
From Forensics to Clinical Research: Expanding the Variant Calling Pipeline for the Precision ID mtDNA Whole Genome Panel

 , ,
, ,  ,
, 
 and
and
Abstract
1. Introduction
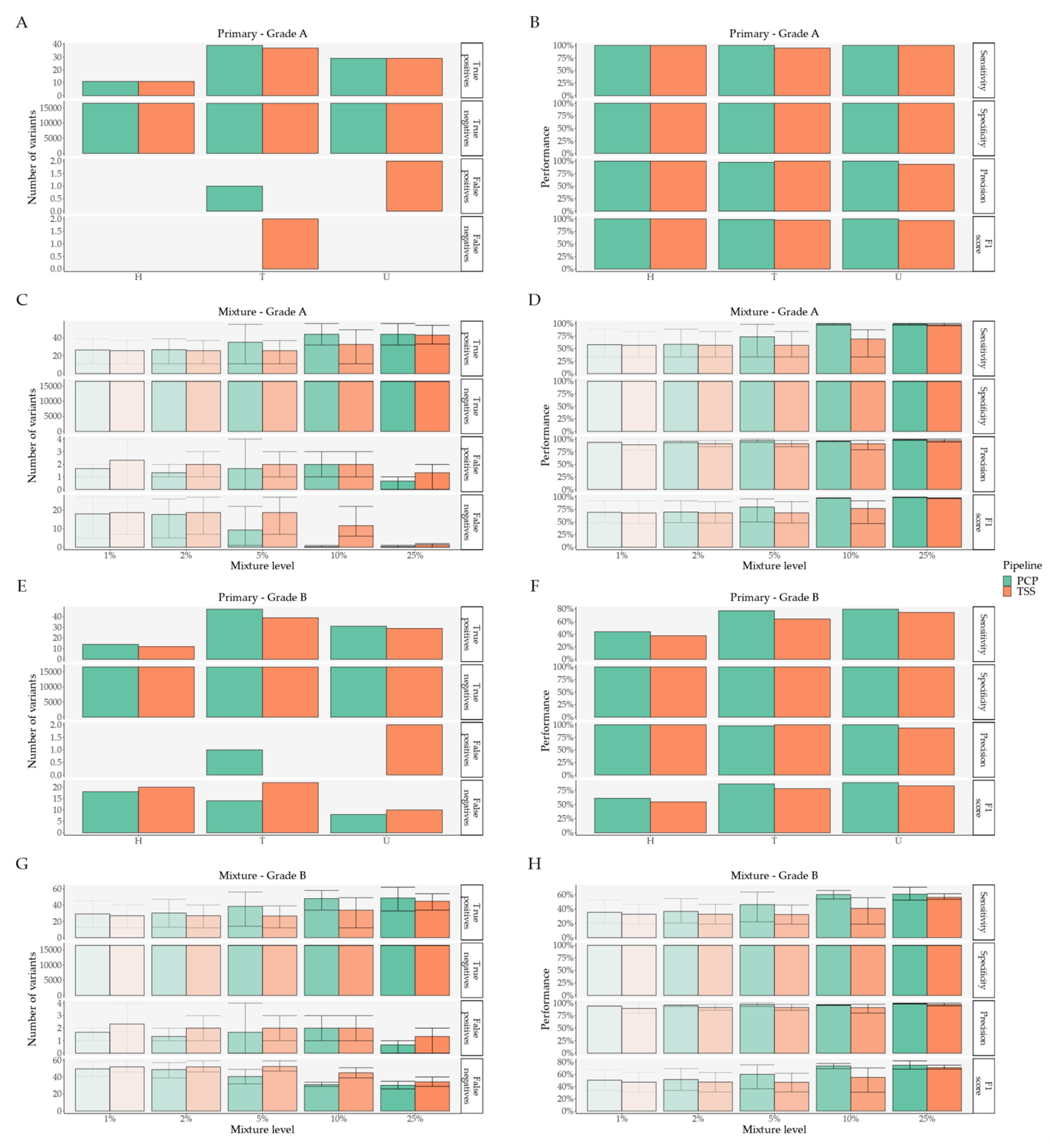
2. Results
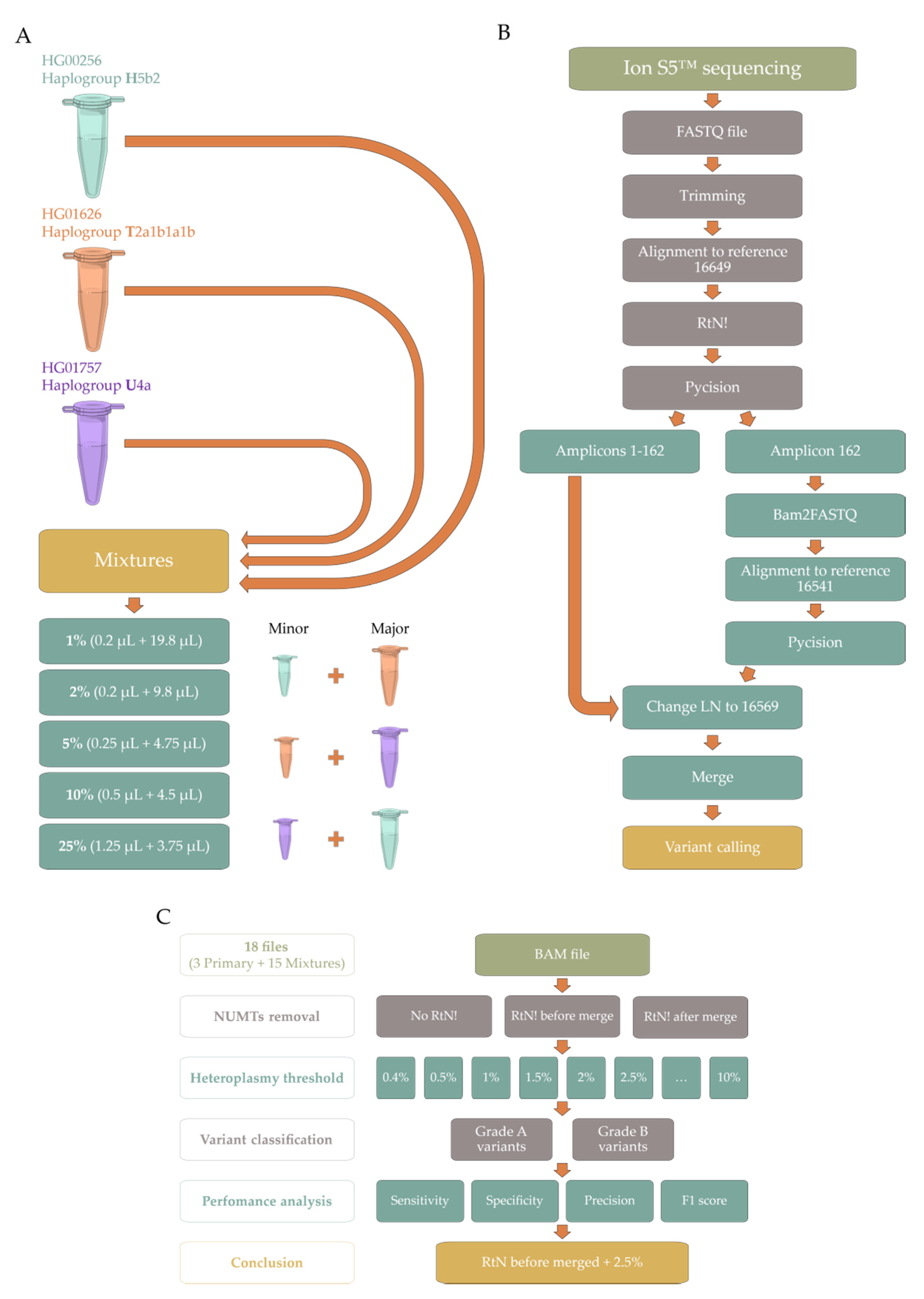
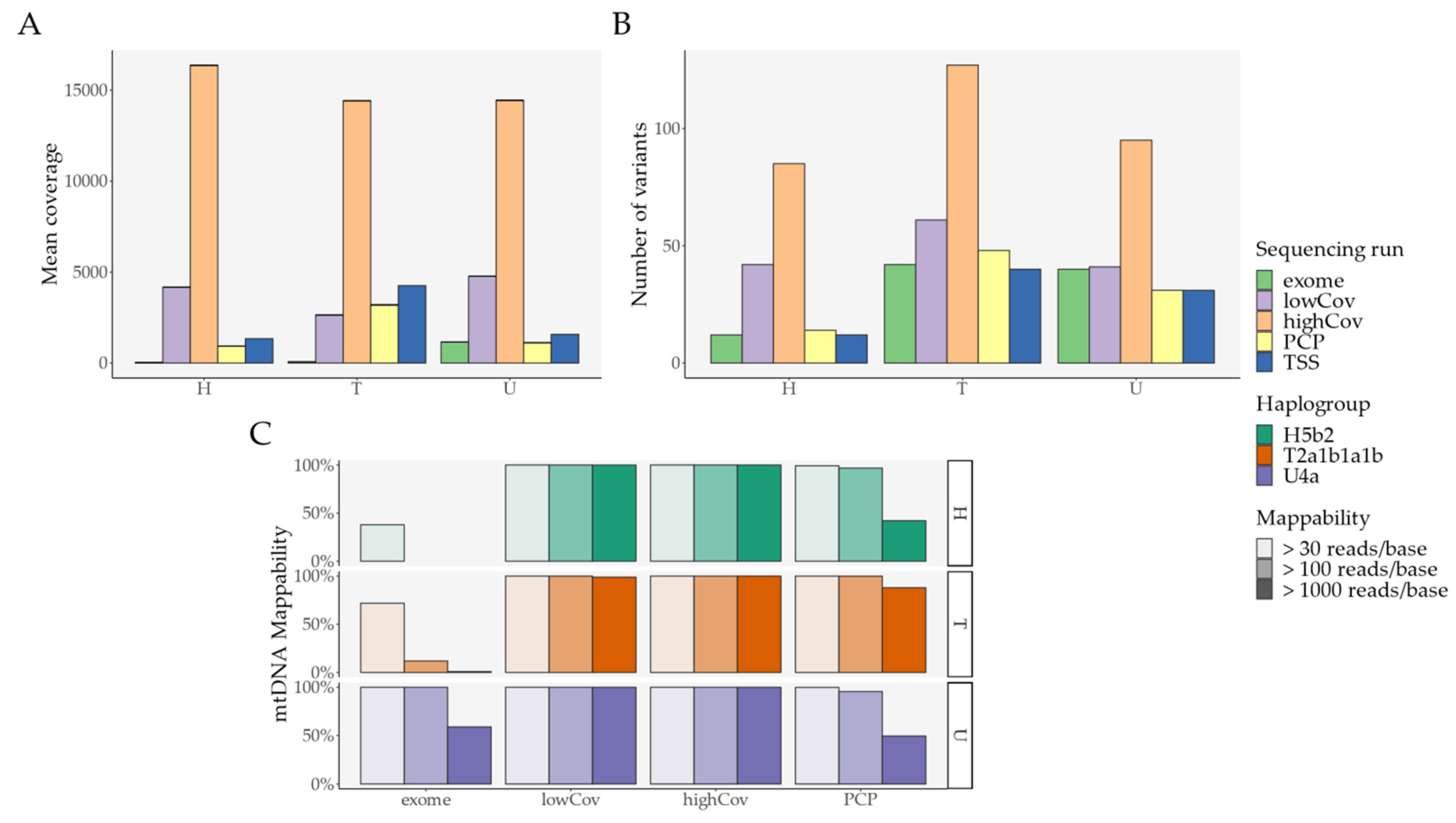
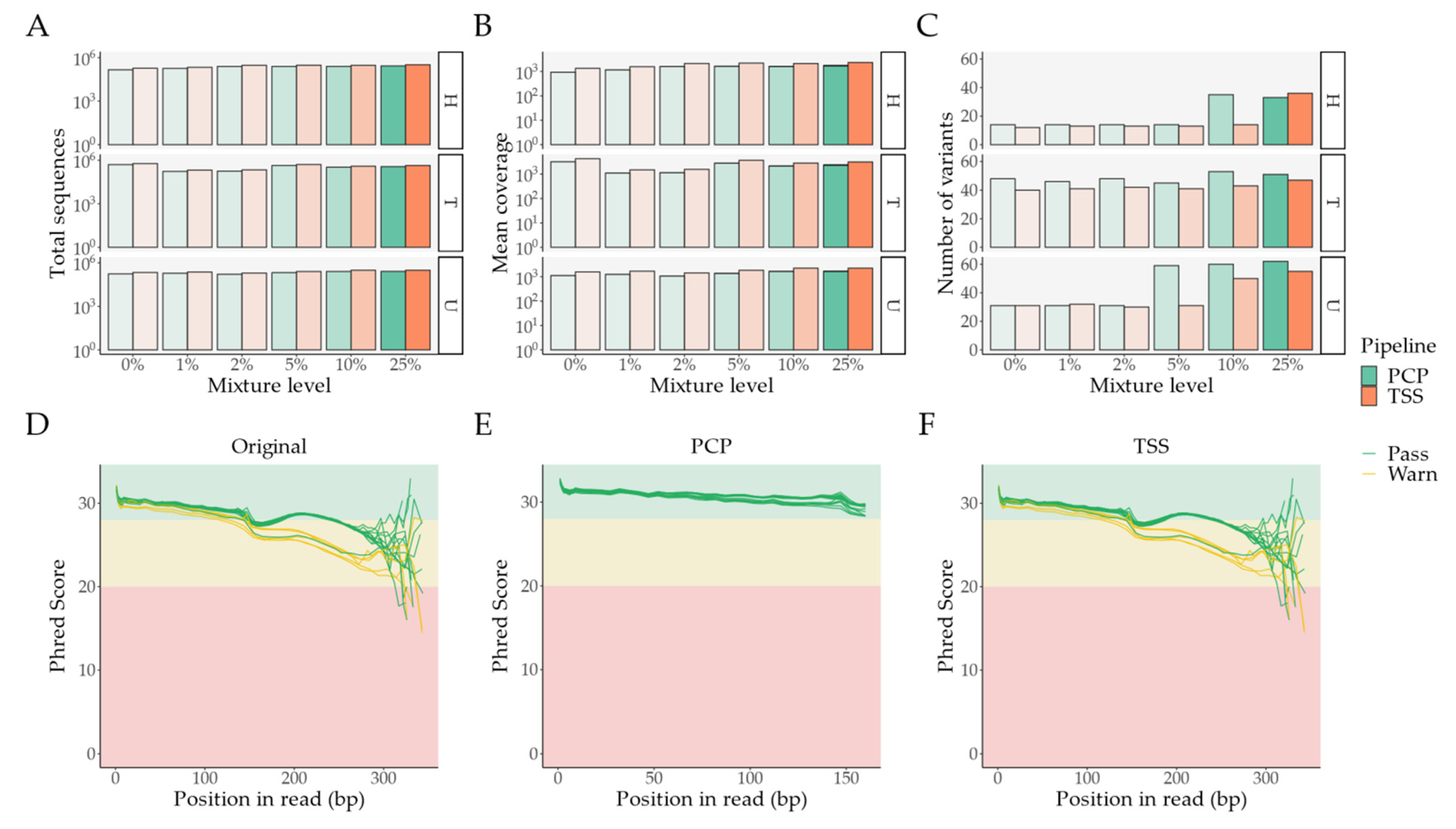
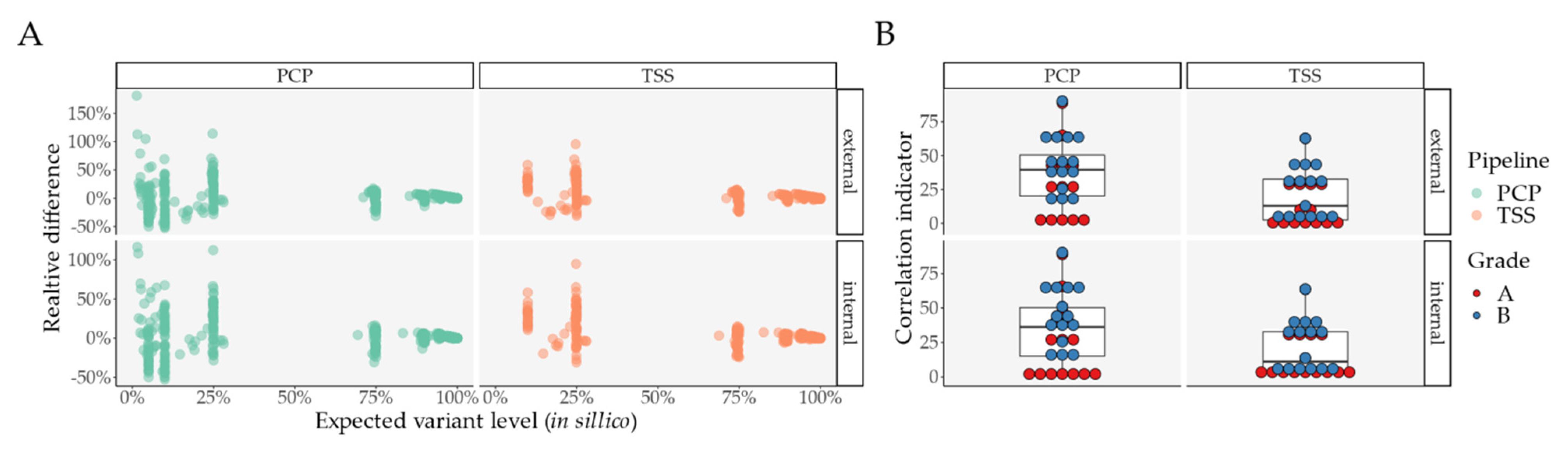
2.1. Pipeline Validation with Samples from the 1000 Genomes Project
- External comparison—the difference to the other Illumina sequencing methods (exome, lowCov, and highCov);
- Internal comparison—the difference within Ion Torrent, taking the primary sequencing as a reference.
2.2. Pipeline Comparison with An Independent Set of 50 Clinical Samples
3. Discussion and Conclusions
4. Materials and Methods
4.1. Sample Acquisition/Collection
- Mixture HT—Minor component: HG00256 (haplogroup H5b2, short identifier H) + Major component: HG01626 (haplogroup T2a1b1a1b, short identifier T);
- Mixture TU—Minor component: HG01626 (haplogroup T2a1b1a1b, short identifier T) + Major component: HG01757 (haplogroup U4a, short identifier U);
- Mixture UH—Minor component: HG01757 (haplogroup U4a, short identifier U) + Major component: HG00256 (haplogroup H5b2, short identifier H).
4.2. DNA Sequencing
4.3. Bioinformatic Processing
4.3.1. PrecisionCallerPipeline (PCP)
4.3.2. Ion Torrent Suite™ Software (TSS)
4.4. Data Analysis
- Grade A variants: homoplasmic variants (mean variant level ≥ 95%) found in both highCov and lowCov, regardless of exome;
- Grade B variants: heteroplasmic variants (mean variant level ≥ 0.4% and ≤ 95%) found in both highCov and lowCov, regardless of exome, or found in highCov plus exome, or lowCov plus exome;
- Grade C variants: found in a single sequencing run;
- Novel variants: found in the Ion Torrent runs only.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant Description | PCP | TSS | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Broad Classification | Detailed Classification | N | Mean Observed VL | Mean Primary VL | Mean Absolute Difference in VLs | N | Mean Observed VL | Mean Primary VL | Mean Absolute Difference in VLs |
| [Min–Max] | [Min–Max] | [Min–Max] | [Min–Max] | [Min–Max] | [Min–Max] | ||||
| Grade A variant found | including exome | 79 | 99.73% [98.50%–100.00%] | 99.61% [97.03%–100.00%] | 0.44% [0.00%–2.73%] | 77 | 99.43% [98.20%–100.00%] | 99.60% [97.03%–100.00%] | 0.65% [0.00%–2.77%] |
| Grade B variant found | including exome | 5 | 26.06% [3.50%–92.50%] | 28.59% [2.57%–94.77%] | 2.91% [0.93%–7.47%] | 2 | 55.65% [19.60%–91.70%] | 58.57% [22.37%–94.77%] | 2.92% [2.77%–3.07%] |
| excluding exome | 8 | 7.07% [2.50%–28.40%] | 6.36% [1.10%–28.35%] | 0.79% [0.05%–2.10%] | 1 | 28.30% | 28.35% | 0.05% | |
| Grade C variant found | from highCov | - | - | - | - | 1 | 21.70% | 0.40% | 21.30% |
| Grade A variant lost | including exome | - | - | - | - | 2 | - | 100.00% [100.00%–100.00%] | - |
| Grade B variant lost | including exome | 4 | - | 0.88% [0.50%–1.27%] | - | 7 | - | 4.19% [0.50%–17.37%] | - |
| excluding exome | 34 | - | 1.10% [0.40%–5.75%] | - | 41 | - | 1.46% [0.40%–6.20%] | - | |
| from lowCov and exome | 2 | - | 0.48% [0.45%–0.50%] | - | 2 | - | 0.48% [0.45%–0.50%] | - | |
| Grade C variant lost | from highCov | 177 | - | 0.82% [0.40%–5.80%] | - | 176 | - | 0.83% [0.40%–5.80%] | - |
| from lowCov | 12 | - | 0.93% [0.40%–3.80%] | - | 12 | - | 0.93% [0.40%–3.80%] | - | |
| from exome | 4 | - | 0.70% [0.40%–1.00%] | - | 4 | - | 0.70% [0.40%–1.00%] | - | |
| Novel variant | Only present in Ion Torrent | 1 | 2.70% | - | - | 2 | 25.15% [19.30%–31.00%] | - | - |
| Variant Description | PCP | TSS | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Macro | Meso | Micro | N | Mean Observed VL | Within Platform | Other Platforms | N | Mean Observed VL | Within Platform | Other Platforms |
| Mean Absolute Difference in VLs | Mean Absolute Difference in VLs | Mean Absolute Difference in VLs | Mean Absolute Difference in VLs | |||||||
| [Min–Max] | [Min–Max] | [Min–Max] | [Min–Max] | [Min–Max] | [Min–Max] | |||||
| Found variants | Found variants | Major Grade A | 270 | 92.51% [51.50%–100.00%] | 3.51% [0.00%–22.90%] | 3.73% [0.00%–23.42%] | 260 | 92.10% [56.80%–100.00%] | 3.40% [0.10%–18.20%] | 3.55% [0.00%–18.15%] |
| Major Grade B | 39 | 20.82% [2.60%–94.30%] | 1.73% [0.16%–6.32%] | 2.09% [0.29%–6.10%] | 15 | 43.18% [11.80%–94.20%] | 2.26% [0.14%–6.29%] | 2.25% [0.08%–4.98%] | ||
| Major Grade C | - | - | - | - | 5 | 28.48% [22.30%–34.10%] | 8.65% [1.03%–13.49%] | 28.11% [21.91%–33.72%] | ||
| Minor Grade A | 133 | 16.95% [2.50%–52.90%] | 4.38% [0.02%–28.00%] | 4.41% [0.06%–28.20%] | 73 | 25.31% [11.00%–48.30%] | 5.73% [0.00%–23.50%] | 5.75% [0.02%–23.60%] | ||
| Minor Grade B | 8 | 7.54% [3.30%–26.00%] | 1.90% [0.32%–3.95%] | 1.72% [0.14%–4.18%] | 1 | 26.30% | 3.38% | 2.61% | ||
| Shared Grade A | 125 | 99.89% [99.20%–100.00%] | 0.09% [0.00%–0.40%] | 0.20% [0.00%–2.41%] | 125 | 99.64% [98.60%–100.00%] | 0.20% [0.00%–1.30%] | 0.45% [0.00%–2.71%] | ||
| Mixture found variants | Major Grade B | 9 | 4.08% [2.60%–5.70%] | - | 0.44% [0.03%–1.69%] | 4 | 12.60% [11.90%–13.20%] | - | 3.12% [0.83%–3.99%] | |
| Lost variants | Mixture lost variants | Major Grade B | 26 | - | 2.77% [1.88%–3.56%] | 2.00% [0.83%–2.72%] | - | - | - | - |
| Minor Grade A | 137 | - | 2.44% [0.98%–24.80%] | 2.44% [0.97%–24.98%] | 187 | - | 3.86% [0.98%–10.00%] | 3.87% [0.97%–10.00%] | ||
| Minor Grade B | 57 | - | 0.48% [0.03%–4.63%] | 0.48% [0.01%–4.74%] | 14 | - | 2.65% [0.20%–9.17%] | 2.78% [0.22%–9.48%] | ||
| Old lost variants | Major Grade A | - | - | - | - | 10 | - | - | 91.40% [75.00%–99.00%] | |
| Major Grade B | 186 | - | - | 0.81% [0.30%–2.70%] | 241 | - | - | 1.44% [0.30%–16.50%] | ||
| Major Grade C | 430 | - | - | 0.72% [0.30%–5.74%] | 425 | - | - | 0.73% [0.30%–5.74%] | ||
| Minor Grade A | - | - | - | - | 10 | - | - | 8.60% [1.00%–25.00%] | ||
| Minor Grade B | 195 | - | - | 0.09% [0.00%–1.44%] | 245 | - | - | 0.16% [0.00%–4.34%] | ||
| Minor Grade C | 430 | - | - | 0.06% [0.00%–0.95%] | 425 | - | - | 0.06% [0.00%–0.95%] | ||
| Shared Grade C | 540 | - | - | 0.86% [0.40%–2.69%] | 540 | - | - | 0.86% [0.40%–2.69%] | ||
| Novel variants | Found variants | Minor novel | - | - | - | - | 5 | 21.24% [16.80%–26.00%] | 18.07% [11.98%–24.07%] | - |
| Shared novel | - | - | - | - | 4 | 31.65% [28.00%–35.10%] | 12.10% [8.10%–15.78%] | - | ||
| Novel variants | Novel variant: mixture | 12 | 27.26% [2.50%–99.20%] | - | - | 9 | 13.92% [10.60%–21.10%] | - | - | |
| Primary novel variants | Mixture lost variants | Major novel | 5 | - | 2.47% [2.02%–2.67%] | - | 5 | - | 28.33% [23.25%–30.69%] | - |
| Minor novel | 5 | - | 0.23% [0.03%–0.68%] | - | 5 | - | 1.15% [0.19%–3.10%] | - | ||
| Shared novel | - | - | - | - | 1 | - | 19.35% | - | ||

References
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA Mutations in Human Disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef]
- Tuppen, H.A.L.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA Mutations and Human Disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Gatk4-Mitochondria-Pipeline. Available online: https://github.com/gatk-workflows/gatk4-mitochondria-pipeline (accessed on 18 August 2021).
- Lee, H.Y.; Song, I.; Ha, E.; Cho, S.-B.; Yang, W.I.; Shin, K.-J. MtDNAmanager: A Web-Based Tool for the Management and Quality Analysis of Mitochondrial DNA Control-Region Sequences. BMC Bioinform. 2008, 9, 483. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Yao, Y.-G. MitoTool: A Web Server for the Analysis and Retrieval of Human Mitochondrial DNA Sequence Variations. Mitochondrion 2011, 11, 351–356. [Google Scholar] [CrossRef]
- Zhidkov, I.; Nagar, T.; Mishmar, D.; Rubin, E. MitoBamAnnotator: A Web-Based Tool for Detecting and Annotating Heteroplasmy in Human Mitochondrial DNA Sequences. Mitochondrion 2011, 11, 924–928. [Google Scholar] [CrossRef]
- Guo, Y.; Li, J.; Li, C.-I.; Shyr, Y.; Samuels, D.C. MitoSeek: Extracting Mitochondria Information and Performing High-Throughput Mitochondria Sequencing Analysis. Bioinformatics 2013, 29, 1210–1211. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vianello, D.; Sevini, F.; Castellani, G.; Lomartire, L.; Capri, M.; Franceschi, C. HAPLOFIND: A New Method for High-Throughput MtDNA Haplogroup Assignment. Hum. Mutat. 2013, 34, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.S.; Lee, H.Y.; Yang, W.I.; Shin, K.-J. MtDNAprofiler: A Web Application for the Nomenclature and Comparison of Human Mitochondrial DNA Sequences. J. Forensic Sci. 2013, 58, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. MtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr. Protoc. Bioinform. 2013, 44, 1.23.1–1.23.26. [Google Scholar] [CrossRef]
- Calabrese, C.; Simone, D.; Diroma, M.A.; Santorsola, M.; Guttà, C.; Gasparre, G.; Picardi, E.; Pesole, G.; Attimonelli, M. MToolBox: A Highly Automated Pipeline for Heteroplasmy Annotation and Prioritization Analysis of Human Mitochondrial Variants in High-Throughput Sequencing. Bioinformatics 2014, 30, 3115–3117. [Google Scholar] [CrossRef]
- Navarro-Gomez, D.; Leipzig, J.; Shen, L.; Lott, M.; Stassen, A.P.M.; Wallace, D.C.; Wiggs, J.L.; Falk, M.J.; van Oven, M.; Gai, X. Phy-Mer: A Novel Alignment-Free and Reference-Independent Mitochondrial Haplogroup Classifier. Bioinformatics 2015, 31, 1310–1312. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Shen, L.; Gonzalez, M.; Leipzig, J.; Lott, M.T.; Stassen, A.P.M.; Diroma, M.A.; Navarro-Gomez, D.; Yeske, P.; Bai, R.; et al. Mitochondrial Disease Sequence Data Resource (MSeqDR): A Global Grass-Roots Consortium to Facilitate Deposition, Curation, Annotation, and Integrated Analysis of Genomic Data for the Mitochondrial Disease Clinical and Research Communities. Mol. Genet. Metab. 2015, 114, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Vellarikkal, S.K.; Dhiman, H.; Joshi, K.; Hasija, Y.; Sivasubbu, S.; Scaria, V. Mit-o-Matic: A Comprehensive Computational Pipeline for Clinical Evaluation of Mitochondrial Variations from next-Generation Sequencing Datasets. Hum. Mutat. 2015, 36, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, H.; Forer, L.; Fuchsberger, C.; Schöpf, B.; Kloss-Brandstätter, A.; Specht, G.; Kronenberg, F.; Schönherr, S. MtDNA-Server: Next-Generation Sequencing Data Analysis of Human Mitochondrial DNA in the Cloud. Nucleic Acids Res. 2016, 44, W64–W69. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstätter, A.; Forer, L.; Specht, G.; Bandelt, H.-J.; Kronenberg, F.; Salas, A.; Schönherr, S. HaploGrep 2: Mitochondrial Haplogroup Classification in the Era of High-Throughput Sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef]
- Ishiya, K.; Ueda, S. MitoSuite: A Graphical Tool for Human Mitochondrial Genome Profiling in Massive Parallel Sequencing. PeerJ 2017, 5, e3406. [Google Scholar] [CrossRef]
- Rueda, M.; Torkamani, A. SG-ADVISER MtDNA: A Web Server for Mitochondrial DNA Annotation with Data from 200 Samples of a Healthy Aging Cohort. BMC Bioinform. 2017, 18, 373. [Google Scholar] [CrossRef]
- Preste, R.; Vitale, O.; Clima, R.; Gasparre, G.; Attimonelli, M. HmtVar: A New Resource for Human Mitochondrial Variations and Pathogenicity Data. Nucleic Acids Res. 2019, 47, D1202–D1210. [Google Scholar] [CrossRef]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and Revision of the Cambridge Reference Sequence for Human Mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Van Oven, M. PhyloTree Build 17: Growing the Human Mitochondrial DNA Tree. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e392–e394. [Google Scholar] [CrossRef]
- Dür, A.; Huber, N.; Parson, W. Fine-Tuning Phylogenetic Alignment and Haplogrouping of MtDNA Sequences. Int. J. Mol. Sci. 2021, 22, 5747. [Google Scholar] [CrossRef]
- Chaitanya, L.; Ralf, A.; van Oven, M.; Kupiec, T.; Chang, J.; Lagacé, R.; Kayser, M. Simultaneous Whole Mitochondrial Genome Sequencing with Short Overlapping Amplicons Suitable for Degraded DNA Using the Ion Torrent Personal Genome Machine. Hum. Mutat. 2015, 36, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Wai, K.T.; Barash, M.; Gunn, P. Performance of the Early Access AmpliSeqTM Mitochondrial Panel with Degraded DNA Samples Using the Ion TorrentTM Platform. Electrophoresis 2018, 39, 2776–2784. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Xu, Z.; Zhao, H.; Tu, Z.; Liu, Z.; Li, W.; Hu, L.; Wan, L. Concordance of Mitochondrial DNA Sequencing Methods on Bloodstains Using Ion PGMTM. Leg. Med. 2018, 32, 27–30. [Google Scholar] [CrossRef]
- Strobl, C.; Eduardoff, M.; Bus, M.M.; Allen, M.; Parson, W. Evaluation of the Precision ID Whole MtDNA Genome Panel for Forensic Analyses. Forensic. Sci. Int. Genet. 2018, 35, 21–25. [Google Scholar] [CrossRef]
- Cuenca, D.; Battaglia, J.; Halsing, M.; Sheehan, S. Mitochondrial Sequencing of Missing Persons DNA Casework by Implementing Thermo Fisher’s Precision ID MtDNA Whole Genome Assay. Genes 2020, 11, 1303. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.; Longobardi, A.; Børsting, C. Sequencing of Mitochondrial Genomes Using the Precision ID MtDNA Whole Genome Panel. Electrophoresis 2018, 39, 2766–2775. [Google Scholar] [CrossRef]
- Faccinetto, C.; Sabbatini, D.; Serventi, P.; Rigato, M.; Salvoro, C.; Casamassima, G.; Margiotta, G.; De Fanti, S.; Sarno, S.; Staiti, N.; et al. Internal Validation and Improvement of Mitochondrial Genome Sequencing Using the Precision ID MtDNA Whole Genome Panel. Int. J. Legal Med. 2021, 135, 2295–2306. [Google Scholar] [CrossRef]
- Strobl, C.; Churchill Cihlar, J.; Lagacé, R.; Wootton, S.; Roth, C.; Huber, N.; Schnaller, L.; Zimmermann, B.; Huber, G.; Lay Hong, S.; et al. Evaluation of Mitogenome Sequence Concordance, Heteroplasmy Detection, and Haplogrouping in a Worldwide Lineage Study Using the Precision ID MtDNA Whole Genome Panel. Forensic Sci. Int. Genet. 2019, 42, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Ming, T.; Wang, M.; Zheng, M.; Zhou, Y.; Hou, Y.; Wang, Z. Exploring of Rare Differences in MtGenomes between MZ Twins Using Massively Parallel Sequencing. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 70–72. [Google Scholar] [CrossRef]
- Woerner, A.E.; Ambers, A.; Wendt, F.R.; King, J.L.; Moura-Neto, R.S.; Silva, R.; Budowle, B. Evaluation of the Precision ID MtDNA Whole Genome Panel on Two Massively Parallel Sequencing Systems. Forensic Sci. Int. Genet. 2018, 36, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Cihlar, J.C.; Amory, C.; Lagacé, R.; Roth, C.; Parson, W.; Budowle, B. Developmental Validation of a MPS Workflow with a PCR-Based Short Amplicon Whole Mitochondrial Genome Panel. Genes 2020, 11, 1345. [Google Scholar] [CrossRef]
- Roth, C.; Parson, W.; Strobl, C.; Lagacé, R.; Short, M. MVC: An Integrated Mitochondrial Variant Caller for Forensics. Aust. J. Forensic Sci. 2019, 51, S52–S55. [Google Scholar] [CrossRef]
- Parson, W.; Gusmão, L.; Hares, D.R.; Irwin, J.A.; Mayr, W.R.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P.M.; et al. DNA Commission of the International Society for Forensic Genetics: Revised and Extended Guidelines for Mitochondrial DNA Typing. Forensic Sci. Int. Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef]
- Cho, S.; Kim, M.Y.; Lee, J.H.; Lee, S.D. Assessment of Mitochondrial DNA Heteroplasmy Detected on Commercial Panel Using MPS System with Artificial Mixture Samples. Int. J. Legal Med. 2018, 132, 1049–1056. [Google Scholar] [CrossRef]
- Churchill, J.D.; Stoljarova, M.; King, J.L.; Budowle, B. Massively Parallel Sequencing-Enabled Mixture Analysis of Mitochondrial DNA Samples. Int. J. Legal Med. 2018, 132, 1263–1272. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Forer, L.; Fendt, L.; Kheirkhah, A.; Salas, A.; Kronenberg, F.; Schoenherr, S. Contamination Detection in Sequencing Studies Using the Mitochondrial Phylogeny. Genome Res. 2021, 31, 309–316. [Google Scholar] [CrossRef]
- Smart, U.; Budowle, B.; Ambers, A.; Soares Moura-Neto, R.; Silva, R.; Woerner, A.E. A Novel Phylogenetic Approach for de Novo Discovery of Putative Nuclear Mitochondrial (PNumt) Haplotypes. Forensic Sci. Int. Genet. 2019, 43, 102146. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Fazzini, F.; Fendt, L.; Schönherr, S.; Forer, L.; Schöpf, B.; Streiter, G.; Losso, J.L.; Kloss-Brandstätter, A.; Kronenberg, F.; Weissensteiner, H. Analyzing Low-Level MtDNA Heteroplasmy—Pitfalls and Challenges from Bench to Benchmarking. Int. J. Mol. Sci. 2021, 22, 935. [Google Scholar] [CrossRef]
- Ring, J.D.; Sturk-Andreaggi, K.; Alyse Peck, M.; Marshall, C. Bioinformatic Removal of NUMT-Associated Variants in Mitotiling next-Generation Sequencing Data from Whole Blood Samples. Electrophoresis 2018, 39, 2785–2797. [Google Scholar] [CrossRef]
- Genomics England Research Consortium; NIHR BioResource; Wei, W.; Pagnamenta, A.T.; Gleadall, N.; Sanchis-Juan, A.; Stephens, J.; Broxholme, J.; Tuna, S.; Odhams, C.A.; et al. Nuclear-Mitochondrial DNA Segments Resemble Paternally Inherited Mitochondrial DNA in Humans. Nat. Commun. 2020, 11, 1740. [Google Scholar] [CrossRef] [PubMed]
- Woerner, A.E.; Cihlar, J.C.; Smart, U.; Budowle, B. Numt Identification and Removal with RtN! Bioinformatics 2020, 36, 5115–5116. [Google Scholar] [CrossRef] [PubMed]
- Cihlar, J.C.; Strobl, C.; Lagacé, R.; Muenzler, M.; Parson, W.; Budowle, B. Distinguishing Mitochondrial DNA and NUMT Sequences Amplified with the Precision ID MtDNA Whole Genome Panel. Mitochondrion 2020, 55, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.; Parson, W. Interpreting NUMTs in Forensic Genetics: Seeing the Forest for the Trees. Forensic Sci. Int. Genet. 2021, 53, 102497. [Google Scholar] [CrossRef]
- Yonova-Doing, E.; Calabrese, C.; Gomez-Duran, A.; Schon, K.; Wei, W.; Karthikeyan, S.; Chinnery, P.F.; Howson, J.M.M. An Atlas of Mitochondrial DNA Genotype-Phenotype Associations in the UK Biobank. Nat. Genet. 2021, 53, 982–993. [Google Scholar] [CrossRef]
- Sosa, M.X.; Sivakumar, I.K.A.; Maragh, S.; Veeramachaneni, V.; Hariharan, R.; Parulekar, M.; Fredrikson, K.M.; Harkins, T.T.; Lin, J.; Feldman, A.B.; et al. Next-Generation Sequencing of Human Mitochondrial Reference Genomes Uncovers High Heteroplasmy Frequency. PLoS Comput. Biol. 2012, 8, e1002737. [Google Scholar] [CrossRef]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive Pathogenicity of Mitochondrial Heteroplasmy in Healthy Human Individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659. [Google Scholar] [CrossRef]
- Li, M.; Schröder, R.; Ni, S.; Madea, B.; Stoneking, M. Extensive Tissue-Related and Allele-Related MtDNA Heteroplasmy Suggests Positive Selection for Somatic Mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 2491–2496. [Google Scholar] [CrossRef]
- Naue, J.; Hörer, S.; Sänger, T.; Strobl, C.; Hatzer-Grubwieser, P.; Parson, W.; Lutz-Bonengel, S. Evidence for Frequent and Tissue-Specific Sequence Heteroplasmy in Human Mitochondrial DNA. Mitochondrion 2015, 20, 82–94. [Google Scholar] [CrossRef]
- Guo, Y.; Li, C.-I.; Sheng, Q.; Winther, J.F.; Cai, Q.; Boice, J.D.; Shyr, Y. Very Low-Level Heteroplasmy MtDNA Variations Are Inherited in Humans. J. Genet. Genom. 2013, 40, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.A.; Wilton, P.R.; Su, M.S.-W.; Paul, I.M.; Arbeithuber, B.; Anthony, K.; Nekrutenko, A.; Nielsen, R.; Makova, K.D. Bottleneck and Selection in the Germline and Maternal Age Influence Transmission of Mitochondrial DNA in Human Pedigrees. Proc. Natl. Acad. Sci. USA 2019, 116, 25172–25178. [Google Scholar] [CrossRef] [PubMed]
- Wachsmuth, M.; Hübner, A.; Li, M.; Madea, B.; Stoneking, M. Age-Related and Heteroplasmy-Related Variation in Human MtDNA Copy Number. PLoS Genet. 2016, 12, e1005939. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive Molecular Characterization of Mitochondrial Genomes in Human Cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Fendt, L.; Fazzini, F.; Weissensteiner, H.; Bruckmoser, E.; Schönherr, S.; Schäfer, G.; Losso, J.L.; Streiter, G.A.; Lamina, C.; Rasse, M.; et al. Profiling of Mitochondrial DNA Heteroplasmy in a Prospective Oral Squamous Cell Carcinoma Study. Cancers 2020, 12, 1933. [Google Scholar] [CrossRef]
- Just, R.S.; Irwin, J.A.; Parson, W. Questioning the Prevalence and Reliability of Human Mitochondrial DNA Heteroplasmy from Massively Parallel Sequencing Data. Proc. Natl. Acad. Sci. USA 2014, 111, 4546–4547. [Google Scholar] [CrossRef]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Reply to Just et al. Mitochondrial DNA Heteroplasmy Could Be Reliably Detected with Massively Parallel Sequencing Technologies. Proc. Natl. Acad. Sci. USA 2014, 111, E4548–E4550. [Google Scholar] [CrossRef] [PubMed]
- Just, R.S.; Irwin, J.A.; Parson, W. Mitochondrial DNA Heteroplasmy in the Emerging Field of Massively Parallel Sequencing. Forensic Sci. Int. Genet. 2015, 18, 131–139. [Google Scholar] [CrossRef]
- Bris, C.; Goudenege, D.; Desquiret-Dumas, V.; Charif, M.; Colin, E.; Bonneau, D.; Amati-Bonneau, P.; Lenaers, G.; Reynier, P.; Procaccio, V. Bioinformatics Tools and Databases to Assess the Pathogenicity of Mitochondrial DNA Variants in the Field of Next Generation Sequencing. Front. Genet. 2018, 9, 632. [Google Scholar] [CrossRef] [PubMed]
- Brandhagen, M.D.; Just, R.S.; Irwin, J.A. Validation of NGS for Mitochondrial DNA Casework at the FBI Laboratory. Forensic Sci. Int. Genet. 2020, 44, 102151. [Google Scholar] [CrossRef] [PubMed]
- Poole, O.V.; Pizzamiglio, C.; Murphy, D.; Falabella, M.; Macken, W.L.; Bugiardini, E.; Woodward, C.E.; Labrum, R.; Efthymiou, S.; Salpietro, V.; et al. Mitochondrial DNA Analysis from Exome Sequencing Data Improves Diagnostic Yield in Neurological Diseases. Ann. Neurol. 2021, 89, 1240–1247. [Google Scholar] [CrossRef]
- Laricchia, K.M.; Lake, N.J.; Watts, N.A.; Shand, M.; Haessly, A.; Gauthier, L.; Benjamin, D.; Banks, E.; Soto, J.; Garimella, K.; et al. Mitochondrial DNA Variation across 56,434 Individuals in GnomAD. bioRxiv 2021. bioRxiv:2021.07.23.453510. [Google Scholar] [CrossRef]
- Bolze, A.; Mendez, F.; White, S.; Tanudjaja, F.; Isaksson, M.; Jiang, R.; Rossi, A.D.; Cirulli, E.T.; Rashkin, M.; Metcalf, W.J.; et al. A Catalog of Homoplasmic and Heteroplasmic Mitochondrial DNA Variants in Humans. bioRxiv 2020. bioRxiv:798264. [Google Scholar] [CrossRef]
- Rausser, S.; Trumpff, C.; McGill, M.A.; Junker, A.; Wang, W.; Ho, S.; Mitchell, A.; Karan, K.R.; Monk, C.; Segerstrom, S.C.; et al. Mitochondrial Phenotypes in Purified Human Immune Cell Subtypes and Cell Mixtures. bioRxiv 2021. bioRxiv:2020.10.16.342923. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Chervova, O.; Conde, L.; Guerra-Assunção, J.A.; Moghul, I.; Webster, A.P.; Berner, A.; Larose Cadieux, E.; Tian, Y.; Voloshin, V.; Jesus, T.F.; et al. The Personal Genome Project-UK, an Open Access Resource of Human Multi-Omics Data. Sci. Data 2019, 6, 257. [Google Scholar] [CrossRef]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Chowdhury, S.; Taft, R.J.; Lebo, M.S.; Buchan, J.G.; Harrison, S.M.; Rowsey, R.; Klee, E.W.; Liu, P.; Worthey, E.A.; et al. Best Practices for the Analytical Validation of Clinical Whole-Genome Sequencing Intended for the Diagnosis of Germline Disease. NPJ Genom. Med. 2020, 5, 47. [Google Scholar] [CrossRef]
- Wong, A.K.; Sealfon, R.S.G.; Theesfeld, C.L.; Troyanskaya, O.G. Decoding Disease: From Genomes to Networks to Phenotypes. Nat. Rev. Genet. 2021. [Google Scholar] [CrossRef]
- Amorim, A.; Fernandes, T.; Taveira, N. Mitochondrial DNA in Human Identification: A Review. PeerJ 2019, 7, e7314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shen, J.; Medico, L.; Platek, M.; Ambrosone, C.B. Length Heteroplasmies in Human Mitochondrial DNA Control Regions and Breast Cancer Risk. Int. J. Mol. Epidemiol. Genet. 2010, 1, 184–192. [Google Scholar] [PubMed]
- Sturk-Andreaggi, K.; Parson, W.; Allen, M.; Marshall, C. Impact of the Sequencing Method on the Detection and Interpretation of Mitochondrial DNA Length Heteroplasmy. Forensic Sci. Int. Genet. 2020, 44, 102205. [Google Scholar] [CrossRef]
- Bamford, J.M.; Sandercock, P.A.; Warlow, C.P.; Slattery, J. Interobserver Agreement for the Assessment of Handicap in Stroke Patients. Stroke 1989, 20, 828. [Google Scholar] [CrossRef] [PubMed]
- Köster, J.; Rahmann, S. Snakemake—A Scalable Bioinformatics Workflow Engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed]
- Aho, A.V.; Kernighan, B.W.; Weinberger, P.J. Awk—A Pattern Scanning and Processing Language. Softw. Pract. Exper. 1979, 9, 267–279. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.; Krueger, F.; Segonds-Pichon, A.; Biggins, L.; Krueger, C.; Wingett, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 18 August 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio, PBC: Boston, MA, USA, 2021. [Google Scholar]
- Chang, W. Extrafont: Tools for Using Fonts; 2014. [Google Scholar]
- Bray, A.; Ismay, C.; Chasnovski, E.; Baume, B.; Cetinkaya-Rundel, M. Infer: Tidy Statistical Inference; 2021. [Google Scholar]
- Ooms, J. Magick: Advanced Graphics and Image-Processing in R; 2021. [Google Scholar]
- Pedersen, T.L. Patchwork: The Composer of Plots; 2020. [Google Scholar]
- Wickham, H.; Bryan, J. Readxl: Read Excel Files; 2019. [Google Scholar]
- Hester, J.; Csárdi, G.; Wickham, H.; Chang, W.; Morgan, M.; Tenenbaum, D. Remotes: R Package Installation from Remote Repositories, Including “GitHub”; 2021. [Google Scholar]
- Wickham, H.; Seidel, D. Scales: Scale Functions for Visualization; 2020. [Google Scholar]
- Wickham, H.; Henry, L.; Pedersen, T.L.; Luciani, T.J.; Decorde, M.; Lise, V. Svglite: An “SVG” Graphics Device; 2021. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. JOSS 2019, 4, 1686. [Google Scholar] [CrossRef]
- European Organization for Nuclear Research. OpenAIRE Zenodo: Research. Shared; 2013. [Google Scholar] [CrossRef]





| Variable | PCP | TSS | ||
|---|---|---|---|---|
| Primary Novel Variants | Mixture Novel Variants | Primary Novel Variants | Mixture Novel Variants | |
| Normalized coverage | ↓ | ↓↓ | ↓↓↓ | ↓↓ |
| Coverage ratio | - | ↑↑↑ | ↑↑↑ | ↑ |
| Number of NUMTs | ↑↑↑ | ↓ | - | ↓↓ |
| LCR prevalence | ↓↓↓ | ↓↓↓ | ↑↑↑ | - |
| Distance to amplicon edge | ↓↓↓ | ↓↓ | ↓ | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes-Figueiredo, F.; Carvalho, F.S.; Fonseca, A.C.; Paul, F.; Ferro, J.M.; Schönherr, S.; Weissensteiner, H.; Morais, V.A. From Forensics to Clinical Research: Expanding the Variant Calling Pipeline for the Precision ID mtDNA Whole Genome Panel. Int. J. Mol. Sci. 2021, 22, 12031. https://doi.org/10.3390/ijms222112031
Cortes-Figueiredo F, Carvalho FS, Fonseca AC, Paul F, Ferro JM, Schönherr S, Weissensteiner H, Morais VA. From Forensics to Clinical Research: Expanding the Variant Calling Pipeline for the Precision ID mtDNA Whole Genome Panel. International Journal of Molecular Sciences. 2021; 22(21):12031. https://doi.org/10.3390/ijms222112031
Chicago/Turabian StyleCortes-Figueiredo, Filipe, Filipa S. Carvalho, Ana Catarina Fonseca, Friedemann Paul, José M. Ferro, Sebastian Schönherr, Hansi Weissensteiner, and Vanessa A. Morais. 2021. "From Forensics to Clinical Research: Expanding the Variant Calling Pipeline for the Precision ID mtDNA Whole Genome Panel" International Journal of Molecular Sciences 22, no. 21: 12031. https://doi.org/10.3390/ijms222112031
APA StyleCortes-Figueiredo, F., Carvalho, F. S., Fonseca, A. C., Paul, F., Ferro, J. M., Schönherr, S., Weissensteiner, H., & Morais, V. A. (2021). From Forensics to Clinical Research: Expanding the Variant Calling Pipeline for the Precision ID mtDNA Whole Genome Panel. International Journal of Molecular Sciences, 22(21), 12031. https://doi.org/10.3390/ijms222112031