cells | not mentioned | Discordant Psoriasis twins’ patients | [38] |
| 811 hypomethylated and 3510 hypermethylated CpG sites; IL17, IRF7, IL7, CXCL1 | bisulfite genome-wide methylation assessment; Genome-wide genotyping; Illumina platform | Skin samples | not mentioned | Psoriasis patients, HLA-Cw*0602 carriers | [39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funes, S.C.; Fernández-Fierro, A.; Rebolledo-Zelada, D.; Mackern-Oberti, J.P.; Kalergis, A.M. Contribution of Dysregulated DNA Methylation to Autoimmunity. Int. J. Mol. Sci. 2021, 22, 11892. https://doi.org/10.3390/ijms222111892
Funes SC, Fernández-Fierro A, Rebolledo-Zelada D, Mackern-Oberti JP, Kalergis AM. Contribution of Dysregulated DNA Methylation to Autoimmunity. International Journal of Molecular Sciences. 2021; 22(21):11892. https://doi.org/10.3390/ijms222111892
Chicago/Turabian StyleFunes, Samanta C., Ayleen Fernández-Fierro, Diego Rebolledo-Zelada, Juan P. Mackern-Oberti, and Alexis M. Kalergis. 2021. "Contribution of Dysregulated DNA Methylation to Autoimmunity" International Journal of Molecular Sciences 22, no. 21: 11892. https://doi.org/10.3390/ijms222111892