
Reconstituting Immune Surveillance in Breast Cancer: Molecular Pathophysiology and Current Immunotherapy Strategies



Abstract
:1. Introduction
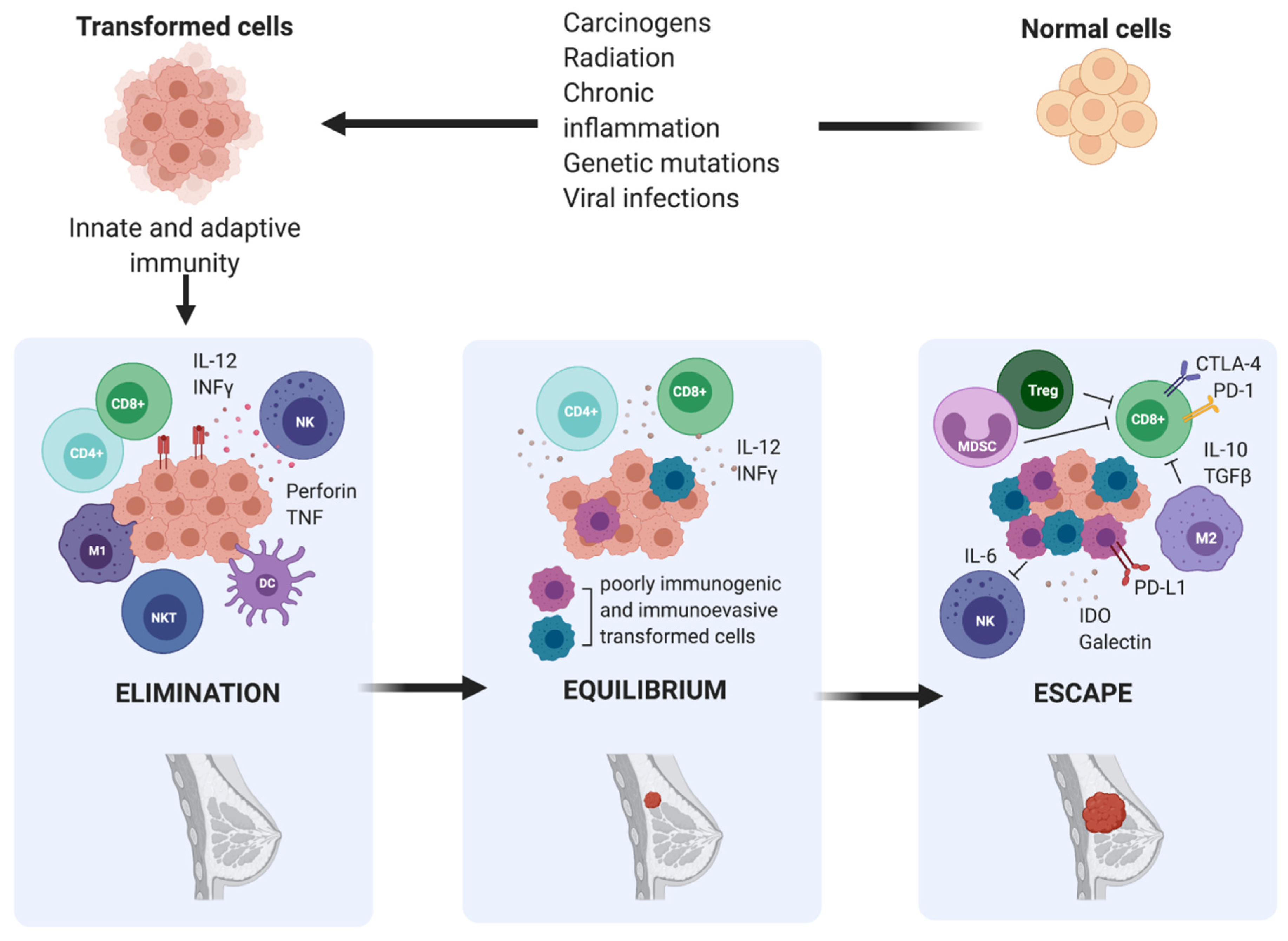
2. Immunoediting in Breast Cancer
3. Immune Evasion in Breast Cancer
3.1. Tumour-Related Immune Evasion
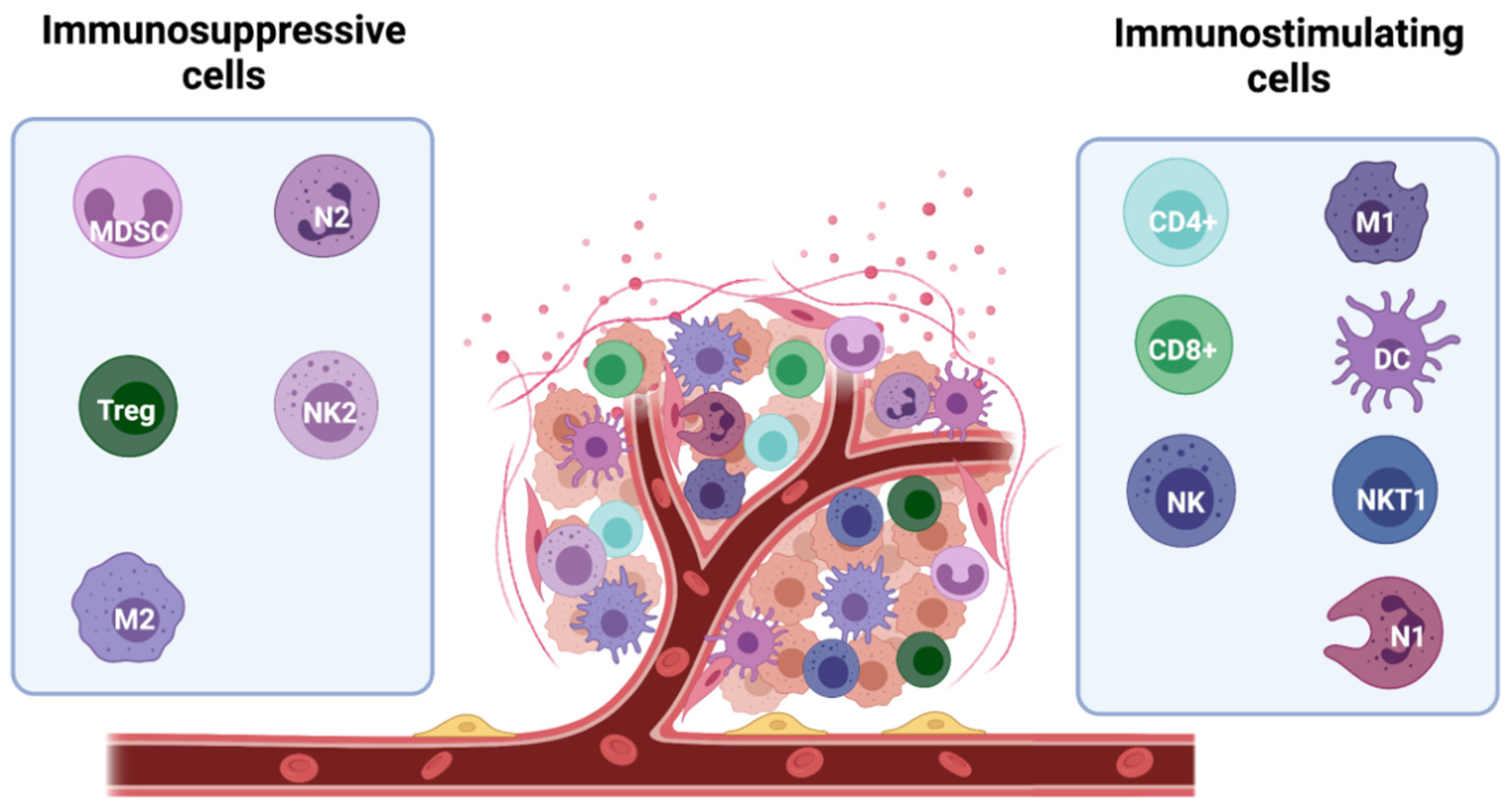
3.2. Immunosuppressive Microenvironment
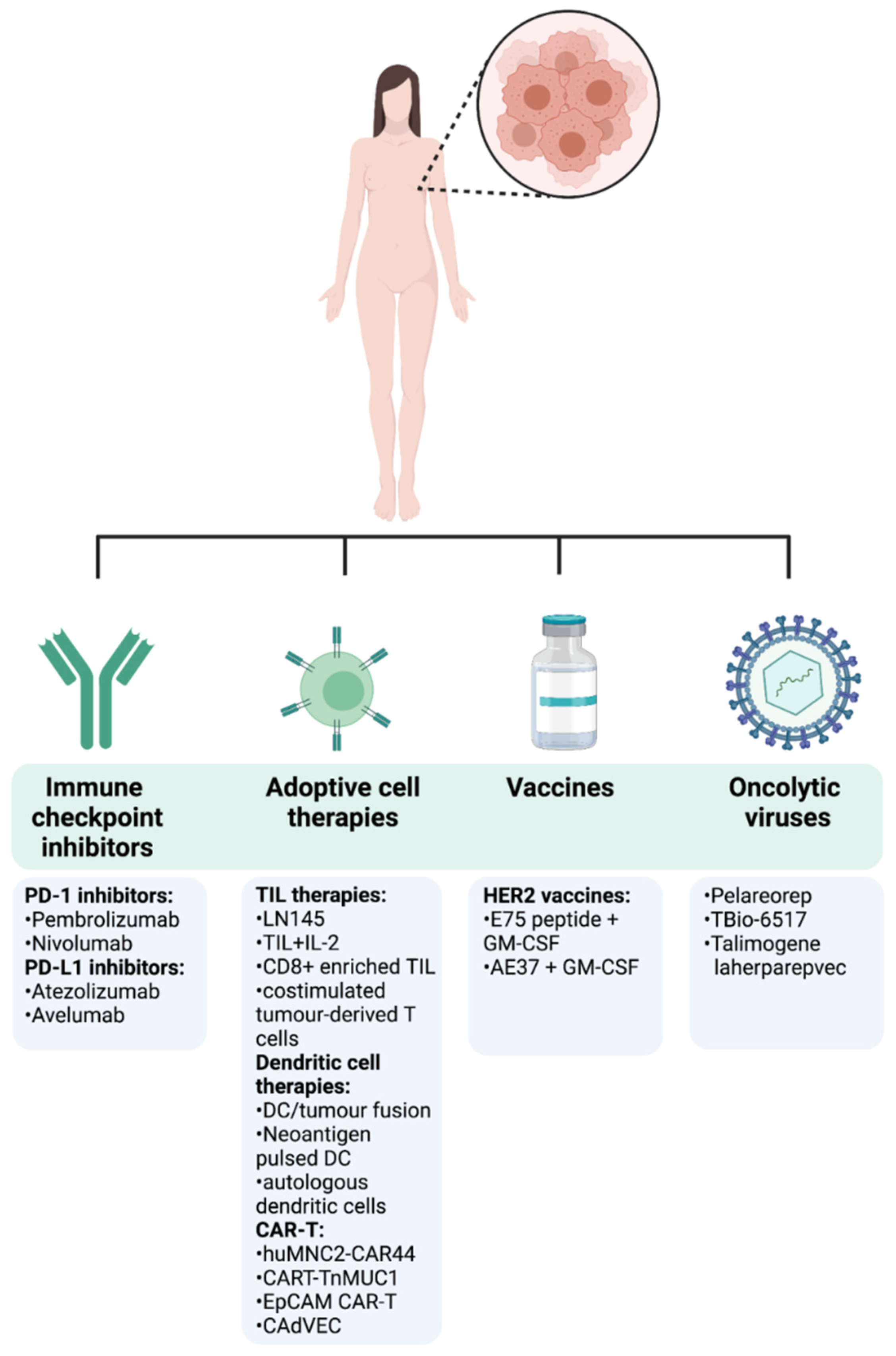
4. Recent Advances in Breast Cancer Immunotherapy
4.1. Clinical Efficacy of Immune Checkpoint Inhibitors in Breast Cancer
4.2. Adoptive Cell Therapy Role in Breast Cancer
4.3. Cancer Vaccines in Breast Cancer
4.4. Oncolytic Viruses in Breast Cancer
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Costa, A.; Norton, L.; Cameron, D.; Cufer, T.; Fallowfield, L.; Francis, P.; Gligorov, J.; Kyriakides, S.; Lin, N.; et al. 1st International consensus guidelines for advanced breast cancer (ABC 1). Breast 2012, 21, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Mariotto, A.B.; Etzioni, R.; Hurlbert, M.; Penberthy, L.; Mayer, M. Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiol. Biomark. Prev. 2017, 26, 809–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef] [PubMed]
- García-Aranda, M.; Redondo, M. Protein Kinase Targets in Breast Cancer. Int. J. Mol. Sci. 2017, 18, 2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 610303. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Zitvogel, L. Cancer immunotherapy in 2017: The breakthrough of the microbiota. Nat. Rev. Immunol. 2018, 18, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Müller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Balko, J.M.; Gameiro, S.R.; Bear, H.D.; Prabhakaran, S.; Fukui, J.; Disis, M.L.; Nanda, R.; Gulley, J.L.; Kalinsky, K.; et al. If we build it they will come: Targeting the immune response to breast cancer. npj Breast Cancer 2019, 5, 37. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Tower, H.; Ruppert, M.; Britt, K. The Immune Microenvironment of Breast Cancer Progression. Cancers 2019, 11, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Spiotto, M.T.; Rowley, D.A.; Schreiber, H. Bystander elimination of antigen loss variants in established tumors. Nat. Med. 2004, 10, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Töpfer, K.; Kempe, S.; Müller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor evasion from T cell surveillance. J. Biomed. Biotechnol. 2011, 2011, 918471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Komohara, Y.; Domenick, N.; Ohno, M.; Ikeura, M.; Hamilton, R.L.; Horbinski, C.; Wang, X.; Ferrone, S.; Okada, H. Expression of antigen processing and presenting molecules in brain metastasis of breast cancer. Cancer Immunol. Immunother. 2012, 61, 789–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhang, C.; Song, Y.; Wang, Z.; Wang, Y.; Luo, F.; Xu, Y.; Zhao, Y.; Wu, Z. Mechanism of immune evasion in breast cancer. Onco Targets Ther. 2017, 10, 1561–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef]
- Bębenek, M.; Duś, D.; Koźlak, J. Prognostic value of the Fas/Fas ligand system in breast cancer. Contemp. Oncol. 2013, 17, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K.; Uchida, Y.; Toge, T. The role of Fas ligand and transforming growth factor beta in tumor progression: Molecular mechanisms of immune privilege via Fas-mediated apoptosis and potential targets for cancer therapy. Cancer 2004, 100, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Meyer, E.; Debatin, K.M. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 2002, 21, 2283–2294. [Google Scholar] [CrossRef] [Green Version]
- Ryan, B.M.; Konecny, G.E.; Kahlert, S.; Wang, H.J.; Untch, M.; Meng, G.; Pegram, M.D.; Podratz, K.C.; Crown, J.; Slamon, D.J.; et al. Survivin expression in breast cancer predicts clinical outcome and is associated with HER2, VEGF, urokinase plasminogen activator and PAI-1. Ann. Oncol. 2006, 17, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, E.; Sahin, A.A.; Chen, J.S.; Krishnamurthy, R.R.; Aggarwal, N.; Brun, A.M.; Sapino, A.; Zhang, F.; Sharma, D.; Yang, X.H.; et al. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene 2002, 21, 8843–8851. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo-Sanz, L.; Muñoz, P. Tumor-Infiltrating Immunosuppressive Cells in Cancer-Cell Plasticity, Tumor Progression and Therapy Response. Cancer Microenviron. 2019, 12, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Jene, N.; Byrne, D.; Millar, E.K.; O’Toole, S.A.; McNeil, C.M.; Bates, G.J.; Harris, A.L.; Banham, A.H.; Sutherland, R.L.; et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res. 2011, 13, R47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, L.M.; Robila, V.; Clark, N.M.; Du, W.; Idowu, M.O.; Rutkowski, M.R.; Bos, P.D. Regulatory T Cells Control the Switch From. Front. Immunol. 2019, 10, 1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Choi, J.; Gyamfi, J.; Jang, H.; Koo, J.S. The role of tumor-associated macrophage in breast cancer biology. Histol. Histopathol. 2018, 33, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e51. [Google Scholar] [CrossRef] [Green Version]
- Segovia-Mendoza, M.; Morales-Montor, J. Immune Tumor Microenvironment in Breast Cancer and the Participation of Estrogen and Its Receptors in Cancer Physiopathology. Front. Immunol. 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielbassa, K.; Vegna, S.; Ramirez, C.; Akkari, L. Understanding the Origin and Diversity of Macrophages to Tailor Their Targeting in Solid Cancers. Front. Immunol. 2019, 10, 2215. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Shen, Y.; Huang, H.; Pan, S.; Jiang, J.; Chen, W.; Zhang, T.; Zhang, C.; Ni, C. A Rosetta Stone for Breast Cancer: Prognostic Value and Dynamic Regulation of Neutrophil in Tumor Microenvironment. Front. Immunol. 2020, 11, 1779. [Google Scholar] [CrossRef] [PubMed]
- Mishalian, I.; Bayuh, R.; Eruslanov, E.; Michaeli, J.; Levy, L.; Zolotarov, L.; Singhal, S.; Albelda, S.M.; Granot, Z.; Fridlender, Z.G. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17—A new mechanism of impaired antitumor immunity. Int. J. Cancer 2014, 135, 1178–1186. [Google Scholar] [CrossRef]
- Kuang, D.M.; Zhao, Q.; Wu, Y.; Peng, C.; Wang, J.; Xu, Z.; Yin, X.Y.; Zheng, L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J. Hepatol. 2011, 54, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Ferrara, N. The Complex Role of Neutrophils in Tumor Angiogenesis and Metastasis. Cancer Immunol. Res. 2016, 4, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef] [Green Version]
- Clappaert, E.J.; Murgaski, A.; Van Damme, H.; Kiss, M.; Laoui, D. Diamonds in the Rough: Harnessing Tumor-Associated Myeloid Cells for Cancer Therapy. Front. Immunol. 2018, 9, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gun, S.Y.; Lee, S.W.L.; Sieow, J.L.; Wong, S.C. Targeting immune cells for cancer therapy. Redox Biol. 2019, 25, 101174. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Woo, S.R.; Zha, Y.; Spaapen, R.; Zheng, Y.; Corrales, L.; Spranger, S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 268–276. [Google Scholar] [CrossRef]
- Kumar, S.; Wilkes, D.W.; Samuel, N.; Blanco, M.A.; Nayak, A.; Alicea-Torres, K.; Gluck, C.; Sinha, S.; Gabrilovich, D.; Chakrabarti, R. ΔNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J. Clin. Investig. 2018, 128, 5095–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergenfelz, C.; Roxå, A.; Mehmeti, M.; Leandersson, K.; Larsson, A.M. Clinical relevance of systemic monocytic-MDSCs in patients with metastatic breast cancer. Cancer Immunol. Immunother. 2020, 69, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef] [Green Version]
- Singh, G.; Singh, S.K.; König, A.; Reutlinger, K.; Nye, M.D.; Adhikary, T.; Eilers, M.; Gress, T.M.; Fernandez-Zapico, M.E.; Ellenrieder, V. Sequential activation of NFAT and c-Myc transcription factors mediates the TGF-beta switch from a suppressor to a promoter of cancer cell proliferation. J. Biol. Chem. 2010, 285, 27241–27250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chrétien, A.S.; Viens, P.; Sabatier, R.; Gonçalves, A. PD-1/PD-L1 Targeting in Breast Cancer: The First Clinical Evidences Are Emerging. A Literature Review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, L.; Dong, C. New B7 Family Checkpoints in Human Cancers. Mol. Cancer Ther. 2017, 16, 1203–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Barriga, V.; Kuol, N.; Nurgali, K.; Apostolopoulos, V. The Complex Interaction between the Tumor Micro-Environment and Immune Checkpoints in Breast Cancer. Cancers 2019, 11, 1205. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.M.; Lee, J.; Koo, J.S. Clinicopathological and prognostic significance of programmed death ligand-1 expression in breast cancer: A meta-analysis. BMC Cancer 2017, 17, 690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, J.B.; Honig, A.; Kapp, M.; Hahne, J.C.; Meyer, S.R.; Dietl, J.; Segerer, S.E. Mechanisms of tumor immune escape in triple-negative breast cancers (TNBC) with and without mutated BRCA 1. Arch. Gynecol. Obs. 2014, 289, 141–147. [Google Scholar] [CrossRef]
- Tringler, B.; Zhuo, S.; Pilkington, G.; Torkko, K.C.; Singh, M.; Lucia, M.S.; Heinz, D.E.; Papkoff, J.; Shroyer, K.R. B7-h4 is highly expressed in ductal and lobular breast cancer. Clin. Cancer Res. 2005, 11, 1842–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. Pillars article: CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994. 1: 405–413. J. Immunol. 2011, 187, 3466–3474. [Google Scholar]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1247135. [Google Scholar] [CrossRef] [Green Version]
- Syed Khaja, A.S.; Toor, S.M.; El Salhat, H.; Faour, I.; Ul Haq, N.; Ali, B.R.; Elkord, E. Preferential accumulation of regulatory T cells with highly immunosuppressive characteristics in breast tumor microenvironment. Oncotarget 2017, 8, 33159–33171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burugu, S.; Gao, D.; Leung, S.; Chia, S.K.; Nielsen, T.O. LAG-3+ tumor infiltrating lymphocytes in breast cancer: Clinical correlates and association with PD-1/PD-L1+ tumors. Ann. Oncol. 2017, 28, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Stamm, H.; Oliveira-Ferrer, L.; Grossjohann, E.M.; Muschhammer, J.; Thaden, V.; Brauneck, F.; Kischel, R.; Müller, V.; Bokemeyer, C.; Fiedler, W.; et al. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology 2019, 8, e1674605. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD47 blockade as another immune checkpoint therapy for cancer. Nat. Med. 2015, 21, 1122–1123. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Shi, X.; Chen, C.; He, H.; Liu, L.; Wu, J.; Yan, H. High expression of CD47 in triple negative breast cancer is associated with epithelial-mesenchymal transition and poor prognosis. Oncol. Lett. 2019, 18, 3249–3255. [Google Scholar] [CrossRef] [Green Version]
- Rugo, H.S.; Delord, J.P.; Im, S.A.; Ott, P.A.; Piha-Paul, S.A.; Bedard, P.L.; Sachdev, J.; Le Tourneau, C.; van Brummelen, E.M.J.; Varga, A.; et al. Safety and Antitumor Activity of Pembrolizumab in Patients with Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 2804–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.W.; De Laurentiis, M.; Nanda, R.; Winer, E.P.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, E.P.; Lipatov, O.; Im, S.A.; Goncalves, A.; Muñoz-Couselo, E.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 499–511. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Kalinsky, K.; Kaklamani, V.G.; D’Adamo, D.R.; Aktan, G.; Tsai, M.L.; O’Regan, R.M.; Kaufman, P.A.; Wilks, S.T.; Andreopoulou, E.; et al. Eribulin Plus Pembrolizumab in Patients with Metastatic Triple-Negative Breast Cancer (ENHANCE 1): A Phase Ib/II Study. Clin. Cancer Res. 2021, 27, 3061–3068. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Nanda, R.; Liu, M.C.; Yau, C.; Shatsky, R.; Pusztai, L.; Wallace, A.; Chien, A.J.; Forero-Torres, A.; Ellis, E.; Han, H.; et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol. 2020, 6, 676–684. [Google Scholar] [CrossRef]
- Miles, D.; Gligorov, J.; André, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 2021, 32, 994–1004. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Avigan, D.; Vasir, B.; Gong, J.; Borges, V.; Wu, Z.; Uhl, L.; Atkins, M.; Mier, J.; McDermott, D.; Smith, T.; et al. Fusion cell vaccination of patients with metastatic breast and renal cancer induces immunological and clinical responses. Clin. Cancer Res. 2004, 10, 4699–4708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal-Estévez, D.A.; Ortíz Barbosa, M.A.; Ortíz-Montero, P.; Cifuentes, C.; Sánchez, R.; Parra-López, C.A. Autologous Dendritic Cells in Combination With Chemotherapy Restore Responsiveness of T Cells in Breast Cancer Patients: A Single-Arm Phase I/II Trial. Front. Immunol. 2021, 12, 669965. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.; Dotti, G. Combining Oncolytic Viruses with Chimeric Antigen Receptor T Cell Therapy. Hum. Gene Ther. 2021, 32, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Lu, B.; Melisko, M.; Price Hiller, J.; Bondarenko, I.; Brunt, A.M.; Sergii, G.; Petrakova, K.; Peoples, G.E. Efficacy and Safety Analysis of Nelipepimut-S Vaccine to Prevent Breast Cancer Recurrence: A Randomized, Multicenter, Phase III Clinical Trial. Clin. Cancer Res. 2019, 25, 4248–4254. [Google Scholar] [CrossRef] [Green Version]
- Clifton, G.T.; Hale, D.; Vreeland, T.J.; Hickerson, A.T.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; Qiao, N.; Philips, A.V.; Lukas, J.J.; et al. Results of a Randomized Phase IIb Trial of Nelipepimut-S + Trastuzumab versus Trastuzumab to Prevent Recurrences in Patients with High-Risk HER2 Low-Expressing Breast Cancer. Clin. Cancer Res. 2020, 26, 2515–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, V.; Ellard, S.L.; Dent, S.F.; Tu, D.; Mates, M.; Dhesy-Thind, S.K.; Panasci, L.; Gelmon, K.A.; Salim, M.; Song, X.; et al. A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: Final analysis of Canadian Cancer Trials Group IND.213. Breast Cancer Res. Treat. 2018, 167, 485–493. [Google Scholar] [CrossRef] [PubMed]
- García-Aranda, M.; Redondo, M. Immunotherapy: A Challenge of Breast Cancer Treatment. Cancers 2019, 11, 1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.; Diamond, J.R.; Hamilton, E.; Pohlmann, P.R.; Tolaney, S.M.; Chang, C.W.; Zhang, W.; Iizuka, K.; Foster, P.G.; Molinero, L.; et al. Atezolizumab Plus nab-Paclitaxel in the Treatment of Metastatic Triple-Negative Breast Cancer With 2-Year Survival Follow-up: A Phase 1b Clinical Trial. JAMA Oncol. 2019, 5, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes-Antrás, J.; Guevara-Hoyer, K.; Baliu-Piqué, M.; García-Sáenz, J.; Pérez-Segura, P.; Pandiella, A.; Ocaña, A. Adoptive Cell Therapy in Breast Cancer: A Current Perspective of Next-Generation Medicine. Front. Oncol. 2020, 10, 605633. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Emerging CAR-T Cell Therapy for the Treatment of Triple-Negative Breast Cancer. Mol. Cancer Ther. 2020, 19, 2409–2421. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghi, S.; Hojati, Z.; Tabatabaeian, H. Cooverexpression of EpCAM and c-myc genes in malignant breast tumours. J. Genet. 2017, 96, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, S.L. Talimogene Laherparepvec: First Global Approval. Drugs 2016, 76, 147–154. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Fong, Y. Viroimmunotherapy for breast cancer: Promises, problems and future directions. Cancer Gene Ther. 2021, 28, 757–768. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Trial Identifier | Phase | Intervention | Breast Cancer Subtype | Start Date (Estimated Completion Date) | Ref |
|---|---|---|---|---|---|---|
| Immune checkpoint inhibitors | ||||||
| PD-1 | NCT02054806 (Keynote-028) | Ib | Pembrolizumab | ER+/HER2-PD-L1+ aBC | 17 February 2014 (30 April 2021) | [84] |
| NCT01848834 (Keynote-012) | Ib | Pembrolizumab | PD-L1 + mTNBC | 7 May 2013 (30 June 2020) | [85] | |
| NCT02447003 (Keynote-086) Cohort A | II | Pembrolizumab | mTNBC ≥ 1 systemic therapy | 11 June 2015 (31 January 2020) | [86] | |
| NCT02447003 (Keynote-086) Cohort B | II | Pembrolizumab | mTNBC PD-L1 + 1st line | 11 June 2015 (31 January 2020) | [87] | |
| NCT02555657 (Keynote-119) | III | Pembrolizumab vs. chemotherapy | mTNBC | 13 October 2015 (10 November 2020) | [88] | |
| NCT03036488 (Keynote-522) | III | Pembrolizumab + Chemotherapy vs. Placebo + Chemotherapy | Stage II/III TNBC 1st line | 7 March 2017 (30 September 2025) | [89] | |
| NCT02513472 (Keynote-150) | Ib/II | Eribulin Mesylate + Pembrolizumab | mTNBC ≤ 2nd line | 28 August 2015 (9 April 2021) | [90] | |
| NCT02499367 (TONIC) | II | Nivolumab Immune induction vs. no induction | mTNBC < 3 lines of therapy | August 2015 (August 2022) | [91] | |
| NCT02819518 (Keynote-355) | III | Pembrolizumab + chemotherapy vs. placebo + chemotherapy | Locally recurrent inoperable TNBC/mTNBC 1st line | 27 July 2016 (12 January 2022) | [92] | |
| NCT01042379 (I-SPY2) | II | Pembrolizumab + chemotherapy vs. placebo + chemotherapy | High-risk, stage II/III BC | 1 March 2010 (December 2031) | [93] | |
| PD-L1 | NCT03125902 (IMpassion-131) | III | Atezolizumab + paclitaxel vs. placebo + paclitaxel | Locally advanced inoperable TNBC/mTNBC 1st line | 25 August 2017 (2 December 2021) | [94] |
| NCT02425891 (IMpassion-130) | III | Atezolizumab + nab-paclitaxel vs. placebo + nab-paclitaxel | Locally advanced/mTNBC 1st line | 23 June 2015 (30 August 2021) | [95] | |
| NCT01772004 (JAVELIN) | Ib | Avelumab | mBC | 31 January 2013 (16 December 2019) | [96] | |
| NCT03197935 (IMpassion-031) | III | Atezolizumab + chemotherapy vs. placebo + chemotherapy | Stage II-III TNBC | 24 July 2017 (21 October 2022) | [97] | |
| Adoptive cell therapies | ||||||
| TIL Therapy | NCT04111510 | II | LN-145 | mTNBC 1–3 lines of therapy | 23 December 2019 (January 2022) | n/a |
| NCT01462903 | I | Tumour infiltrating lymphocytes + IL-2 | Breast Carcinoma | September 2011 (December 2014) | n/a | |
| NCT01174121 | II | CD8+ Enriched TIL vs. unselected TIL vs. unselected TIL + pembrolizumab | Metastatic BC ≥ 2 lines of therapy | 26 August 2010 (27 December 2024) | n/a | |
| NCT00301730 | I | Costimulated tumour-derived T cells | mBC | October 2005 (n/a) | n/a | |
| Dendritic cell Therapy | n/a | I | DC/tumour fusion | mBC | July 1999 (March 2002) | [98] |
| NCT04105582 | I | Neo-antigen pulsed DC | BC | 1 August 2019 (1 March 2022) | n/a | |
| NCT03630809 | II | HER2 DC1 Vaccine | HER2+ | 10 January 2019 (December 2024) | n/a | |
| NCT03450044 | I/II | Autologous dendritic cells + chemotherapy | IDC TNM IIA-IV | January 2014 (August 2018) | [99] | |
| NCT04348747 | IIa | anti-HER2/3 dendritic cell vaccine + Celecoxib + Pembrolizumab + IFN alpha-2b + Rintatolimod | Brain metastases from TNBC or HER2 + BC | 1 October 2021 (1 October 2024) | n/a | |
| CAR-T | NCT04020575 | I | huMNC2-CAR44 CAR T cells | Metastatic HR+ (≥3 lines), HER2+ (≥3 lines), TNBC (≥2 lines) | 15 January 2020 (15 January 2035) | n/a |
| NCT04025216 | I | CART-TnMUC1 | mTNBC | 10 October 2019 (31 October 2036) | n/a | |
| NCT02915445 | I | CAR-T cells recognizing EpCAM | EpCAM + BC | July 2016 (July 2022) | n/a | |
| NCT03740256 | I | CAdVEC | HER2 + BC | 14 December 2020 (30 December 2038) | [100] | |
| Cancer vaccines | ||||||
| HER2 Vaccine | NCT01479244 (PRESENT) | III | E75 peptide + GM-CSF or placebo + GM-CSF | T1-T3 HER2 IHC 1+/2 + node + BC | November 2011 (21 September 2016) | [101] |
| NCT01570036 | II | E75 peptide (KIFGSLAFL) vaccine + GM-CSF vs. placebo + GM-CSF | Disease-free after HER2 1+/2 + BC | 21 May 2013 (28 September 2018) | [102] | |
| NCT00524277 | II | AE37 + GM-CSF vs. GP2 + GM-CSF vs. placebo + GM-CSF | Disease-free after Lymph node+ or high-risk lymph node-HER2 + BC | January 2007 (31 March 2017) | n/a | |
| Oncolytic viruses | ||||||
| Oncolytic virus | NCT01656538 | II | Pelareorep + paclitaxel vs. paclitaxel | Advanced BC/mBC | 30 July 2012 (14 February 2018) | [103] |
| NCT04215146 (BRACELET-1) | II | Pelareorep + paclitaxel + avelumab vs. pelareorep + paclitaxel vs. paclitaxel | HR+/HER2-endocrine refractory mBC | 10 June 2020 (January 2024) | n/a | |
| NCT04102618 (AWARE-1) | Early I | Pelareorep + letrozole vs. pelareorep + letrozole + atezolizumab vs. pelareorep + atezolizumab vs. pelareorep + atezolizumab + trastuzumab | HR+/HER2-, TNBC, HER2+/HR+, HER2+/HR- | 29 March 2019 (December 2020) | n/a | |
| NCT04301011 (RAPTOR) | I/IIa | TBio-6517 vs. TBio-6517 + Pembrolizumab | Locally advanced/metastatic BC | 2 June 2020 (30 December 2022) | n/a | |
| NCT04185311 | I | talimogene laherparepvec, nivolumab, ipilimumab | Localized TN or ER+ HER2-BC | 10 July 2019 (1 July 2023) | n/a | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cilibrasi, C.; Papanastasopoulos, P.; Samuels, M.; Giamas, G. Reconstituting Immune Surveillance in Breast Cancer: Molecular Pathophysiology and Current Immunotherapy Strategies. Int. J. Mol. Sci. 2021, 22, 12015. https://doi.org/10.3390/ijms222112015
Cilibrasi C, Papanastasopoulos P, Samuels M, Giamas G. Reconstituting Immune Surveillance in Breast Cancer: Molecular Pathophysiology and Current Immunotherapy Strategies. International Journal of Molecular Sciences. 2021; 22(21):12015. https://doi.org/10.3390/ijms222112015
Chicago/Turabian StyleCilibrasi, Chiara, Panagiotis Papanastasopoulos, Mark Samuels, and Georgios Giamas. 2021. "Reconstituting Immune Surveillance in Breast Cancer: Molecular Pathophysiology and Current Immunotherapy Strategies" International Journal of Molecular Sciences 22, no. 21: 12015. https://doi.org/10.3390/ijms222112015
APA StyleCilibrasi, C., Papanastasopoulos, P., Samuels, M., & Giamas, G. (2021). Reconstituting Immune Surveillance in Breast Cancer: Molecular Pathophysiology and Current Immunotherapy Strategies. International Journal of Molecular Sciences, 22(21), 12015. https://doi.org/10.3390/ijms222112015