6. Materials and Methods
6.1. Reagents and Constructs
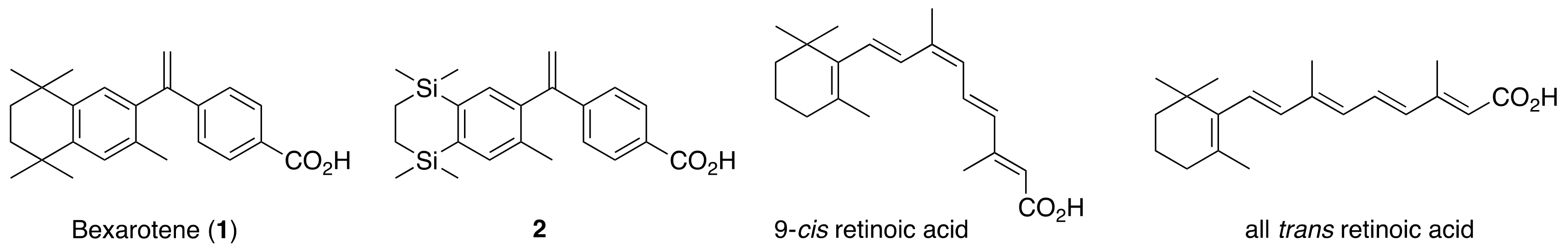
Bexarotene was from LC Laboratories. ATRA was from Sigma Aldrich. The ApoA1-Luciferase and pBABE-RXRA plasmids were a gift from Vivek Arora, Washington University. MSCV–Gal4 DBD–RXRA LBD–IRES–mCherry (Gal4-RXRA) has been previously described [
82].
6.2. Molecular Modeling
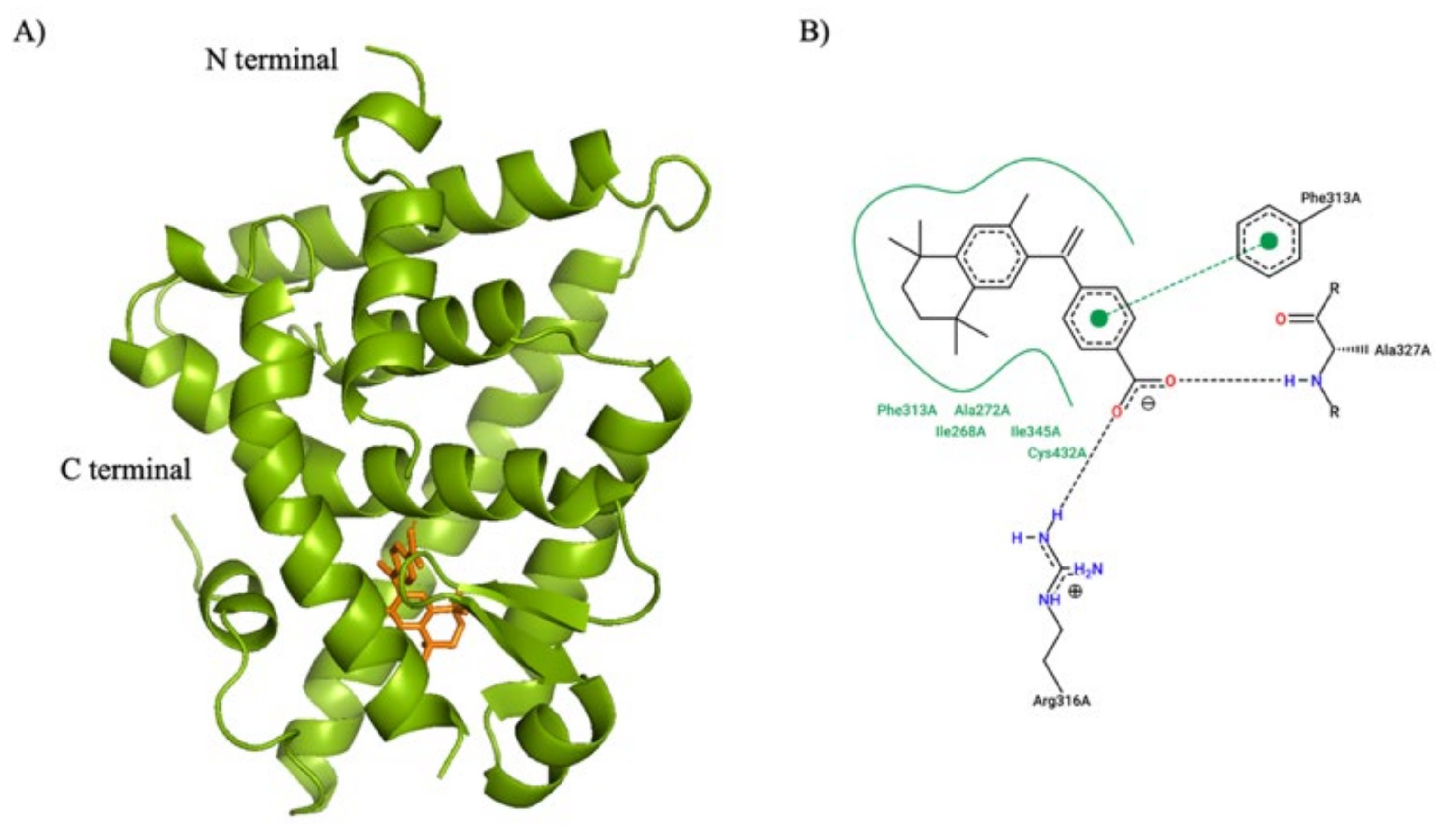
The three-dimensional structures of the compounds reported herein were generated using ChemDraw 3D (PerkinElmer Informatics), energy minimized, and exported in the Protein Data Bank (PDB) format. The human RXR alpha ligand binding domain structure model was obtained from the PDB (PDB code: 1FBY, [
83]). The crystallized ligand, 9-cis retinoic acid, was removed from the protein model prior to docking simulations. Furthermore, 9-cis retinoic acid was also used as a positive control in the docking studies presented here. Both the protein and ligand models were prepared using MGLTools (version 1.5.7) [
84] and screened virtually using AutoDock Vina [
85]. The search space volume (4,032 Å
3) was determined using MGLTools (center_x = 12.848, center_y = 29.174, center_z = 50.269, size_x = 16, size_y = 14, size_z = 18). The exhaustiveness was set to 8.
6.3. Cell Culture
UAS-GFP x KMT2A-MLLT3 cells were produced as described [
82] and cultured in vitro using expansion medium (RPMI1640 medium, 15% FBS, Scf (50 ng/mL), IL3 (10 ng/mL), L-glutamine (2 mM), sodium pyruvate (1 mM), HEPES buffer (10 mM), penicillin/streptomycin (100 units/mL), and β-mercaptoethanol (50 µM)).
6.4. UAS/Gal4 Assay
UAS-GFP x KMT2A-MLLT3 cells were transduced with retroviruses MSCV-Gal4 (DNA binding domain, DBD)—RXRA (ligand binding domain, LBD)—IRES—mCherry. Cells were treated, and after 48 h, GFP measured by a ZE5 Flow Cytometer (Biorad).
6.5. Luciferase Detection
293T cells were transfected using Lipofectamine 2000 (Invitrogen). Six hours after transfection, the cells were collected and plated into a 48-well plate in 1% BSA media in triplicate and treated with compounds. After 40 h incubation, the cells were harvested and assayed for luciferase (Luc Assay System with Reporter Lysis Buffer, Promega) in a Beckman Coulter LD400 plate reader.
6.6. LXRE Assay
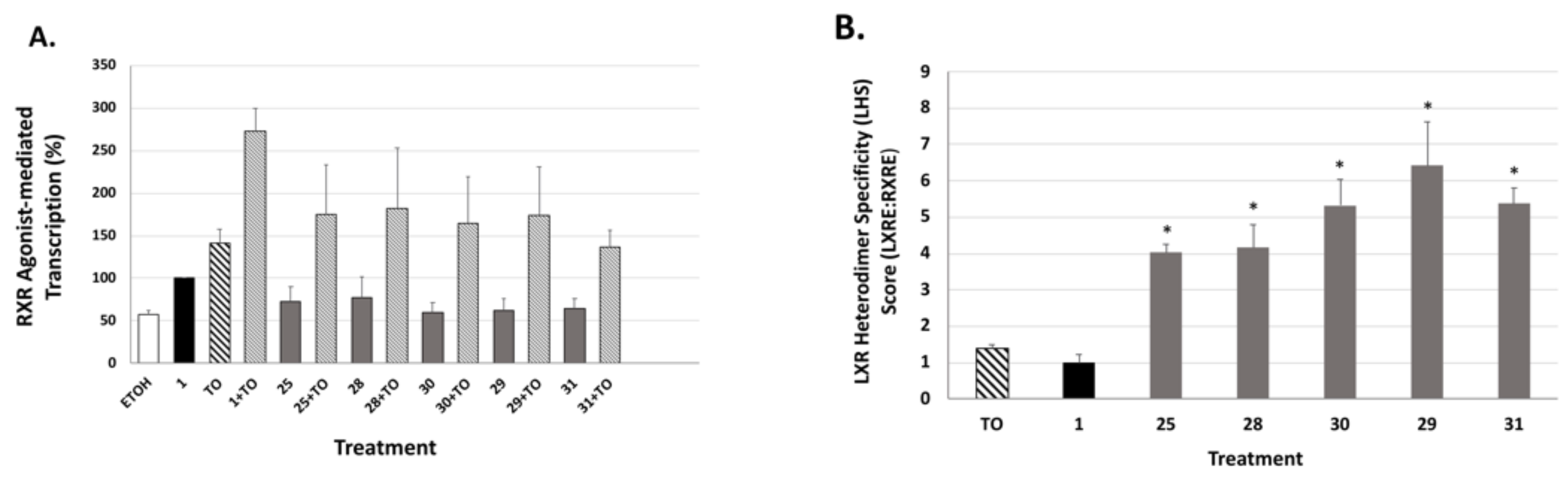
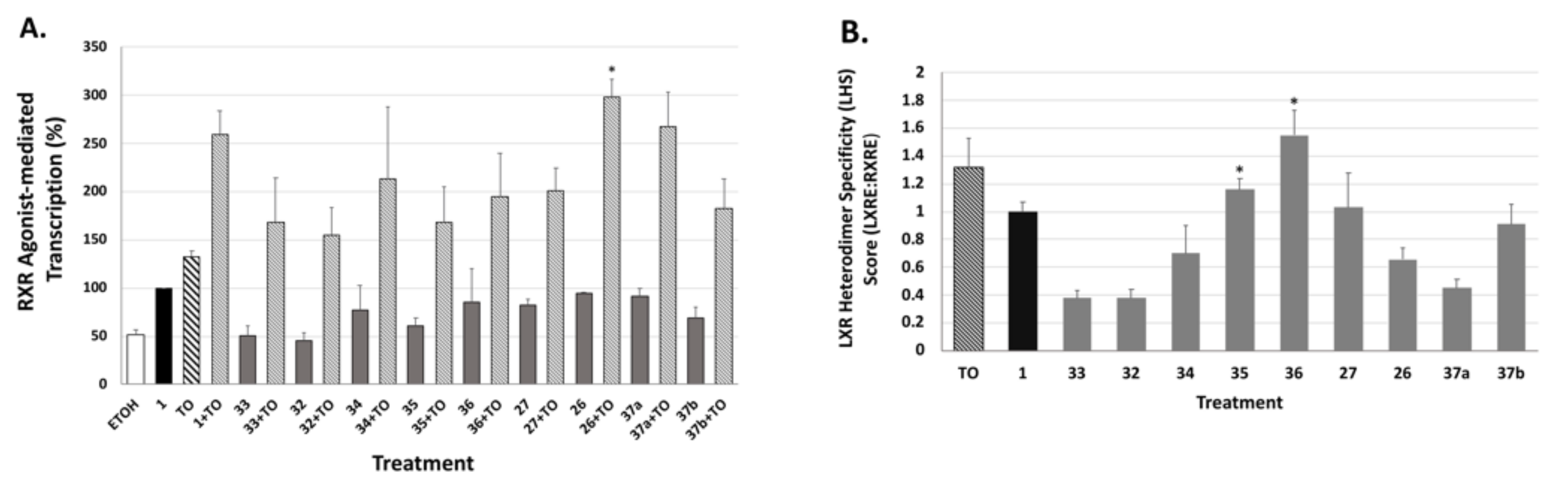
The LXRE-mediated assays were performed using human embryonic cells (HEK293) seeded at a density of 60,000 cells/well in a 24-well plate and maintained in DMEM (Hyclone) supplemented with 10% fetal bovine serum, 100 µg/mL streptomycin, 100 U/mL penicillin (Invitrogen, Carlsbad, CA, USA) at 37 degrees Celsius, 5% CO2 for 24 h. The cells were co-transfected with 250 ng of an LXRE-luciferase reporter gene, 50 ng of pSG5-human RXRα, and 20 ng of renilla control plasmid. The transfections were conducted using 1.25 µL of polyethylenimine (PEI) (Polysciences, Inc., Warrington, PA, USA) for 16–22 h. After transfection, the cells were treated with either ethanol vehicle control (0.1%), reference compound bexarotene (1) or analog, and/or T0901317 (an LXR ligand) at the indicated concentrations. After 24 h post treatment, the cells were lysed and the transcriptional activity mediated by the LXRE was measured using the Dual Luciferase Assay System (Promega, Madison, WI) in a Sirius FB12 luminometer (Berthold Detection Systems, Pforzheim, Germany) according to the manufacturer’s protocol. The data are a compilation of between three and six independent assays with each treatment group dosed in triplicate for each independent assay. The transcription efficiency on the LXRE was measured in comparison to the reference compound bexarotene (1) set to 100%. Bars on all graphs indicate standard deviation of the replicate experiments.
6.7. RARE Assay
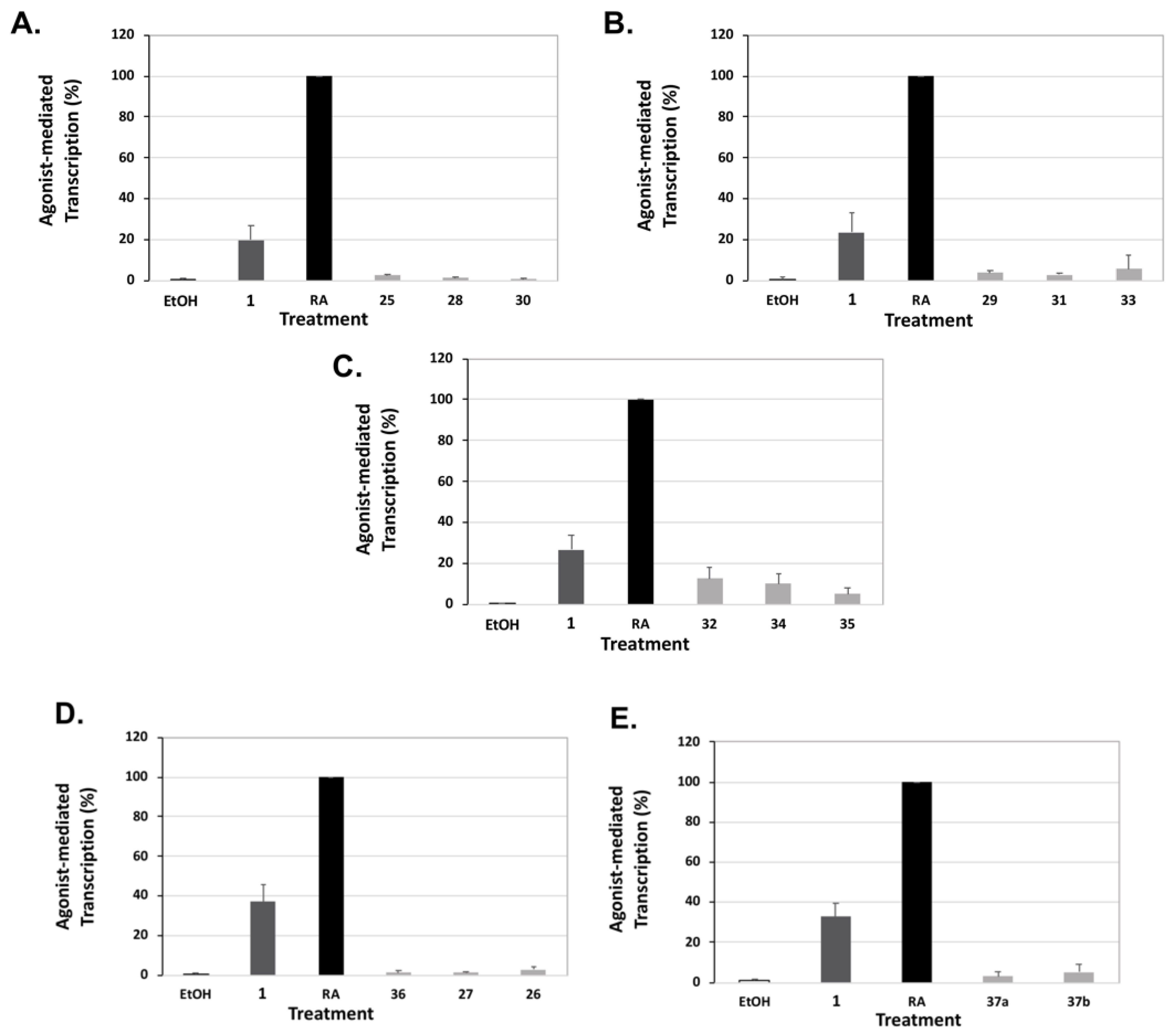
Human embryonic kidney cells (HEK293) were plated at 60,000 cells per well in a 24-well plate and maintained as described above. After 24 h, the cells were transfected with 250 ng pTK-DR5(X2)-Luc, 25 ng pCMX-human RARα, and 20 ng renilla utilizing 1.25 µL polyethylenimine (PEI) per well for 24 h. The sequence of the double DR5 RARE is: 5′-AAAGGTCACCGAAAGGTCACCATCCCGGGAGGTCACCGAAAGGTCACC-3′ (DR5 responsive elements underlined). The cells were treated with ethanol vehicle (0.1%), all-trans-retinoic acid (RA, the ligand for RAR), or the indicated rexinoid at a final concentration of 10 nM. After 24 h of treatment, the retinoid activity was measured as described above (dual luciferase assay). The activity of compound 1 or analog divided by the activity of all-trans-RA (expressed as a percentage) represents the RARE activity. Three independent assays were conducted with triplicate samples for each treatment group. The value for RA was set to 100%.
6.8. Cell Viability and Growth Analysis
UAS-GFP x KMT2A-MLLT3 cells were plated at 10,000 cells per well in 96-well plates with indicated compounds in 200 µL. These cells doubled every 8–10 h. After 48 h, 10 µL were replated in new media with indicated compounds re-applied in 200 µL. After an additional 96 h, the number of viable cells in 50 µL was determined using a ZE5 flow cytometer (Bio-rad) using forward scatter/side scatter and PE exclusion to isolate viable cells.
6.9. Data Analysis
Statistical analysis was performed using Prism (Graphpad). T-test was performed, as appropriate. Error bars represent standard deviation. Data points without error bars have standard deviations below Graphpad’s limit to display. For
Figure 8,
Figure 9 and
Figure 10, data are expressed as means ± SD. Statistical differences between two groups (generally the bexarotene control group versus bexarotene analog group) were determined by a two-sided Student’s
t-test. A
p-value of less than 0.05 was considered significant.
6.10. Mutagenicity and Toxicity Assay
All compounds were tested for toxicity and mutagenicity using a Saccharomyces cerevisiae based assay as described previously [
58]. Toxicity was assessed in this assay (
Table 1), comparing growth on plates to control treatments. Compounds were solubilized in DMSO at increasing concentrations and cells were incubated with the compounds for 3 h before plating on selective media or YPD to assess toxicity and mutagenicity. Cytotoxicity was assessed as described [
86]. Growth of colonies on the full nutrient YPD plate for each treatment was compared to the DMSO only control. The concentration at which 50% cell death (as indicated by colony count compared to DMSO only control) +/−10% cell death is reported as the 50% killing rate. The highest concentration tested was 11 µg/µL.
6.11. HPLC
All tested compounds were assessed on a Waters Acquity UPLC with QDA and PDA detectors. Compounds were assayed in ESI-mode on an ACE Excel C18-PFP (1.7 µm, 50 mm × 2.1 mm) column using a 0.1% formic acid/water:acetonitrile gradient over 5 min. HPLC traces for compounds
25–
36,
37a, and
37b are available in the
Supplementary Materials.
6.12. NMR and High Resolution Mass Spectrometry
A 400 MHz Bruker Avance III spectrometer was used to acquire
1H NMR and
13C NMR spectra. Chemical shifts (δ) are listed in ppm against residual non-deuterated solvent peaks in a given deuterated solvent (e.g., CHCl
3 in CDCl
3) as an internal reference. Coupling constants (J) are reported in Hz, and the abbreviations for splitting include: s, single; d, doublet; t, triplet; q, quartet; p, pentet; m, multiplet; br, broad. All
13C NMR spectra were acquired on a Bruker instrument at 100.6 MHz. Chemical shifts (δ) are listed in ppm against deuterated solvent carbon peaks as an internal reference. High resolution mass spectra were recorded using either a JEOL GCmate (2004), a JEOL LCmate (2002) high resolution mass spectrometer or an ABI Mariner (1999) ESI-TOF mass spectrometer. NMR spectra are available in the
Supplementary Materials.
6.13. General Procedures
Removal of volatile solvents transpired under reduced pressure using a Büchi rotary evaporator and is referred to as removing solvents in vacuo. Thin layer chromatography was conducted on precoated (0.25 mm thickness) silica gel plates with 60F-254 indicator (Merck). Column chromatography was conducted using 230–400 mesh silica gel (E. Merck reagent silica gel 60). All tested compounds were analyzed for purity by NMR as well as HPLC analysis and were found to be >95% pure.
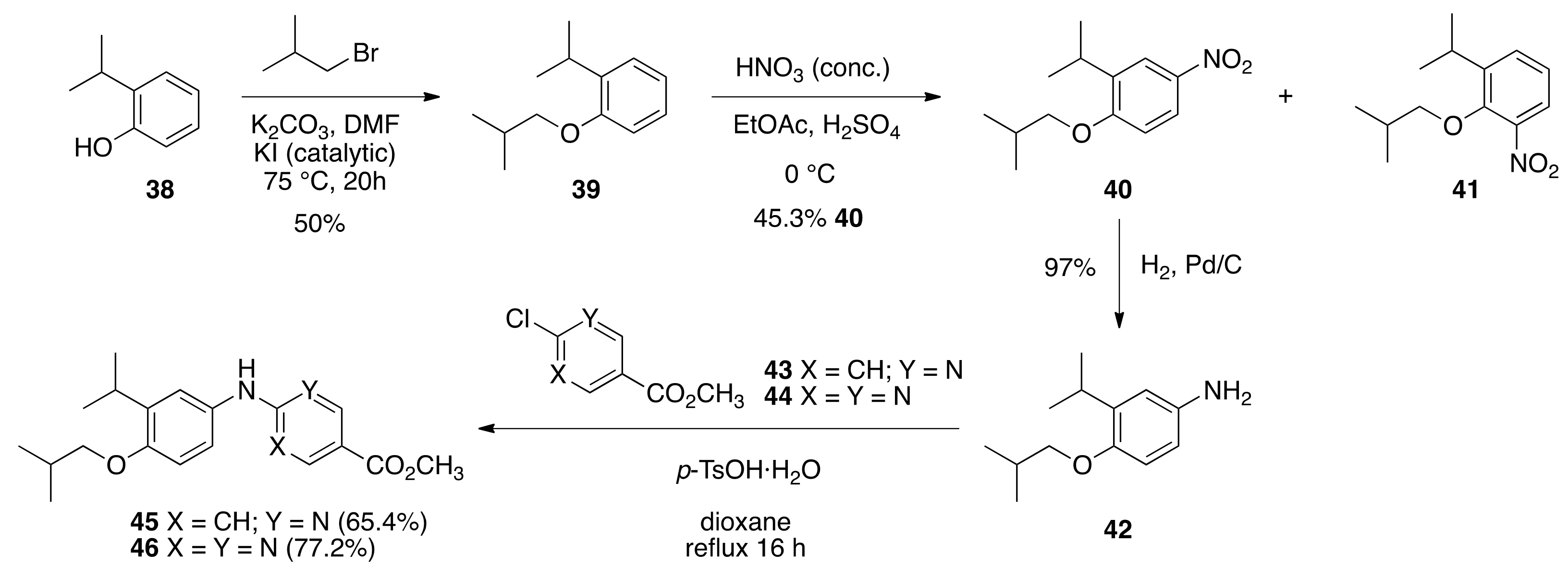
6.14. 1-isobutoxy-2-isopropylbenzene
(39) To a solution of 2-isopropylphenol (38) (12.5 mL, 92.9 mmols) and 1-bromo-2-methylpropane (20.5 mL, 189 mmols) in DMF (50 mL) was added finely ground potassium carbonate (13.9 g, 101 mmols) and potassium iodide (0.652 g, 3.9 mmols), and the reaction was stirred for 20 h at 70–75 °C. The reaction solution was then poured into water and extracted with ethyl acetate. The organic layers were washed with brine, dried over sodium sulfate, and concentrated in vacuo to provide a crude oil that was purified by column chromatography (1% ethyl acetate in hexanes) to give 39 as a colorless oil (8.7947 g, 50%): 1H NMR (400 MHz, CDCl3) δ 7.23 (d, J = 7.6, 1H), 7.16 (td, J = 8.0, 2.4, 1H), 6.93 (t, J = 7.6, 1H), 6.85 (d, J = 8.0, 1H), 3.76 (d, J = 6.4, 2H), 3.39 (hept, J = 6.8, 1H), 2.15 (nonet, J = 6.8, 1H), 1.27 (d, J = 6.8, 6H), 1.08 (d, J = 6.4, 6H); 13C NMR (100.6 MHz, CDCl3) δ156.3, 136.9, 126.4, 125.9, 120.2, 74.2, 28.5, 26.9, 22.6, 19.4; IR (neat) 2959, 1599, 1491, 1236 cm−1; GC-MS-CI (M + NH4)+ calcd for C13H24NO 210.1858, found 210.1850.
6.15. 1-isobutoxy-2-isopropyl-4-nitrobenzene (40)
To a solution of 1-isobutoxy-2-isopropylbenzene (39) (17.208 g, 89.486 mmols) in ethyl acetate (100 mL) at 0 °C was added concentrated (>90%) nitric acid (50.5 mL, 1.2 mols). The reaction was stirred at 0 °C for 40 min at which point it was carefully poured into water and extracted with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate to give a crude oil that consisted of 40 and 41 in a 3:1 ratio—TLC separates these isomers after four elutions in 1% ethyl acetate:hexanes (41 Rf ~ 0.5 and 40 Rf ~ 0.45). This crude oil was purified by column chromatography (0.7% to 1% to 5% ethyl acetate in hexanes) to give 40 (9.6225 g, 45.3%) as a pale yellow oil: 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 2.4, 1H), 8.06 (dd, J = 8.8, 2.8, 1H), 6.84 (d, J = 9.2, 1H), 3.83 (d, J = 6.4, 2H), 3.35 (hept, J = 6.8, 1H), 2.16 (nonet, J = 6.8, 1H), 1.25 (d, J = 6.8, 6H), 1.07 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ161.5, 141.2, 138.0, 123.3, 121.9, 110.2, 28.3, 27.0, 22.1, 19.2; IR (neat) 2962, 1588, 1512, 1336, 1251 cm−1; ES-MS (M + Na)+ calcd for C13H19NO3Na 260.1263, found 260.1256.
6.16. Methyl 6-((4-isobutoxy-3-isopropylphenyl)amino)nicotinate (45)
A solution of 1-isobutoxy-2-isopropyl-4-nitrobenzene (40) (2.0064 g, 8.455 mmols) in ethyl acetate (183 mL) was passed through a 10% Pd/C cartridge at 1.0 mL/min in the ThalesNano H-cube® at 65 °C and 2–5 bar pressure. The resulting solution was concentrated in vacuo to give 4-isobutoxy-3-isopropylaniline (42) (1.7057 g, 97%) as a yellow oil that was used without further purification: 1H NMR (400 MHz, CDCl3) δ 6.66 (d, J = 8.4, 1H), 6.63 (d, J = 2.8, 1H), 6.50 (dd, J = 8.4, 2.8, 1H), 3.64 (d, J = 6.4, 2H), 3.63 (br s, 1H), 3.31 (hept, J = 6.8, 1H), 2.09 (nonet, J = 6.8, 1H), 1.20 (d, J = 6.8, 6H), 1.03 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 149.8, 138.9, 138.2, 114.3, 113.1, 112.6, 75.2, 28.5, 26.8, 22.6, 19.4. To a solution of 42 (1.783 g, 8.60 mmols) and methyl 6-chloronicotinate (1.6567 g, 9.655 mmols) in dioxane (15.0 mL) was added para-toluenesulfonic acid monohydrate (1.7977 g, 9.45 mmols) and the reaction was refluxed overnight in an oil bath at 111 °C. The reaction was cooled to room temperature, and then the mixture was poured into water, extracted with ethyl acetate, and the organic layers were washed with brine, dried over sodium sulfate and concentrated to give a crude oil that was purified by column chromatography (150 mL SiO2, 6% ethyl acetate:hexanes) to give 45 (1.9259 g, 65.4%) as a white crystalline solid, m.p. 123–124 °C: 1H NMR (400 MHz, CDCl3) δ 8.76 (dd, J = 2.4, 0.8, 1H), 8.00 (dd, J = 8.8, 2.4, 1H), 7.77 (br s, 1H), 7.11 (s, 1H), 7.09 (dd, J = 7.6, 2.8, 1H), 6.82 (dd, J = 7.6, 0.8, 1H), 6.65 (dd, J = 8.8, 0.8, 1H), 3.86 (s, 3H), 3.74 (d, J = 6.0, 2H), 3.36 (hept, J = 6.8, 1H), 2.13 (nonet, J = 6.8, 1H), 1.22 (d, J = 6.8, 6H), 1.06 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 166.1, 160.1, 154.1, 151.1, 138.9, 138.4, 131.0, 122.2, 121.9, 115.9, 111.6, 105.5, 51.6, 28.4, 26.9, 22.5, 19.3; IR (neat) 3235, 2953, 1721, 1612, 1598, 1496, 1277, 1115 cm−1; ES-MS (M + H)+ calcd for C20H27N2O3 343.2022, found 343.2024.
6.17. Methyl 2-((4-isobutoxy-3-isopropylphenyl)amino)pyrimidine-5-carboxylate(46)
To a solution of 42 (1.7057 g, 8.23 mmols) and methyl 2-chloropyrimidine-5-carboxylate (1.5852 g, 9.1859 mmols) in dioxane (15.0 mL) was added para-toluenesulfonic acid monohydrate (1.7197 g, 9.04 mmols) and the reaction was refluxed overnight in an oil bath at 111 °C. The reaction was cooled to room temperature, and then the mixture was poured into water, extracted with ethyl acetate, and the organic layers were washed with brine, dried over sodium sulfate, and concentrated to give a crude oil that was purified by column chromatography (150 mL SiO2, 10% ethyl acetate:hexanes) to give 46 (2.1821 g, 71.3%) as a white crystalline solid, m.p. 122–124.2 °C: 1H NMR (400 MHz, CDCl3) δ 8.92 (s, 2H), 8.36 (br s, 1H), 7.44 (dd, J = 8.8, 2.4, 1H), 7.27 (d, J = 2.4, 1H), 6.83 (d, J = 8.8, 1H), 3.89 (s, 3H), 3.74 (d, J = 6.0, 2H), 3.37 (hept, J = 6.8, 1H), 2.12 (nonet, J = 6.8, 1H), 1.24 (d, J = 6.8, 6H), 1.06 (d, J = 6.4, 6H); 13C NMR (100.6 MHz, CDCl3) δ 164.7, 161.7, 160.0, 153.5, 137.7, 130.4, 120.1, 120.1, 114.4, 111.3, 74.5, 51.8, 28.4, 26.9, 22.5, 19.3; IR (neat) 3261, 2956, 1721, 1597, 803 cm−1; ES-MS (M + Na)+ calcd for C29H25N3O3Na 366.1794, found 366.1801.
6.18. Methyl 2-fluoro-4-((4-isobutoxy-3-isopropylphenyl)amino)benzoate (50)
To a solution of
42 (1.3452 g, 6.49 mmol), methyl 2-fluoro-4-iodobenzoate
47 [
74] (1.9807 g, 7.07 mmols), Cs
2CO
3 (5.5562 g, 17.08 mmol), and rac-BINAP (0.3386 g, 0.55 mmol) in toluene (8.6 mL) in a 100 mL round-bottomed flask was added Pd
2(dba)
3 (0.319 g, 1.82 mmol). The solution was sparged with nitrogen for 5 min, then a reflux condenser was fitted to the flask, the atmosphere was evacuated and back-filled with nitrogen (three times), and then the reaction was heated to reflux with stirring in an oil bath (125–120 °C) for 22 h. After cooling the reaction to room temperature, excess cesium carbonate and other solid particulates were filtered and washed with ethyl acetate, and the organic filtrate was concentrated in vacuo to obtain a crude product that was purified by column chromatography (150 mL SiO
2, 6% ethyl acetate:hexanes to 12% ethyl acetate: hexanes) to give
50 (2.147 g, 88.9%) as a crystalline solid, m.p. 93.4–100.9 °C:
1H NMR (400 MHz, CDCl
3) δ 7.84–7.75 (m, 1H), 7.01 (br s, 1H), 6.97–6.72 (m, 1H), 6.80 (d, J = 8.4, 1H), 6.54 (d, J = 8.4, 1H), 6.47 (d, J = 13.6, 1H), 3.86 (s, 3H), 3.75 (d, J = 6.0, 2H), 3.35 (hept, J = 6.8, 1H), 2.13 (nonet, J = 6.8, 1H), 1.21 (d, J = 6.8, 6H), 1.06 (d, J = 6.8, 6H);
13C NMR (100.6 MHz, CDCl
3) δ 165.3, 164.9, 164.9, 164.5, 164.4, 164.2, 161.6, 155.2, 153.9, 152.0, 151.9, 139.4, 138.5, 136.9, 133.6, 133.0, 131.9, 128.9, 128.3, 125.7, 125.5, 122.1, 121.7, 116.9, 112.1, 112.0, 111.9, 111.7, 109.7, 109.4, 109.3, 107.4, 107.3, 100.6, 100.3, 74.6, 51.6, 28.4, 26.9, 22.5, 22.4, 19.3; IR (neat) 3343, 2960, 1686, 1618, 1603, 1498, 1439 cm
−1; ES-MS (M + Na)+ calcd for C
21H
26NFO
3Na 382.1794, found 382.1795.
6.19. Methyl 4-((4-isobutoxy-3-isopropylphenyl)amino)benzoate (51)
To a solution of 42 (1.1807 g, 5.695 mmol), methyl 4-iodobenzoate 48 (1.6531 g, 6.308 mmols), Cs2CO3 (4.9777 g, 15.28 mmol), and rac-BINAP (0.3097 g, 0.50 mmol) in toluene (7.4 mL) in a 100 mL round-bottomed flask was added Pd2(dba)3 (0.2868 g, 1.64 mmol). The solution was sparged with nitrogen for 5 min, then a reflux condenser was fitted to the flask, the atmosphere was evacuated and back-filled with nitrogen (three times), and the reaction was heated to reflux with stirring in an oil bath (125–120 °C) for 22 h. After cooling the reaction to room temperature, excess cesium carbonate and other solid particulates were filtered and washed with ethyl acetate, and the organic filtrate was concentrated in vacuo to give a crude product that was purified by column chromatography (150 mL SiO2, 6% ethyl acetate:hexanes to 12% ethyl acetate: hexanes) to give 51 (1.0494 g, 54%) as a crystalline solid, m.p. 101.1–103.8 °C: 1H NMR (400 MHz, CDCl3) δ 8.13 (d, J = 8.4, 1H), 7.88 (br s, 1H), 7.69 (d, J = 8.4, 1H), 7.06 (d, J = 8.8, 1H), 6.80 (d, J = 8.4, 4H), 3.86 (s, 3H), 3.74 (d, J = 6.4, 2H), 3.35 (hept, J = 7.2, 1H), 2.13 (nonet, J = 6.8, 1H), 1.21 (d, J = 7.2, 6H), 1.06 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 167.0, 151.2, 131.5, 131.5, 131.4, 131.4, 130.8, 130.2, 129.7, 127.2, 121.4, 113.1, 74.6, 51.5, 28.5, 26.9, 22.5, 19.3; IR (neat) 3379, 2958, 1684, 1612, 1591, 1283, 1177 cm−1; ES-MS (M + Na)+ calcd for C21H27NO3Na 364.1889, found 364.1896.
6.20. Methyl 4-((4-isobutoxy-3-isopropylphenyl)amino)-3-nitrobenzoate (52)
To a solution of
42 (1.68 g, 8.10 mmol), methyl 4-iodo-3-nitrobenzoate
49 [
68] (2.63 g, 8.57 mmols), Cs
2CO
3 (5.44 g, 16.7 mmol), and rac-BINAP (0.4286 g, 0.7472 mmol) in toluene (10.8 mL) in a 100 mL round-bottomed flask was added Pd
2(dba)
3 (0.404 g, 2.31 mmol). The solution was sparged with nitrogen for 5 min, then a reflux condenser was fitted to the flask, the atmosphere was evacuated and back-filled with nitrogen (three times), and the reaction was heated to reflux with stirring in an oil bath (125–120 °C) for 22 h. After cooling the reaction to room temperature, excess cesium carbonate and other solid particulates were filtered and washed with ethyl acetate, and the organic filtrate was concentrated in vacuo to give a crude product that was purified by column chromatography (150 mL SiO
2, 4% ethyl acetate:hexanes to 12% ethyl acetate: hexanes) to give
52 (2.5619 g, 81.8%) as a crystalline solid, m.p. 78.1–82.3 °C:
1H NMR (400 MHz, CDCl
3) δ 9.71 (br s, 1H), 8.90 (d, J = 2.0, 1H), 7.90 (ddd, J = 9.2, 2.0, 0.4, 1H), 7.07 (d, J = 2.8, 1H), 7.04 (dd, J = 8.8, 2.8, 4H), 7.00 (d, J = 8.8, 1H), 6.86 (d, J = 8.4, 1H), 3.89 (s, 3H), 3.77 (d, J = 6.4, 2H), 3.37 (hept, J = 6.8, 1H), 2.13 (nonet, J = 6.8, 1H), 1.22 (d, J = 6.8, 6H), 1.07 (d, J = 6.8, 6H);
13C NMR (100.6 MHz, CDCl
3) δ 165.4, 155.2, 147.2, 138.9, 135.7, 131.5, 129.5, 129.2, 128.9, 128.3, 124.0, 123.9, 118.2, 115.5, 111.7, 74.5, 52.0, 28.4, 26.9, 22.4, 19.3; IR (neat) 3340, 2958, 1716, 1705, 1622, 1212, 757 cm
−1; ES-MS (M + Na)+ calcd for C
21H
26N
2O
5Na 409.1740, found 409.1750.
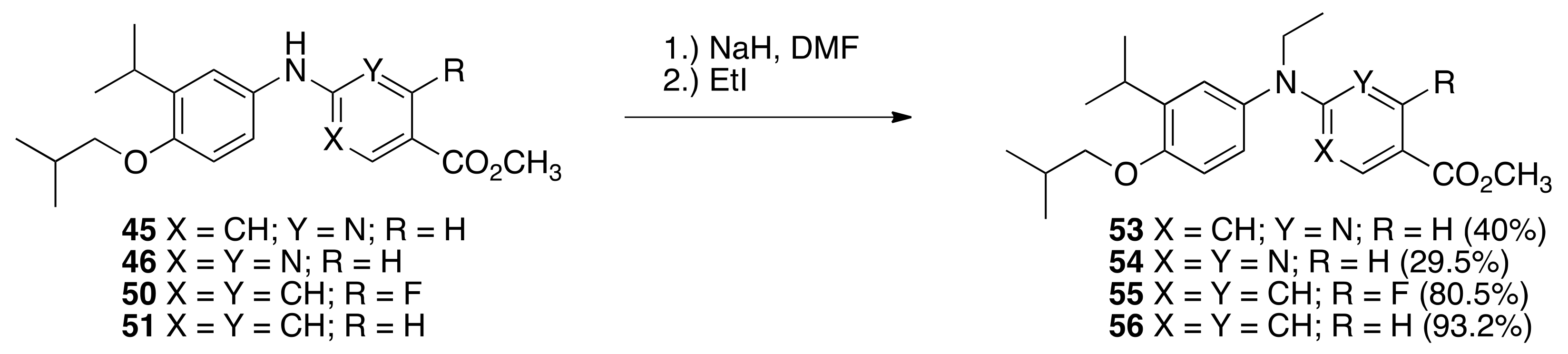
6.21. Methyl 6-(ethyl(4-isobutoxy-3-isopropylphenyl)amino)nicotinate (53)
To a flame-dried, 100 mL round-bottomed flask equipped with a magnetic stir bar was added a 60% dispersion of sodium hydride in mineral oil (0.2351 g, 5.88 mmol). The dispersion of sodium hydride was washed with hexanes (3 mL, twice) and dried under vacuum and suspended in 3.0 mL of DMF under nitrogen. To this solution of sodium hydride in DMF was added a solution of 45 (0.8331 g, 2.433 mmol) in DMF (9.2 mL), and the reaction was stirred for 15 min, and then ethyl iodide (0.30 mL, 3.8 mmol) was added, and the reaction was stirred for 1 h. The reaction was poured into water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo to yield a crude product that was purified by column chromatography (150 mL SiO2, 6% ethyl acetate:hexanes) to give 53 (0.3629 g, 40%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.83 (dd, J = 2.0, 0.4, 1H), 7.77 (dd, J = 8.8, 2.4, 1H), 7.01 (d, J = 2.4, 1H), 6.96 (dd, J = 8.4, 2.4, 1H), 6.86 (d, J = 8.4, 1H), 6.14 (dd, J = 8.8, 0.4, 1H), 4.00 (q, J = 7.2, 2H), 3.85 (s, 3H), 3.76 (d, J = 6.4, 2H), 3.36 (hept, J = 6.8, 1H), 2.14 (nonet, J = 6.8, 1H), 1.21 (t, J = 7.2, 3H), 1.20 (d, J = 6.8, 6H), 1.08 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 165.3, 162.8, 159.7, 155.1, 138.1, 135.3, 125.4, 125.3, 112.7, 111.2, 74.2, 51.6, 46.4, 28.4, 27.1, 22.4, 19.4, 12.7; IR (neat) 2959, 1711, 1596, 1495, 1263 cm−1; ES-MS (M)+ calcd for C22H30N2O3 370.2256, found 370.2242.
6.22. Methyl 2-(ethyl(4-isobutoxy-3-isopropylphenyl)amino)pyrimidine-5-carboxylate (54)
To a flame-dried, 100 mL round-bottomed flask equipped with a magnetic stir bar was added a 60% dispersion of sodium hydride in mineral oil (0.2377 g, 5.95 mmol). The dispersion of sodium hydride was washed with hexanes (3 mL, twice) and dried under vacuum and suspended in 3.0 mL of DMF under nitrogen. To this solution of sodium hydride in DMF was added a solution of 46 (0.8442 g, 2.458 mmol) in DMF (9.2 mL), and the reaction was stirred for 15 min, and then ethyl iodide (0.30 mL, 3.8 mmol) was added, and the reaction was stirred for 1 h. The reaction was poured into water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo to yield a crude product that was purified by column chromatography (150 mL SiO2, 6% ethyl acetate:hexanes) to give 54 (0.2698 g, 29.5%) as a white crystalline solid, m.p. 122.8–125.8 °C: 1H NMR (400 MHz, CDCl3) δ 8.83 (br s, 2H), 7.03 (d, J = 2.8, 1H), 6.99 (dd, J = 8.4, 2.8, 1H), 6.85 (d, J = 8.4, 1H), 4.02 (q, J = 6.8, 2H), 3.86 (s, 3H), 3.75 (d, J = 6.4, 2H), 3.35 (hept, J = 6.8, 1H), 2.13 (nonet, J = 6.8, 1H), 1.24 (t, J = 6.8, 3H), 1.23 (d, J = 6.8, 6H), 1.06 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 165.3, 162.8, 159.7, 155.1, 138.1, 135.3, 125.4, 125.3, 112.7, 111.2, 74.2, 51.6, 46.4, 28.4, 27.1, 22.4, 19.4, 12.7; IR (neat) 2960, 1708, 1595, 1494, 1284, 805 cm−1; ES-MS (M + Na)+ calcd for C21H29N3O3Na 394.2107, found 394.2109.
6.23. Methyl 4-(ethyl(4-isobutoxy-3-isopropylphenyl)amino)-2-fluorobenzoate (55)
To a flame-dried, 100 mL round-bottomed flask equipped with a magnetic stir bar was added a 60% dispersion of sodium hydride in mineral oil (0.2461 g, 6.16 mmol). The dispersion of sodium hydride was washed with hexanes (3 mL, twice) and dried under vacuum and suspended in 3.0 mL of DMF under nitrogen. To this solution of sodium hydride in DMF was added a solution of 50 (0.8918 g, 2.301 mmol) in DMF (9.2 mL), and the reaction was stirred for 15 min, and then ethyl iodide (0.30 mL, 3.8 mmol) was added, and the reaction was stirred for 1 h. The reaction was poured into water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo to yield a crude product that was purified by column chromatography (150 mL SiO2, 6% ethyl acetate:hexanes) to give 55 (0.7741 g, 80.5%) as a white crystalline solid, m.p. 66–71 °C: 1H NMR (400 MHz, CDCl3) δ 7.70 (t, J = 8.8, 1H), 6.98 (d, J = 2.4, 1H), 6.93 (dd, J = 8.8, 2.4, 1H), 6.84 (d, J = 8.8, 1H), 6.32 (dd, J = 8.8, 2.4, 1H), 6.22 (dd, J = 15.2, 2.4, 1H), 3.85 (s, 3H), 3.76 (d, J = 6.0, 2H), 3.69 (q, J = 7.2, 2H), 3.35 (hept, J = 6.8, 1H), 2.14 (nonet, J = 6.8, 1H), 1.22 (t, J = 7.2, 3H), 1.22 (d, J = 6.8, 6H), 1.08 (d, J = 6.4, 6H); 13C NMR (100.6 MHz, CDCl3) δ 165.1, 165.1, 162.6, 155.0, 154.1, 154.0, 139.0, 137.2, 133.1, 133.0, 126.0, 125.9, 111.9, 108.1, 105.2, 105.1, 99.8, 99.6, 74.4, 51.5, 46.9, 28.5, 27.0, 22.5, 19.4, 12.2; IR (neat) 2957, 1707, 1692, 1620, 1495, 1299, 763 cm−1; ES-MS (M)+ calcd for C23H30FNO3 387.2210, found 387.2200.
6.24. Methyl 4-(ethyl(4-isobutoxy-3-isopropylphenyl)amino)benzoate (56)
To a flame-dried, 100 mL round-bottomed flask equipped with a magnetic stir bar was added a 60% dispersion of sodium hydride in mineral oil (0.2417 g, 6.05 mmol). The dispersion of sodium hydride was washed with hexanes (3 mL, twice) and dried under vacuum and suspended in 3.0 mL of DMF under nitrogen. To this solution of sodium hydride in DMF was added a solution of 51 (0.8581 g, 2.513 mmol) in DMF (9.2 mL), and the reaction was stirred for 15 min, and then ethyl iodide (0.30 mL, 3.8 mmol) was added, and the reaction was stirred for 1 h. The reaction was poured into water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo to yield a crude product that was purified by column chromatography (150 mL SiO2, 6% ethyl acetate:hexanes) to give 56 (0.8655 g, 93.2%) as a white crystalline solid, m.p. 86.8–89.9 °C: 1H NMR (400 MHz, CDCl3) δ 7.80 (dd, J = 7.2, 2.4, 1H), 7.01 (d, J = 2.8, 1H), 6.95 (dd, J = 8.4, 2.4, 1H), 6.84 (d, J = 8.4, 1H), 6.58 (dd, J = 7.2, 2.0, 1H), 3.84 (s, 3H), 3.76 (d, J = 6.0, 2H), 3.71 (q, J = 7.2, 2H), 3.35 (hept, J = 7.2, 1H), 2.14 (nonet, J = 6.8, 1H), 1.24 (t, J = 6.8, 3H), 1.21 (d, J = 7.2, 6H), 1.08 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 167.3, 154.7, 152.2, 138.8, 137.9, 131.0, 126.0, 125.9, 117.3, 112.0, 111.8, 74.4, 51.4, 46.7, 28.5, 27.0, 22.5, 19.4, 12.3; IR (neat) 2958, 1698, 1609, 1598, 1495, 1269, 1179, 767 cm−1; ES-MS (M)+ calcd for C23H31NO3Na 392.2202, found 392.2196.
6.25. Methyl 3-amino-4-((4-isobutoxy-3-isopropylphenyl)amino)benzoate (57)
A solution of 52 (1.2000 g, 3.105 mmols) in ethyl acetate (63 mL) was passed through a 10% Pd/C cartridge at 1.0 mL/minute in the ThalesNano H-cube® at 65 °C and 2–5 bar pressure. The resulting solution was concentrated in vacuo to give 57 (quantitative yield) as a yellow oil that was used without further purification: 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 2.0, 1H), 7.45 (dd, J = 5.6, 2.0, 1H), 6.95 (d, J = 8.8, 1H), 6.94 (d, J = 2.8, 1H), 6.83 (dd, J = 8.8, 2.8, 1H), 6.78 (d, J = 8.8, 1H), 5.52 (br s, 1H), 3.85 (s, 3H), 3.72 (d, J = 6.4), 3.50 (br s, 2H), 3.35 (hept, J = 6.8, 1H), 2.13 (nonet, J = 6.8, 1H), 1.20 (d, J = 6.8, 6H), 1.06 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 167.2, 152.4, 139.3, 138.3, 134.7, 134.2, 122.9, 121.6, 119.6, 118.8, 114.1, 111.9, 60.3, 51.6, 28.5, 26.9, 22.5, 21.0, 19.4, 14.1; IR (neat) 3379, 2959, 1697, 1496, 1296 cm−1; ES-MS (M + Na)+ calcd for C21H28N2O3Na 379.1998, found 379.1998.
6.26. Methyl 1-(4-isobutoxy-3-isopropylphenyl)-1H-benzo[d][1,2,3]triazole-5-carboxylate (58)
To a solution of 57 (1.0839 g, 3.04 mmols) in THF (14.0 mL) at 0 °C was added a 1:1 solution of concentrated sulfuric acid and water (14.0 mL) followed by a solution of NaNO2 (0.3292 g, 4.77 mmols) in water (14.0 mL), and the reaction was allowed to warm to room temperature and stirred for 1 h. The reaction was poured into ethyl acetate, extracted, and the organic layers were combined, washed with saturated sodium bicarbonate and brine, dried over sodium sulfate, filtered, and concentrated to give crude 58, which was purified by column chromatography (150 mL SiO2, 4–6% ethyl acetate:hexanes) to give 58 (0.9791 g, 87.6%) as a crystalline solid, m.p. 86–89 °C: 1H NMR (400 MHz, CDCl3) δ 8.83 (d, J = 0.8, 1H), 8.19 (dd, J = 8.8, 1.6, 1H), 7.65 (dd, J = 8.8, 0.4, 1H), 7.53 (d, J = 2.8, 1H), 7.45 (dd, J = 8.8, 2.8, 1H), 6.99 (d, J = 8.8, 1H), 3.98 (s, 3H), 3.82 (d, J = 6.0, 2H), 3.44 (hept, J = 6.8, 1H), 2.18 (nonet, J = 6.4, 1H), 1.28 (d, J = 6.8, 6H), 1.09 (d, J = 6.4, 6H); 13C NMR (100.6 MHz, CDCl3) δ 166.4, 156.8, 145.9, 138.9, 134.8, 129.0, 128.6, 126.4, 122.9, 121.5, 121.4, 111.4, 110.2, 52.4, 28.4, 27.1, 22.3, 19.3; IR (neat) 2962, 1711, 1616, 1505, 1471, 1239, 750 cm−1; ES-MS (M + Na)+ calcd for C21H25N3O3Na 390.1794, found 390.1794.
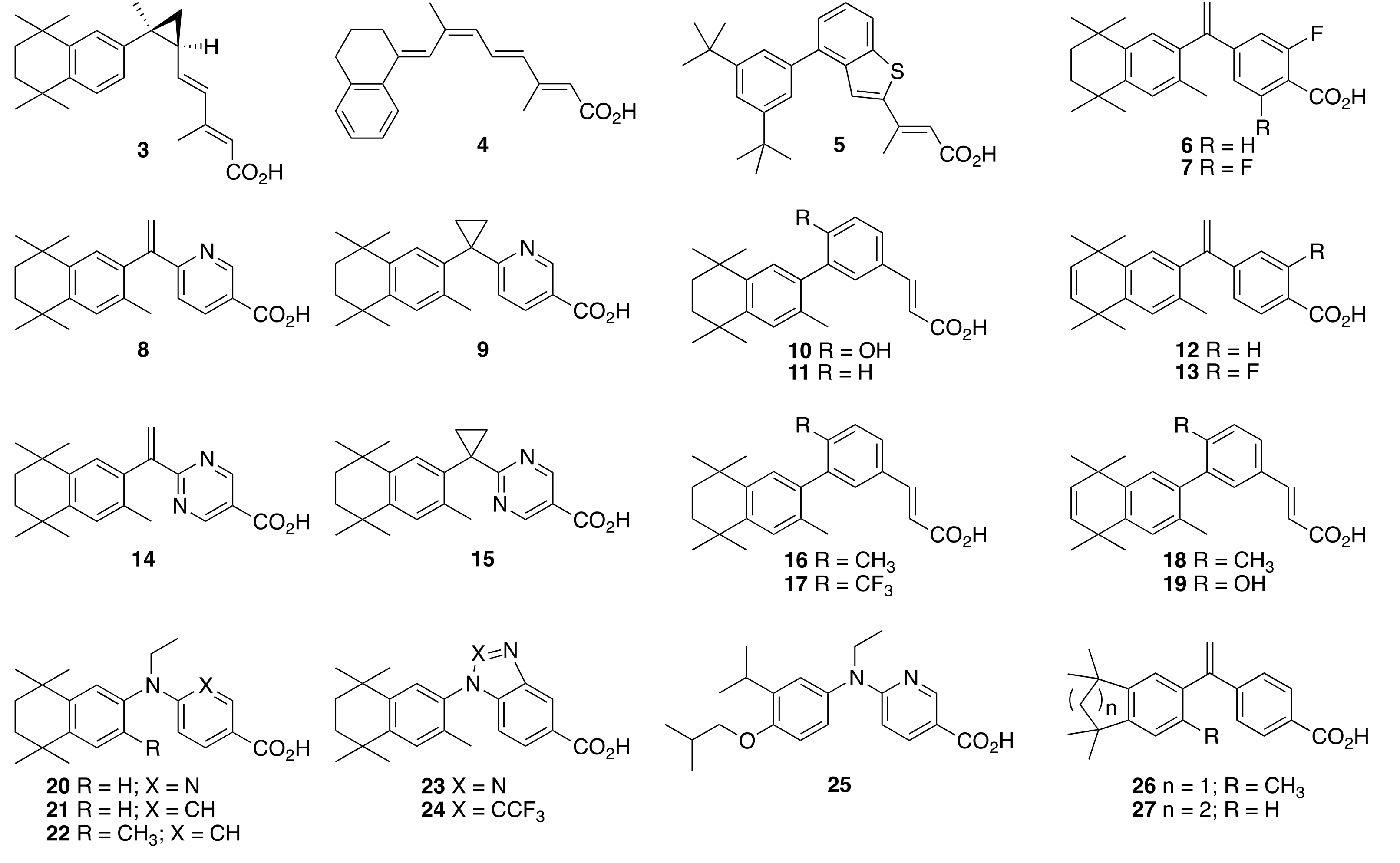
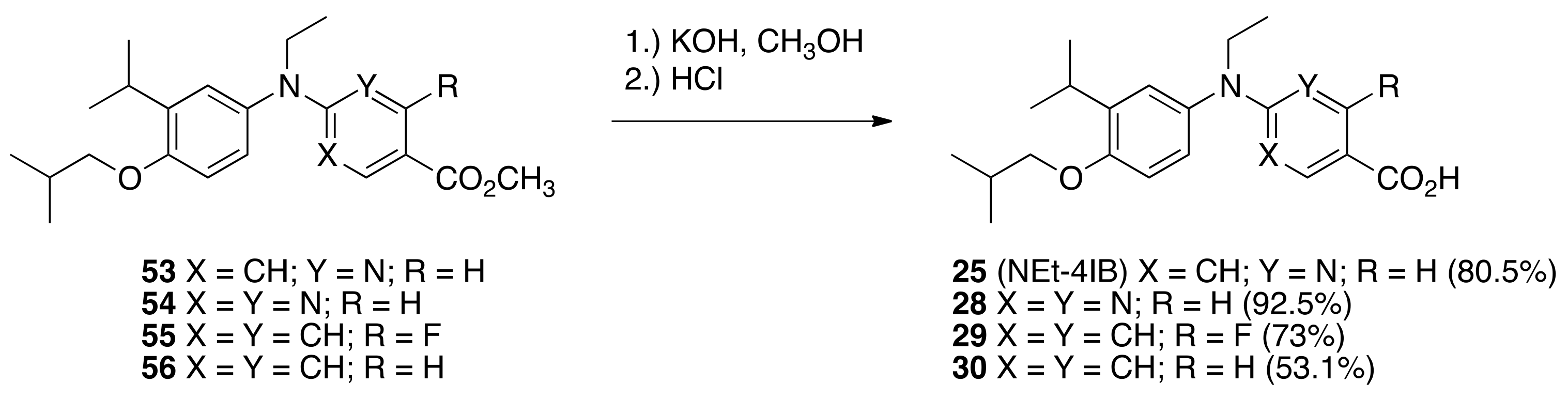
6.27. 6-(ethyl(4-isobutoxy-3-isopropylphenyl)amino)nicotinic acid (25) (NEt-4IB)
To a solution of 53 (0.9265 g, 2.501 mmols) in methanol (9.0 mL) was added a solution of KOH (0.4545 g, 8.100 mmols) in water (0.56 mL) and the solution was heated to reflux with stirring for 1 h. The solution was then cooled to room temperature, quenched with 1 N HCl (80 mL), extracted with ethyl acetate, the organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo to give a crude product that was purified by column chromatography (25 mL SiO2, 20–52% ethyl acetate:hexanes) to give 25 (0.7184 g, 80.5%) as a crystalline solid, m.p. 158.5–168.2 °C: 1H NMR (400 MHz, CDCl3) δ10.90 (br s, 1H), 8.92 (d, J = 2.4, 1H), 7.82 (dd, J = 9.2, 2.4, 1H), 7.03 (d, J = 2.4, 1H), 6.98 (dd, J = 8.4, 2.4, 1H), 6.87 (d, J = 8.4, 1H), 6.17 (d, J = 9.2, 1H), 4.03 (q, J = 7.2, 2H), 3.77 (d, J = 6.4, 2H), 3.36 (hept, J = 6.8, 1H), 2.15 (nonet, J = 6.4, 1H), 1.24 (t, J = 7.2, 3H), 1.22 (d, J = 7.2, 6H), 1.08 (d, J = 6.4, 6H); 13C NMR (100.6 MHz, CDCl3) δ 171.4, 160.9, 155.4, 151.6, 139.1, 137.9, 135.6, 126.0, 125.9, 113.3, 111.9, 107.5, 74.4, 45.6, 28.4, 27.0, 22.5, 19.3, 12.9; IR (neat) 2959, 1661, 1592, 1495, 1271, 785 cm−1; ES-MS (M–H)- calcd for C21H27N2O3 355.2022, found 355.2022.
6.28. 2-(ethyl(4-isobutoxy-3-isopropylphenyl)amino)pyrimidine-5-carboxylic acid (28)
To a solution of 54 (0.7580 g, 2.04 mmols) in methanol (7.3 mL) was added a solution of KOH (0.3846 g, 6.854 mmols) in water (0.46 mL) and the solution was heated to reflux with stirring for 1 h. The solution was then cooled to room temperature, quenched with 1 N HCl (80 mL), and the resulting precipitate was filtered to give a crude product that was purified by column chromatography (25 mL SiO2, 20–60% ethyl acetate:hexanes) to give 28 (0.6753 g, 92.6%) as a crystalline solid, m.p. 201.5-202.8 °C: 1H NMR (400 MHz, CDCl3) δ10.65 (br s, 1H), 8.88 (br s, 2H), 7.03 (d, J = 2.4, 1H), 7.00 (dd, J = 8.4, 2.4, 1H), 6.86 (d, J = 8.4, 1H), 4.05 (q, J = 7.2, 2H), 3.74 (d, J = 6.0, 2H), 3.35 (hept, J = 6.8, 1H), 2.13 (nonet, J = 6.4, 1H), 1.27 (t, J = 7.2, 3H), 1.25 (d, J = 6.8, 6H), 1.08 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 169.8, 162.7, 160.4, 155.3, 138.3, 135.0, 125.5, 125.3, 112.0, 111.3, 74.2, 46.6, 28.4, 27.1, 22.4, 19.4, 12.7; IR (neat) 2961, 1663, 1589, 1518, 1495, 1271, 807 cm−1; ES-MS (M–H)- calcd for C20H26N3O3 356.1974, found 356.1965.
6.29. 4-(ethyl(4-isobutoxy-3-isopropylphenyl)amino)-2-fluorobenzoic acid (29)
To a solution of 55 (0.6837 g, 1.764 mmols) in methanol (6.0 mL) was added a solution of KOH (0.3493 g, 6.225 mmols) in water (0.41 mL) and the solution was heated to reflux with stirring for 1 h. The solution was then cooled to room temperature, quenched with 1 N HCl (80 mL), and the resulting precipitate was filtered to give a crude product that was purified by column chromatography (25 mL SiO2, 10–60% ethyl acetate:hexanes) to give 29 (0.4821 g, 73%) as a crystalline solid, m.p. 184–186 °C: 1H NMR (400 MHz, CDCl3) δ10.01 (br s, 1H), 7.77 (t, J = 9.2, 1H), 6.99 (d, J = 2.4, 1H), 6.94 (dd, J = 8.4, 2.4, 1H), 6.85 (d, J = 8.8, 1H), 6.34 (d, J = 2.4, 1H), 6.32 (d, J = 2.4, 1H), 3.76 (d, J = 6.4, 2H), 3.70 (q, J = 7.2, 2H), 3.74 (d, J = 7.2, 2H), 3.35 (hept, J = 6.8, 1H), 2.15 (nonet, J = 6.8, 1H), 1.24 (t, J = 7.2, 3H), 1.22 (d, J = 6.8, 6H), 1.08 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 169.7, 165.8, 163.3, 155.2, 154.8, 154.7, 139.1, 137.0, 133.7, 126.0, 125.9, 111.9, 108.1, 104.1, 104.0, 99.8, 99.5, 74.4, 47.0, 28.4, 27.0, 22.5, 19.3, 12.2; IR (neat) 2958, 1667, 1613, 1600, 1496, 1299, 1273, 1244, 836 cm−1; ES-MS (M–H)- calcd for C22H27FNO3 372.1975, found 372.1982.
6.30. 4-(ethyl(4-isobutoxy-3-isopropylphenyl)amino)benzoic acid (30)
To a solution of 56 (0.7035 g, 1.904 mmols) in methanol (6.8 mL) was added a solution of KOH (0.3626 g, 6.462 mmols) in water (0.43 mL) and the solution was heated to reflux with stirring for 1 h. The solution was then cooled to room temperature, quenched with 1 N HCl (80 mL), and the resulting precipitate was filtered to give a crude product that was purified by column chromatography (25 mL SiO2, 10–60% ethyl acetate:hexanes) to give 30 (0.3594 g, 53.1%) as a crystalline solid, m.p. 179.4–181.0 °C: 1H NMR (400 MHz, CDCl3) δ10.89 (br s, 1H), 7.88 (d, J = 8.8, 2H), 7.03 (d, J = 2.8, 1H), 6.97 (dd, J = 8.8, 2.8, 1H), 6.86 (d, J = 8.4, 1H), 6.60 (d, J = 9.2, 2H), 3.77 (d, J = 6.4, 2H), 3.75 (q, J = 7.2, 2H), 3.36 (hept, J = 6.8, 1H), 2.15 (nonet, J = 6.8, 1H), 1.25 (t, J = 7.2, 3H), 1.23 (d, J = 7.2, 6H), 1.09 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 172.4, 154.8, 152.9, 138.9, 137.7, 131.8, 126.0, 116.3, 112.0, 111.8, 74.4, 46.8, 28.5, 27.0, 22.5, 19.4, 12.3; IR (neat) 2955, 1664, 1593, 1268, 1273, 1181, 773 cm−1; ES-MS (M–H)- calcd for C22H28NO3 354.2069, found 354.2077.
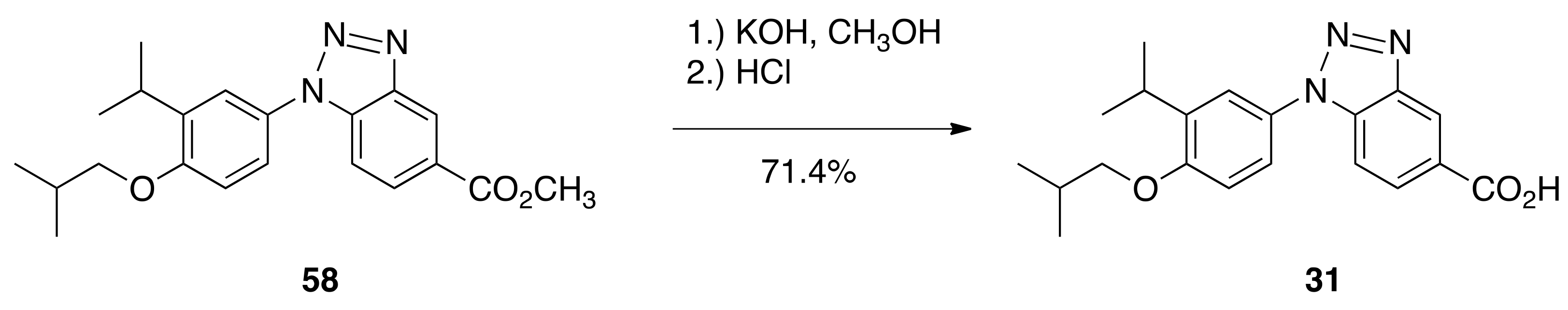
6.31. 1-(4-isobutoxy-3-isopropylphenyl)-1H-benzo[d][1,2,3]triazole-5-carboxylic acid (31)
To a solution of 58 (0.7544 g, 2.053 mmols) in methanol (5.2 mL) was added a solution of KOH (0.3795 g, 6.764 mmols) in water (0.45 mL) and the solution was heated to reflux with stirring for 1 h. The solution was then cooled to room temperature, quenched with 1 N HCl (80 mL), and the resulting precipitate was filtered to give a crude product that was purified by column chromatography (150 mL SiO2, 20–60% ethyl acetate:hexanes) to give 31 (0.5182 g, 71.4%) as a crystalline solid, m.p. 177.3–178.7 °C: 1H NMR (400 MHz, CDCl3) δ10.91 (br s, 1H), 8.98 (t, J = 0.8, 1H), 8.27 (dd, J = 8.8, 1.6, 1H), 7.71 (dd, J = 8.8, 0.4, 1H), 7.55 (d, J = 2.4, 1H), 7.47 (dd, J = 8.8, 2.4, 1H), 7.01 (d, J = 8.8, 1H), 3.84 (d, J = 6.4, 2H), 3.45 (hept, J = 6.8, 1H), 2.19 (nonet, J = 6.8, 1H), 1.30 (d, J = 6.8, 6H), 1.10 (d, J = 6.8, 6H); 13C NMR (100.6 MHz, CDCl3) δ 171.3, 156.9, 145.8, 139.0, 135.3, 129.0, 128.9, 125.7, 124.0, 121.6, 121.5, 111.5, 110.4, 74.7, 28.4, 27.1, 22.4, 19.3; IR (neat) 2955, 1675, 1613, 1495, 1302, 1242, 1028, 814 cm−1; ES-MS (M–H)- calcd for C20H22N3O3 352.1661, found 352.1657.
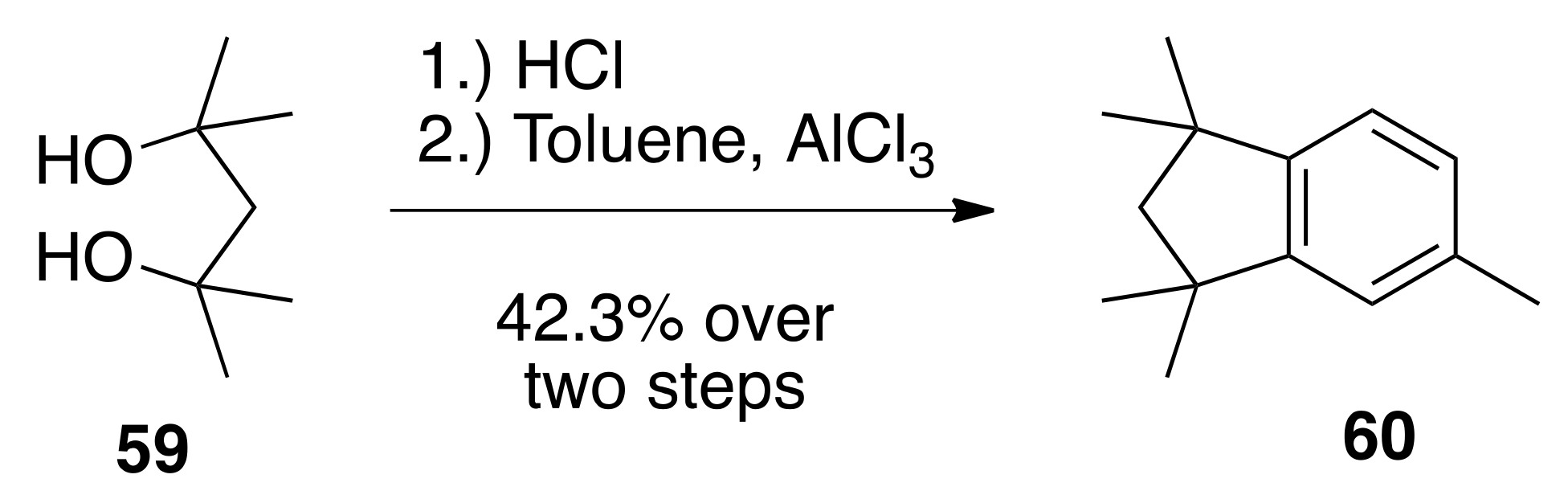
6.32. 1,1,3,3,5-. pentamethyl-2,3-dihydro-1H-indene (60)
To 2,4-dimethylpentane-2,4-diol (59) (5.00 g, 37.8 mmols) in a 100 mL round bottom flask was added concentrated hydrochloric acid (50.0 mL). The reaction was slightly diluted with water and extracted with hexanes. The hexanes was concentrated and the crude product was run through a column of silica gel (25 mL) in hexanes. The fractions containing the product were combined and concentrated to give crude 2,4-dichloro-2,4-dimethylpentane as a colorless oil (2.94 g, 46%) that was used without further purification. The crude 2,4-dichloro-2,4-dimethylpentane (2.94 g, 17.4 mmols) was dissolved in dichloromethane (10.0 mL) in a 100 mL round bottom flask and toluene (18.2 mL) was added. To this solution was slowly added aluminum chloride (1.80 g). The reaction was stirred at reflux in an oil bath for 15 min, then cooled to room temperature and poured into ice. The organics were extracted with ethyl acetate, and the organic layers were dried over sodium sulfate, filtered, and concentrated to give a crude oil that was purified by column chromatography (silica gel; hexanes) to give 60 (3.024 g, 92%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.03–7.04 (m, 2H), 6.95 (s, 1H), 2.37 (s, 3H), 1.93 (s, 2H), 1.32 (s, 6H), 1.31 (s, 6H).
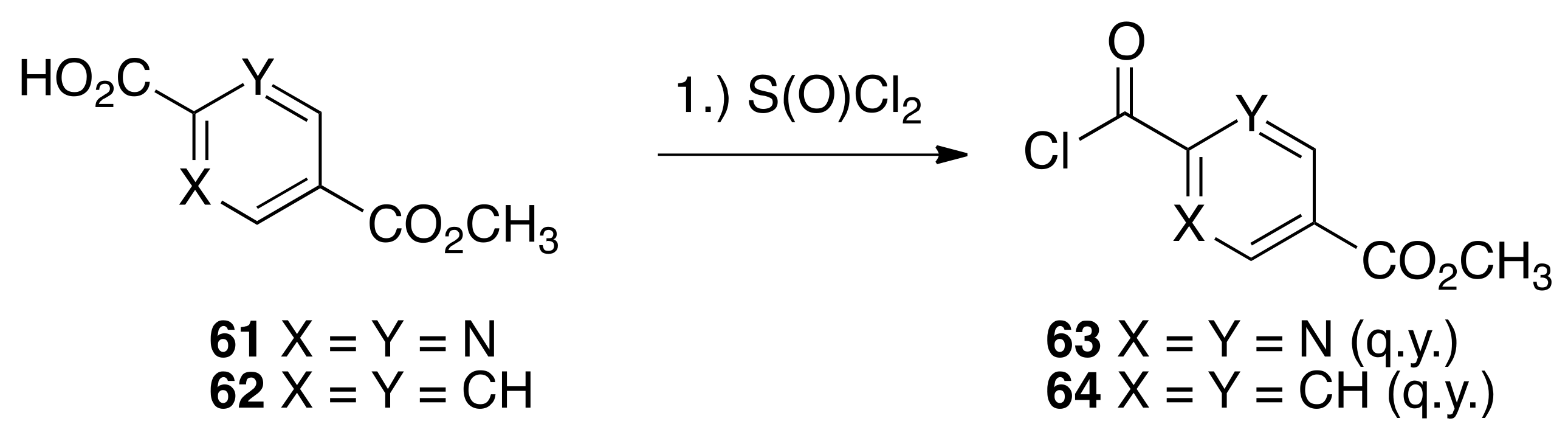
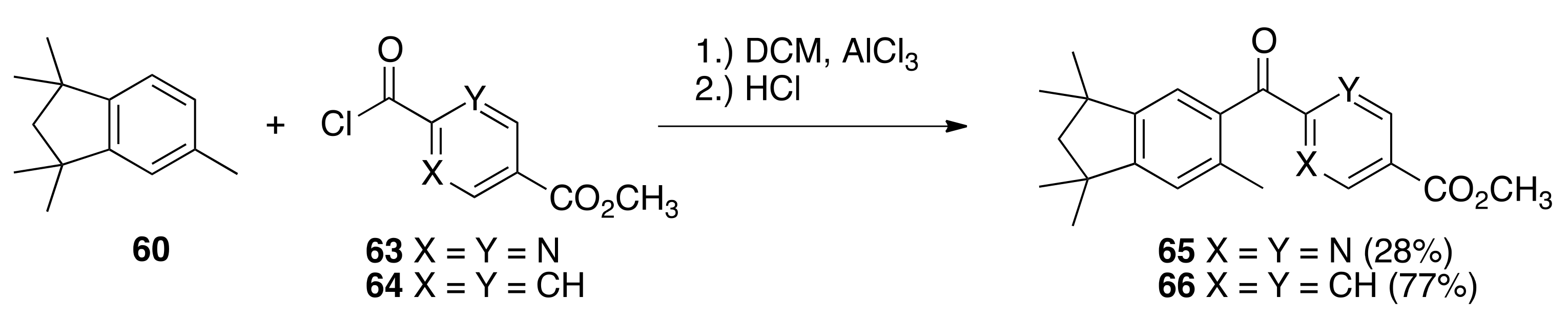
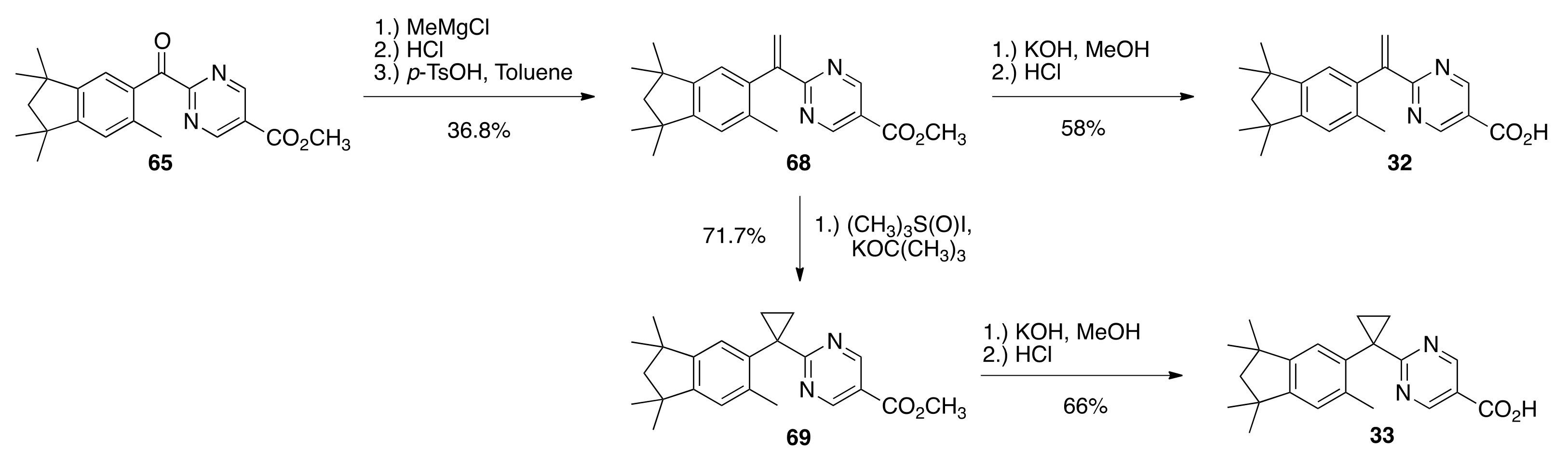
6.33. Methyl 2-(1,1,3,3,6-pentamethyl-2,3-dihydro-1H-indene-5-carbonyl)pyrimidine-5-carboxylate (65)
To a solution of 60 (3.05 g, 16.0 mmols) and methyl 2-(chlorocarbonyl)pyrimidine-5-carboxylate 63 (3.19 g, 15.9 mmols) in dichloromethane (35 mL) in a 100 mL round bottom flask was slowly added aluminum chloride (5.6 g) and the resulting mixture was stirred in an oil bath at reflux for 15 min. The reaction solution was cooled to room temperature and quenched by pouring onto 100 mL of an ice water solution. The solution was extracted with ethyl acetate, and the combined organic layers were dried over sodium sulfate, filtered, and concentrated to give a crude product that was purified by column chromatography (silica gel; 1:9 ethyl acetate:hexanes to 1:4 ethyl acetate:hexane) to give pure 65 (1.5869 g, 28%) as an orange, crystalline solid (98.1–103.2 °C): 1H NMR (400 MHz, CDCl3) δ 9.42 (s, 2H), 7.17 (s, 1H), 7.03 (s, 1H), 4.02 (s, 3H), 2.44 (s, 3H), 1.92 (s, 2H), 1.31 (s, 6H), 1.23 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ193.2, 166.0, 163.5, 158.5, 156.6, 148.6, 138.9, 133.5, 126.1, 126.0, 124.1, 56.5, 52.9, 42.8, 42.2, 31.3, 31.1, 21.3. ES-MS (M + Na)+ calcd for C21H24N2O3Na 375.1685, found 375.1668.
6.34. Methyl 4-(1,1,3,3,6-pentamethyl-2,3-dihydro-1 H-indene-5-carbonyl)benzoate (66)
To a solution of 60 (4.8058 g, 25.5 mmols) and methyl 4-(chlorocarbonyl)benzoate 64 (3.214 g, 16.18 mmols) in dichloromethane (35 mL) in a 100 mL round bottom flask was slowly added aluminum chloride (5.54 g) and the resulting mixture was stirred in an oil bath at reflux for 15 min. The reaction solution was cooled to room temperature and quenched by pouring onto 100 mL of an ice water solution. The solution was extracted with ethyl acetate, and the combined organic layers were dried over sodium sulfate, filtered, and concentrated to give a crude product that was purified by column chromatography (150 mL silica gel; 2.5% ethyl acetate:hexanes) to give pure 66 (4.4007 g, 77.6%) as a white, crystalline solid (120.2–122.2 °C): 1H NMR (400 MHz, CDCl3) δ 8.11 (dd, J = 6.8, 2.0, 2H), 7.86 (dd, J = 6.8, 2.0, 2H), 7.05 (s, 1H), 7.03 (s, 1H), 3.95 (s, 3H), 2.35 (s, 3H), 1.94 (s, 2H), 1.34 (s, 6H), 1.26 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ198.1, 166.3, 154.5, 148.4, 141.8, 136.3, 136.2, 133.4, 129.9, 129.5, 125.3, 123.7, 56.5, 52.4, 42.6, 42.2, 31.3, 31.2, 20.3. ES-MS (M + H)+ calcd for C23H27O3 351.1960, found 351.1959.
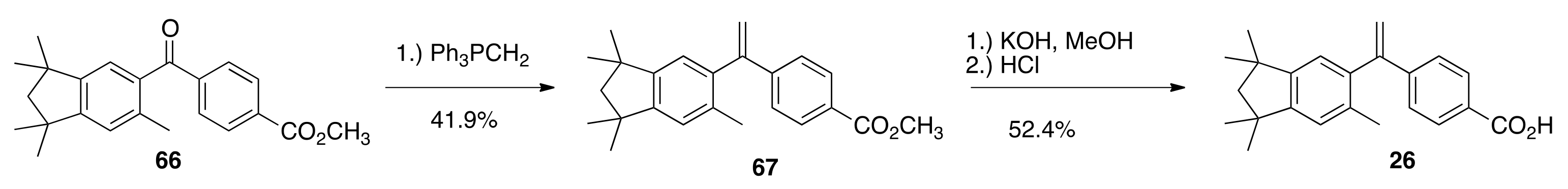
6.35. Methyl 4-(1-(1,1,3,3,6-pentamethyl-2,3-dihydro-1 H-inden-5-yl)vinyl)benzoate (67)
To a solution of diisopropylamine (0.66 mL, 4.71 mmols) in THF (2 mL) in a 100 mL round bottom flask was added a 1.6 M solution of n-butyl lithium in hexanes (2.7 mL, 4.32 mmols) at room temperature with stirring. After 15 min of stirring, methyltriphenylphosphonium bromide (1.15 g, 3.22 mmol) was added and the heterogeneous solution was stirred for 20 min after which time the solution became homogeneous and bright canary yellow. This yellow ylide solution was added to a solution of 66 (0.7867 g, 2.24 mmol) in THF (4 mL), and the resulting reaction solution was stirred for 1 h and then poured into water (50 mL) and extracted with ethyl acetate. The combined organic extracts were washed with water, then brine, dried over sodium sulfate, filtered, and concentrated to give a crude oil that was purified by column chromatography (150 mL silica gel; 2.5% ethyl acetate:hexanes to 5% ethyl acetate:hexanes) to give pure 67 (0.3272 g, 41.9%) as a white, crystalline solid (120.2–122.2 °C): 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.0, 2H), 7.35 (d, J = 8.8, 2H), 6.95 (s, 1H), 6.91 (s, 1H), 5.83 (d, J = 1.6, 1H), 5.33 (d, J = 1.2, 1H), 3.91 (s, 3H), 1.99 (s, 3H), 1.94 (s, 2H), 1.34 (s, 6H), 1.31 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ166.9, 150.9, 149.4, 148.9, 145.5, 139.3, 134.2, 129.6, 128.9, 126.5, 123.9, 116.8, 56.8, 52.0, 42.4, 42.3, 31.6, 31.5, 20.2. GC-MS CI (M)+ calcd for C24H28O2 348.2089, found 348.2082.
6.36. Methyl 4-(1-(1,1,3,3,6-pentamethyl-2,3-dihydro-1 H-inden-5-yl)vinyl)benzoate (26)
To a 100 mL round bottom flask charged with 67 (0.4262 g, 1.223 mmols) and a small magnetic Teflon stir bar was added methanol (5.0 mL), followed by a solution of potassium hydroxide (0.2242 g, 3.996 mmols) in water (0.30 mL). A reflux condenser was fitted to the flask, and the solution was refluxed in an oil bath at 85 °C for 1.3 h. After cooling to room temperature, 1 N HCl (84 mL) was poured into the reaction, and the crude precipitate was filtered and dried to give crude 26 (0.3713 g, 90.7%) which was dissolved in a minimum of warm ethyl acetate and purified by column chromatography (150 mL silica, 50% ethyl actate:hexanes) to give pure 26 (0.2174, 52.4%) as a white, crystalline solid (197.0–198.8 °C): 1H NMR (400 MHz, CDCl3) δ 8.04 (dd, J = 8.8, 2.0, 2H), 7.38 (d, J = 8.8, 2H), 6.95 (s, 1H), 6.92 (s, 1H), 5.86 (d, J = 1.6, 1H), 5.36 (d, J = 0.8, 1H), 2.00 (s, 3H), 1.94 (s, 2H), 1.34 (s, 6H), 1.32 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ171.8, 150.9, 149.2, 148.9, 146.4, 139.2, 134.2, 130.3, 128.0, 126.6, 124.0, 117.2, 56.8, 42.4, 42.3, 31.6, 31.5, 20.2. ES-MS- (M-H)- calcd for C23H25O2 333.1855, found 333.1867.
6.37. Methyl 2-(1-(1,1,3,3,6-pentamethyl-2,3-dihydro-1H-inden-5-yl)vinyl)pyrimidine-5-carboxylate (68)
A solution of 65 (2.18 g, 6.18 mmols) in toluene (22.0 mL) in a 100 mL round bottom flask was cooled to −10 °C under nitrogen with stirring and a 3.0 M solution methyl magnesium chloride (2.72 mL, 8.16 mmols) was added dropwise. After 15 min of stirring, the reaction solution was warmed to room temperature and stirred for an additional 35 min. The reaction mixture was then quenched by the slow addition of 1.0 N hydrochloric acid (14.0 mL, 14.0 mmols). The mixture was extracted with ethyl acetate, and the organic layers were washed with water and saturated sodium chloride, then dried over sodium sulfate, filtered, and concentrated in a 300 mL round bottom flask to give a crude alcohol product that was used without further purification. The alcohol product was dissolved in toluene (98.0 mL) and p-TsOH·H2O (1.197 g) was added, and the reaction flask was fitted with a Dean Stark trap and a water condenser. The vessel was evacuated and back-filled with nitrogen three times, and then heated to reflux in an oil bath at 130 °C and stirred for 3 h, during which time water collected in the Dean Stark trap. The reaction was cooled to room temperature, poured into water, and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate, filtered, and concentrated to give a crude product that was purified by column chromatography (silica gel; 2.5% ethyl acetate: hexanes to 5% ethyl acetate:hexanes) to give pure 68 (0.7969 g, 36.8%) as a white solid (182.9–185.5 °C): 1H NMR (400 MHz, CDCl3) δ 9.25 (s, 2H), 6.98 (s, 1H), 6.94 (s, 1H), 6.84 (d, J = 2.0, 1H), 5.81 (d, J = 2.0, 1H), 3.96 (s, 3H), 2.02 (s, 3H), 1.92 (s, 2H), 1.33 (s, 6H), 1.31 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ168.7, 164.3, 158.2, 150.9, 148.8, 148.7, 137.6, 134.2, 126.6, 123.9, 123.8, 121.1, 56.9, 52.5, 42.4, 42.3, 31.6, 31.5, 20.3; IR (neat) 2953, 1722.25 cm−1. ES-MS (M + H)+ calcd for C22H27N2O2 351.2072, found 351.2068.
6.38. 2-(1-(1,1,3,3,6-pentamethyl-2,3-dihydro-1H-inden-5-yl)vinyl)pyrimidine-5-carboxylic Acid (32)
To a solution of 68 (0.6637 g, 1.8938 mmols) in methanol (12.0 mL) in a 100 mL round bottom flask was added a solution of potassium hydroxide (0.3032 g, 5.40 mmols) in water (0.45 mL). The resulting reaction solution was refluxed with stirring for 1 h in an oil bath at 85 °C. After cooling the reaction solution to room temperature, 1 N HCl (90 mL) was added. The resulting precipitate was filtered and washed with cold water and dried to give crude 32 (0.6143 g, 96.4%). The crude 32 was dissolved in hot ethyl acetate (16.0 mL), hexanes (51 mL) were added, and the homogenous solution was concentrated, filtered, and washed with hexanes to give pure 32 (0.3695 g, 58%) as a white solid (182.7–188.2 °C): 1H NMR (400 MHz, CDCl3) δ 9.31 (s, 2H), 6.98 (s, 1H), 6.95 (s, 1H), 6.87 (d, J = 2.0, 1H), 5.87 (d, J = 1.6, 1H), 2.03 (s, 3H), 1.91 (s, 2H), 1.30 (s, 12H); 13C NMR (100.6 MHz, CDCl3) δ 169.0, 168.0, 158.8, 151.1, 148.9, 148.3, 137.2, 134.2, 127.5, 124.0, 123.9, 120.7, 56.8, 42.4, 42.3, 31.5, 31.4, 20.3; IR (neat) 2954, 1715 cm−1. ES-MS (M + H)+ calcd for C21H25N2O2 337.1916, found 337.1913.
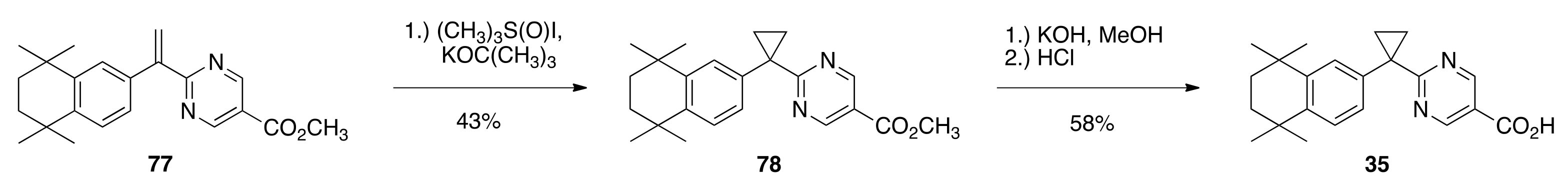
6.39. Methyl 2-(1-(1,1,3,3,6-pentamethyl-2,3-dihydro-1H-inden-5-yl)cyclopropyl)pyrimidine-5-carboxylate (69)
To a suspension of trimethylsulfoxonium iodide (0.760 g, 3.45 mmols) in DMSO (2.5 mL) in a 50 mL 2-neck round bottom flask was added a 20 wt% solution of potassium tert-butoxide in THF (1.94 mL, 3.45 mmols) with stirring at 35 °C. The reaction mixture was stirred for 5 min and then a solution of 68 (0.8061 g, 2.30 mmols) in THF (9.9 mL) was added. The reaction was stirred for 1 h at 35 °C, then allowed to cool to room temperature, at which point 1 N hydrochloric acid (10.0 mL) was added. The resulting solution was extracted with ethyl acetate, the combined organic layers were washed with saturated sodium chloride, dried over sodium sulfate, filtered, and concentrated to give a crude off-white solid that was purified by column chromatography (silica gel; 2.5% ethyl acetate:hexanes to 10% ethyl acetate: hexanes) to give 69 (0.6009 g, 71.7%) as a white solid (236.4–242.4 °C): 1H NMR (400 MHz, CDCl3) δ 9.08 (s, 2H), 7.06 (s, 1H), 6.92 (s, 1H), 3.92 (s, 3H), 2.16 (s, 3H), 1.91 (s, 2H), 1.90 (t, J = 2.8, 2H), 1.49 (t, J = 3.2, 2H), 1.32 (s, 6H), 1.29 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 176.8, 164.6, 157.8, 149.8, 148.5, 138.2, 137.3, 124.5, 123.8, 119.9, 56.9, 52.3, 42.3, 42.2, 32.0, 31.6, 31.5, 23.8, 21.8, 19.8; IR (neat) 2963, 1680 cm−1. ES-MS (M + H)+ calcd for C23H29N2O2 365.2229, found 365.2213.
6.40. 2-(1-(1,1,3,3,6-pentamethyl-2,3-dihydro-1H-inden-5-yl)cyclopropyl)pyrimidine-5-carboxylic Acid (33)
To a solution of 69 (0.5324 g, 1.4607 mmols) in methanol (9.4 mL) in a 100 mL round bottom flask was added a solution of potassium hydroxide (0.2492 g, 4.44 mmols) in water (0.34 mL). The resulting reaction solution was refluxed with stirring for 1 h in an oil bath at 85 °C. After cooling the reaction solution to room temperature, 1 N HCl (90 mL) was added. The resulting precipitate was filtered and washed with cold water and dried to give crude 33 (0.4932 g, 96.3%). The crude 33 was dissolved in hot ethyl acetate (28.0 mL), hexanes (20 mL) were added, and the homogenous solution was concentrated, filtered, and washed with hexanes to give pure 33 (0.3402 g, 66%) as a white solid (261.6–267.3 °C): 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 2H), 7.05 (s, 1H), 6.92 (s, 1H), 2.17 (s, 3H), 1.93 (m, 2H), 1.89 (s, 2H), 1.53 (m, 2H), 1.29 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 177.1, 167.3, 159.0, 158.5, 158.4, 150.4, 148.9, 137.3, 124.7, 124.1, 119.7, 56.8, 42.4, 42.3, 32.0, 31.6, 31.5, 31.4, 23.9, 22.8, 19.8; IR (neat) 2954, 1721 cm−1. ES-MS (M + H)+ calcd for C22H27N2O2 351.2072, found 351.2067.
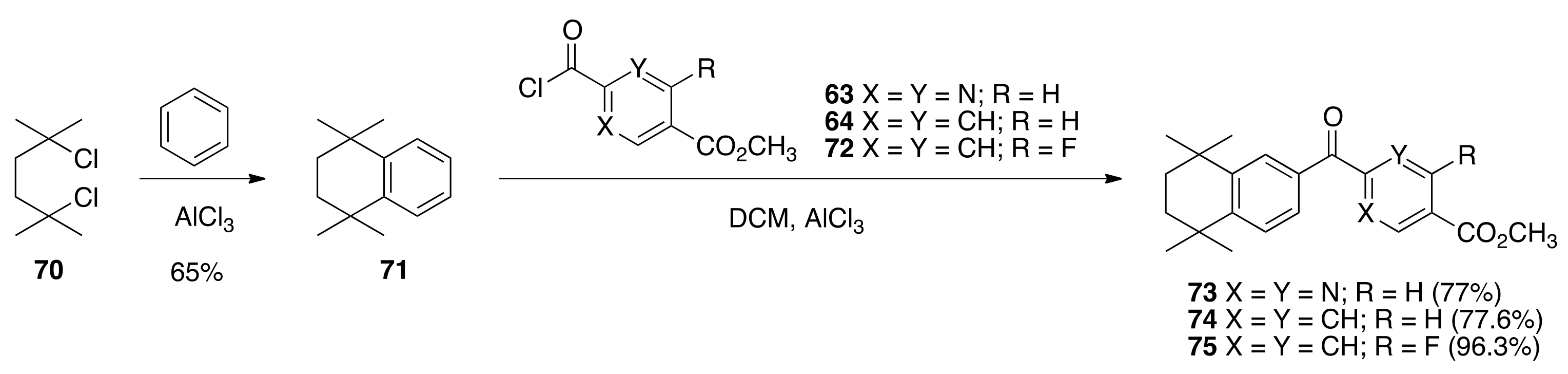
6.41. 1,1,4,4-. tetramethyl-1,2,3,4-tetrahydronaphthalene (71)
The procedure of Bruson and Kroeger was followed. To a solution of 2,5-dichloro-2,5-dimethylhexane (70) (11.36 g, 62.04 mmols) in benzene (280 mL) was added aluminum chloride (1.5 g) in a 500 mL round bottom flask equipped with a stir bar and water condenser and the reaction was heated to 75–82 °C for 24 h with stirring under nitrogen. After cooling to room temperature, the reaction solution was poured into 1 N HCl (450 mL) and extracted with benzene. The combined organic layers were washed with water, saturated sodium bicarbonate, water, and finally saturated sodium chloride. The combined organic layers were dried over sodium sulfate, concentrated to a crude oil that was then vacuum distilled with a short-path distillation head at an oil bath temperature of 95–100 °C, and a head temperature of 78 °C for the major fraction, at 0.2–0.3 mm Hg to give pure 71 (7.5693g, 65%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.35 (m, 2H), 7.16 (m, 2H), 1.72 (s, 4H), 1.32 (s, 12H); 13C NMR (100.6 MHz, CDCl3) δ 144.7, 126.4, 125.5, 35.0, 34.1, 31.8; IR (neat) 3021, 1715 cm−1.
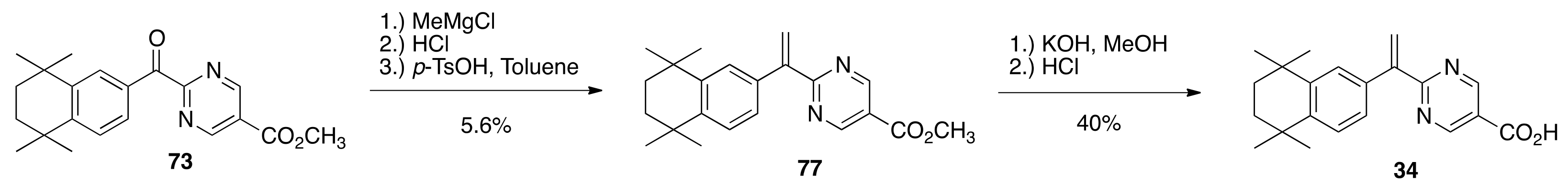
6.42. Methyl 2-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalene-2-carbonyl)pyrimidine-5-carboxylate (73)
To a solution of 71 (5.3945 g, 28.646 mmols) and 63 (5.37 g, 26.772 mmols) in dichloromethane (60 mL) was slowly added aluminum chloride (8.8 g) and the resulting mixture was stirred at reflux in an oil bath at 55 °C for 15 min. The solution was then cooled to room temperature and poured into 200 mL of an ice water solution. The resulting mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated sodium chloride, dried over sodium sulfate, filtered, and concentrated to give a crude product that was purified by column chromatography (silica gel, 15% ethyl acetate:hexanes to 20% ethyl acetate: hexanes) to give pure 73 (5.9627 g, 77%) as a bright canary yellow crystalline solid (79.1–83.4 °C): 1H NMR (400 MHz, CDCl3) δ 9.43 (s, 2H), 8.02 (d, J = 1.6, 1H), 7.63 (dd, J = 7.6, 2.0, 1H), 7.39 (d, J = 8.0, 1H), 4.03 (s, 3H), 1.70 (s, 4H), 1.29 (s, 12H); 13C NMR (100.6 MHz, CDCl3) δ 190.3, 165.3, 163.5, 158.3, 152.1, 145.5, 131.9, 129.3, 128.0, 126.7, 124.1, 52.9, 34.8, 34.7, 34.6, 34.4, 31.7, 31.5; IR (neat) 2954, 1721 cm−1. ES-MS (M + H)+ calcd for C21H25N2O3 353.1865, found 353.1864.
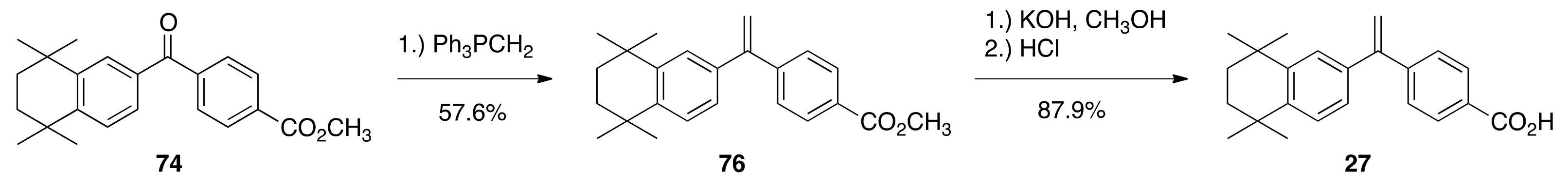
6.43. Methyl 4-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalene-2-carbonyl)benzoate (74)
To a 250 mL flask, 4-(methoxycarbonyl)benzoic acid (3.535 g, 19.62 mmols) was treated with thionyl chloride (30.0 mL), resulting in a quantitative crude yield of methyl 4-(chlorocarbonyl)-benzoate (64) after the removal of thionyl chloride. Acid chloride 64 and compound 71 (4.0404 g, 21.45) were dissolved in 30 mL of DCM and aluminum chloride (4.18 g, 31.34 mmols) was slowly added followed by heating in an oil bath at 55 °C for 15 min. The reaction was quenched in 100 mL of ice water, the product was extracted with ethyl acetate, dried over sodium sulfate, and purified by column chromatography (300 mL silica gel, 2.5% ethyl acetate:hexanes to 20% ethyl acetate: hexanes) to give pure 74 (5.3353 g, 77.6%) as an off-white crystalline solid, mp 136.9–139.8 °C: 1 H NMR (400 MHz, CDCl3) δ 8.16 (dd, J = 6.8, 2.0, 2H), 7.83 (dd, J = 6.8, 2.0, 2H), 7.78 (d, J = 2.0, 1H), 7.53 (dd, J = 8.4, 2.0, 1H), 7.40 (d, J = 8.4, 1H), 3.96 (s, 3H), 1.72 (s, 4H), 1.31 (s, 6H), 1.29 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 195.9, 166.4, 150.7, 145.2, 141.8, 134.1, 132.8, 129.6, 129.3, 128.8, 127.3, 126.7, 52.4, 34.8, 34.7, 34.7, 34.4, 31.7, 31.6. ES-MS (M + H)+ calcd for C23H27O3 351.1960, found 351.1978.
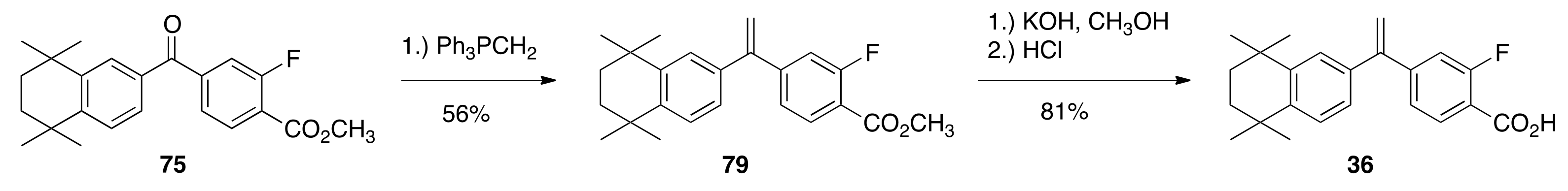
6.44. Methyl 2-fluoro-4-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalene-2-carbonyl)benzoate (75)
To a 250 mL flask, 2-fluoro-terephthalic acid 4-methyl ester (2.4067 g, 12.146 mmol) was treated with thionyl chloride (20.0 mL), resulting in a quantitative crude yield of methyl 4-(chlorocarbonyl)-2-fluorobenzoate (72). Acid chloride 72 and compound 71 (2.68 g, 14.243 mmol) were dissolved in 30 mL DCM and aluminum chloride (4.24 g) was slowly added, followed by heating in an oil bath at 55 °C for 15 min. The reaction was quenched in 100 mL of ice water, the product was extracted with ethyl acetate, dried over sodium sulfate, and purified by column chromatography (2.5% ethyl acetate/hexane; 7.5% ethyl acetate/hexane; 20% ethyl acetate/hexane) to give pure ketone 75 (4.3126 g, 96.3%) as white solid, mp 94.4–96.8 °C: 1 H NMR (400 MHz, CDCl3) δ 8.03 (t, J = 7.6, 1H), 7.77 (d, J = 2.0, 1H), 7.58 (dd, J = 8.0, 1.6, 1H), 7.53 (dd, J = 11.2, 1.6, 1H), 7.50 (d, J = 2.0, 1H), 7.41 (d, J = 8.0, 1H), 3.97 (s, 3H), 1.72 (s, 4H), 1.29 (s, 6H), 1.24 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 194.3, 194.2, 164.3, 164.3, 162.7, 160.1, 151.1, 145.5, 143.6, 143.6, 133.5, 132.0, 128.8, 127.2, 126.8, 124.9, 124.9, 121.4, 121.3, 118.2, 118.0, 52.6, 34.8, 34.7, 34.6, 34.4, 31.7, 31.5. ES-MS (M + Na)+ calcd for C23H25FO3Na 391.1685, found 391.1671.
6.45. Methyl 4-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoate (76)
To a 250 mL flask, ketone 74 (0.750 g, 2.14 mmol) was combined with dry THF (7.0 mL) at room temperature and a triphenylphosphonium methylide solution—made from the addition of methyltriphenylphosphonium bromide (1.15 g, 3.22 mmol) to a solution of n-butyl lithium (2.7 mL, 1.6 M in hexanes, 4.32 mmol) and diisopropyl amine (0.66 mL, 4.67 mmol) in THF (5 mL)—was added with stirring. The reaction was stirred for thirty minutes, poured into water, and extracted with ethyl acetate. The organic extracts were washed with water, dried over sodium sulfate, and purified by column chromatography (150 mL SiO2) with 2.5% ethyl acetate/hexanes; 5% ethyl acetate/hexane; 20% ethyl acetate/hexane to give pure 76 (0.4293 g, 57.6%) as a colorless, waxy solid, mp 112.1−113.9 °C: 1 H NMR (400 MHz, CDCl3) δ 8.02 (dd, J = 8.4, 2.0, 2H), 7.44 (dd, J = 8.4, 2.0, 2H), 7.27 (d, J = 8.4, 1H), 7.24 (d, J = 2.0, 1H), 7.08 (dd, J = 8.0, 2.0, 1H), 5.54 (d, J = 1.2, 1H), 5.48 (d, J = 1.2, 1H), 3.93 (s, 3H), 1.70 (s, 4H), 1.30 (s, 6H), 1.24 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 166.9, 149.3, 146.3, 144.8, 144.6, 137.5, 129.3, 129.1, 128.2, 126.4, 126.3, 125.3, 114.9, 52.0, 35.0, 34.9, 34.2, 34.1, 31.7. ES-MS (M + H)+ calcd for C24H29O2 349.2168, found 349.2168.
6.46. 4-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoic acid (27)
Compound 27 was synthesized according to the method of Boehm and co-workers. Compound 76 (1.5597 g, 4.476 mmols) was suspended in methanol (28.0 mL) and a solution of potassium hydroxide (0.7420 g, 13.22 mmols) in water (1.08 mL) was added. The reaction was refluxed in an oil bath at 85 °C. After 70 min of reflux, the solution was cooled to room temperature and the crude product was precipitated with 1 N hydrochloric acid (200 mL) and filtered to give (1.4655 g, 97.9%) of crude 27. It was then purified by column chromatography (150 mL SiO2, with 2.5% ethyl acetate/hexanes; 5% ethyl acetate/hexane; 20% ethyl acetate/hexane) to give pure 27 (1.3152 g, 87.9%) as tint yellow solid, mp 211.3−215.8 °C: 1 H NMR (400 MHz, CDCl3) δ 10.33 (br s, 1H), 8.10 (dd, J = 6.8, 2.0, 2H), 7.48 (dd, J = 6.8, 2.0, 2H), 7.29 (d, J = 8.4, 1H), 7.25 (d, J = 2.0, 1H), 7.08 (dd, J = 8.4, 2.0, 1H), 5.57 (d, J = 0.8, 1H), 5.51 (d, J = 1.2, 1H), 1.70 (s, 4H), 1.31 (s, 6H), 1.25 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 172.1, 149.3, 147.2, 144.9, 144.7, 137.4, 130.0, 128.4, 128.3, 126.4, 126.3, 125.3, 115.2, 35.1, 35.0, 34.2, 34.1, 31.8. ES-MS- (M-H)- calcd for C23H25O2 333.1855, found 333.1872.
6.47. Methyl 2-(1-(5,5,8,8-tetrahydronaphthalen-2-yl)vinyl)pyrimidine-5-carboxylate (77)
A solution of 73 (5.2603 g, 14.926 mmols) in toluene (53.0 mL) in a 250 mL round bottom flask was cooled to −10 °C under nitrogen with stirring and a 3.0 M solution methyl magnesium chloride (6.56 mL, 19.68 mmols) was added dropwise. After 15 min of stirring, the reaction solution was warmed to room temperature and stirred for an additional 35 min. The reaction mixture was then quenched by the slow addition of 1.0 N hydrochloric acid (35.0 mL, 35.0 mmols). The mixture was extracted with ethyl acetate, and the organic layers were washed with water and saturated sodium chloride, then dried over sodium sulfate, filtered, and concentrated in a 300 mL round bottom flask to give a crude alcohol product that was used without further purification. The alcohol product was dissolved in toluene (110.0 mL) and p-TsOH·H2O (5.7782 g, 33.56 mmol) was added, and the reaction flask was fitted with a Dean Stark trap and a water condenser. The vessel was evacuated and back-filled with nitrogen three times, and then heated to reflux in an oil bath at 130 °C and stirred for 3 h, during which time water collected in the Dean Stark trap. The reaction was cooled to room temperature, poured into water, and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate, filtered, and concentrated to give a crude product that was purified by column chromatography (silica gel; 2.5% ethyl acetate:hexanes to 5% ethyl acetate:hexanes) to give pure 77 (0.2936 g, 5.6%) as a white solid (171.3–174.1 °C): 1H NMR (400 MHz, CDCl3) δ 9.28 (s, 2H), 7.35 (d, J = 2.0, 1H), 7.31 (d, J = 8.0, 1H), 7.17 (dd, J = 8.4, 2.0, 1H), 6.58 (d, J = 1.6, 1H), 5.93 (d, J = 1.6, 1H), 3.98 (s, 3H), 1.70 (s, 4H), 1.30 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 168.9, 164.3, 158.0, 147.7, 144.7, 144.4, 135.5, 126.8, 126.2, 125.8, 124.4, 121.5, 52.5, 36.6, 35.1, 35.0, 34.2, 34.1, 31.8, 31.7, 24.6; IR (neat) 2952, 1721.95 cm−1. ES-MS (M + Na)+ calcd for C22H26N2O2Na 373.1892, found 373.1902.
6.48. 2-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)pyrimidine-5-carboxylic acid (34)
To a solution of 77 (0.6585 g, 1.879 mmols) in methanol (12.0 mL) in a 100 mL round bottom flask was added a solution of potassium hydroxide (0.3046 g, 5.43 mmols) in water (0.45 mL). The resulting reaction solution was refluxed with stirring for 1 h in an oil bath at 85 °C. After cooling the reaction solution to room temperature, 1 N HCl (90 mL) was added. The resulting precipitate was filtered and washed with cold water and dried to give crude 34 (0.5526 g, 87%). The crude 34 was dissolved in hot ethyl acetate (17.0 mL), hexanes (50 mL) were added, and the homogenous solution was concentrated, filtered, and washed with hexanes to give pure 34 (0.2572 g, 40%) as a white solid (224.0–227.8 °C): 1H NMR (400 MHz, CDCl3) δ 9.35 (s, 2H), 7.35 (d, J = 1.6, 1H), 7.32 (d, J = 8.0, 1H), 7.17 (dd, J = 8.4, 2.0, 1H), 6.62 (d, J = 1.2, 1H), 5.98 (d, J = 1.6, 1H), 1.69 (s, 4H), 1.29 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 169.4, 158.7, 147.5, 144.9, 144.5, 135.3, 126.8, 126.3, 125.8, 125.2, 120.7, 35.1, 35.0, 34.2, 34.1, 31.8, 31.7; IR (neat) 3009, 1715.95 cm−1. ES-MS (M + Na)+ calcd for C21H24N2O2Na 359.1736, found 359.1720.
6.49. Methyl 2-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)cyclopropyl)pyrimidine-5-carboxylate (78)
To a suspension of trimethylsulfoxonium iodide (0.3204 g, 1.45 mmols) in DMSO (1.05 mL) in a 50 mL 2-neck round bottom flask was added a 20 wt% solution of potassium tert-butoxide in THF (0.84 mL, 1.49 mmols) with stirring at 35 °C. The reaction mixture was stirred for 5 min and then a solution of 78 (0.3408 g, 0.97 mmols) in THF (4.8 mL) was added. The reaction was stirred for 1 h at 35 °C, then allowed to cool to room temperature, at which point 1 N hydrochloric acid (5.0 mL) was added. The resulting solution was extracted with ethyl acetate, the combined organic layers were washed with saturated sodium chloride, dried over sodium sulfate, filtered, and concentrated to give a crude off-white solid that was purified by column chromatography (silica gel; 2.5% ethyl acetate:hexanes to 10% ethyl acetate:hexanes) to give 78 (0.1533 g, 43%) as a white solid (168.7–171.8 °C): 1H NMR (400 MHz, CDCl3) δ 9.08 (s, 2H), 7.30 (d, J = 1.6, 1H), 7.27 (d, J = 8.0, 1H), 7.15 (dd, J = 8.0, 2.0, 1H), 3.92 (s, 3H), 1.79 (m, 2H), 1.68 (s, 4H), 1.50 (m, 2H), 1.29 (s, 6H), 1.26 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 177.0, 164.6, 157.7, 144.5, 143.1, 138.2, 129.5, 128.8, 127.6, 126.2, 120.4, 120.0, 115.2, 52.4, 36.5, 35.1, 35.0, 34.2, 34.0, 33.0, 31.8, 24.6, 20.2; IR (neat) 2954, 1721 cm−1. ES-MS (M + Na)+ calcd for C23H28N2O2Na 387.2048, found 387.2048.
6.50. 2-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)cyclopropyl)pyrimidine-5-carboxylic acid (35)
To a solution of 78 (0.1316 g, 0.361 mmols) in methanol (2.5 mL) in a 100 mL round bottom flask was added a solution of potassium hydroxide (0.0803 g, 1.43 mmols) in water (0.18 mL). The resulting reaction solution was refluxed with stirring for 1 h in an oil bath at 85 °C. After cooling the reaction solution to room temperature, 1 N HCl (70 mL) was added. The resulting precipitate was filtered and washed with cold water and dried to give crude 78 (0.1041 g, 82%). The crude 78 was dissolved in hot ethyl acetate (5.0 mL), and the homogenous solution was concentrated, filtered, and washed with hexanes to give pure 78 (0.0734 g, 58%) as a white solid (251.5–254.6 °C): 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 2H), 7.30 (d, J = 1.6, 1H), 7.29 (d, J = 8.4, 1H), 7.15 (dd, J = 8.0, 2.0, 1H), 1.81 (m, 2H), 1.68 (s, 4H), 1.53 (m, 2H), 1.28 (s, 6H), 1.26 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 177.6, 167.7, 158.8, 158.3, 144.7, 143.4, 137.6, 128.9, 127.8, 126.4, 119.4, 35.1, 35.0, 34.2, 34.0, 33.2, 31.9, 31.8, 20.9, 20.9; IR (neat) 2957, 1679.64 cm−1. ES-MS (M + Na)+ calcd for C22H26N2O2Na 373.1892, found 373.1898.
6.51. Methyl 2-fluoro-4-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoate (79)
To a 250 mL flask, ketone product 75 (0.7971 g, 2.16 mmol) was combined with dry THF (4.0 mL) at room temperature and a triphenylphosphonium methylide solution—made from the addition of methyltriphenylphosphonium bromide (1.15 g, 3.22 mmol) to a solution of n-butyl lithium (2.7 mL, 1.6 M in hexanes, 4.32 mmol) and diisopropyl amine (66 mL, 4.67 mmol) in THF (2 mL)—was added with stirring. The reaction was stirred for thirty minutes, poured into water, and extracted with ethyl acetate. The organic extracts were washed with water, dried over sodium sulfate, and purified by column chromatography (150 mL SiO2) with 2.5% ethyl acetate/hexanes to 5% ethyl acetate/hexane giving pure compound 79 (0.4454 g, 56%) as yellow solid, mp 80.8–96.5 °C: 1H NMR (400 MHz, CDCl3) δ 7.90 (t, J = 7.6, 2H), 7.28 (d, J = 8.0, 1H), 7.22 (dd, J = 8.0, 1.6, 1H), 7.21 (dd, J = 2.0, 1H), 7.14 (dd, J = 12.4, 1.2, 1H), 7.05 (dd, J = 8.4, 2.0, 1H), 5.54 (d, J = 0.8, 1H), 5.50 (d, J = 0.8, 1H), 1.69 (s, 4H), 1.30 (s, 6H), 1.24 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 164.8, 164.7, 163.0, 160.4, 148.5, 148.4, 148.3, 148.2, 145.0, 144.8, 136.9, 131.8, 126.5, 126.2, 125.3, 123.7, 123.7, 117.3, 117.2, 116.7, 116.4, 115.7, 52.2, 35.0, 34.9, 34.2, 34.1, 31.7. ES-MS (M + Na)+ calcd for C24H27FO2Na 389.1893, found 389.1892.
6.52. 2-fluoro-4-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoic Acid (36)
Compound 36 was synthesized according to the method of Boehm and co-workers. Compound 79 (2.0486 g, 5.59 mmols) and methanol (36.0 mL) were added to a solution of potassium hydroxide (0.9348 g, 16.66 mmols) in water (1.34 mL). The reaction was refluxed, and after 70 min of reflux, the solution was cooled to room temperature and the crude product was precipitated with 1 N hydrochloric acid (560 mL) to give crude product (1.7523 g) 36. It was then purified by column chromatography (150 mL SiO2) in 50% ethyl acetate/hexane to give pure 36 (1.5974 g, 81.0%) as white crystalline solid, mp 165.6–183.0 °C: 1 H NMR (400 MHz, CDCl3) δ 8.01 (t, J = 8.0, 2H), 7.29 (d, J = 8.4, 1H), 7.27 (dd, J = 8.4, 1.6, 1H), 7.23 (d, J = 2.0, 1H), 7.18 (dd, J = 12.0, 1.2, 1H), 7.06 (dd, J = 8.4, 2.0, 1H), 5.58 (d, J = 0.4, 1H), 5.54 (d, J = 0.4, 1H), 1.70 (s, 4H), 1.31 (s, 6H), 1.26 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 169.5, 169.4, 163.8, 161.2, 149.7, 149.6, 148.2, 148.2, 145.1, 144.9, 136.8, 132.4, 126.5, 126.3, 125.3, 123.9, 123.8, 116.8, 116.6, 116.3, 116.2, 116.1, 35.0, 34.9, 34.2, 34.1, 31.8, 31.7. ES-MS (M + Na)+ calcd for C23H25FO2Na 375.1736, found 375.1753.
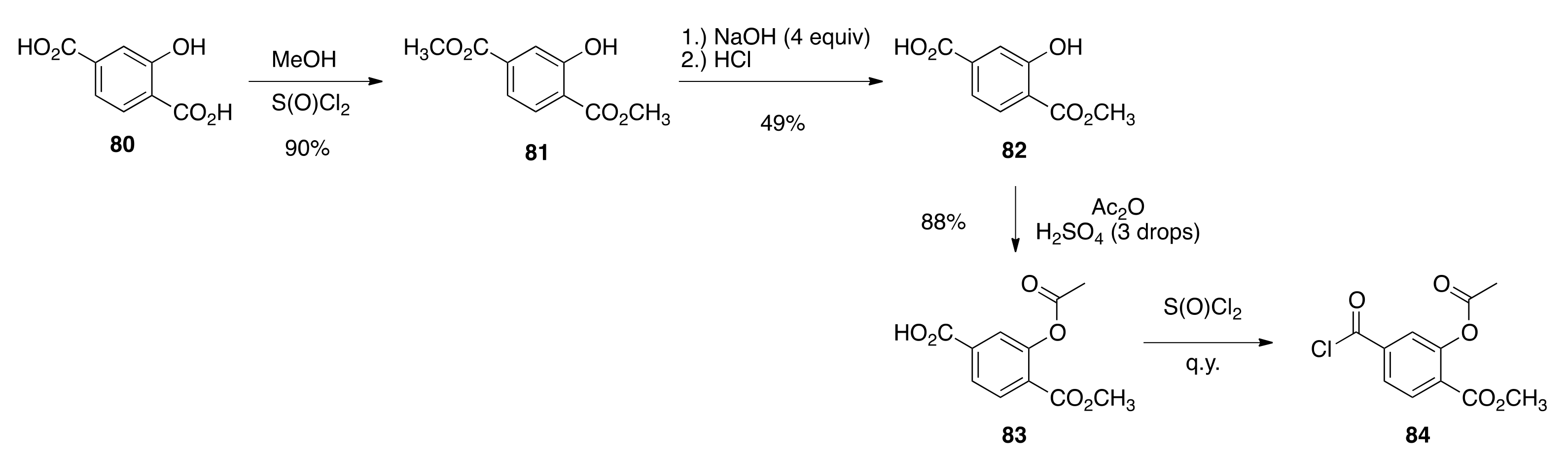
6.53. Dimethyl-2-hydroxyterepthalate (81)
To a solution of hydroxyl-terepthalic acid (80) (9.93 g, 54.5 mmols) in methanol (189 mL) in a 500 mL round bottom flask equipped with a magnetic stir bar and cooled to 0 °C in an ice bath was added thionyl chloride (14.5 mL, 200 mmols) dropwise with stirring. After addition, the flask was equipped with a reflux condenser, placed under a nitrogen atmosphere, and warmed to reflux in an oil bath set at 85 °C and boiled for 2.5 h. The solution was allowed to cool to room temperature, and most of the methanol was removed in vacuo. The crude product was dissolved in ethyl acetate, and the solvent was washed with water followed by brine and then dried over sodium sulfate. The solvent was filtered and the ethyl acetate was removed in vacuo to provide a crude product that was dissolved in warm ethyl acetate (20 mL) and purified by column chromatography (250 mL SiO2) with 10% ethyl acetate/hexanes to give 81 (10.24 g, 90%) as white solid, m.p. 92.2–94.8 °C: 1H NMR (400 MHz, CDCl3) δ 10.75 (s, 1H), 7.89 (d, J = 8.4, 1H), 7.62 (d, J = 1.6, 1H), 7.51 (dd, J = 8.4, 1.6, 1H), 3.97 (s, 3H), 3.92 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 169.9, 165.9, 161.2, 136.3, 130.0, 119.6, 118.8, 115.6, 52.6, 52.4. ES-MS (M + Na)+ calcd for C10H10O5Na 233.0426, found 233.0416.
6.54. 3-Hydroxy-4-(methoxycarbonyl)benzoic Acid (82)
The method of Zhang and co-workers was followed. Sodium hydroxide (0.7966 g, 19.9 mmols) was dissolved in water (32 mL), the solution was cooled to 0 °C, and a finely ground powder of dimethyl-2-hydroxyterepthalate (81) (1.0119 g, 4.81 mmols) was added to the solution. The solution was stirred for 1.5 h at 0 °C, and then a solution of 1 N hydrochloric acid was added (12 mL, 12 mmol)m which brought the solution to pH = 9.0, and the insoluble precipitate was filtered off. To the filtrate, an additional amount of 1 N hydrochloric acid (9.5 mL, 9.5 mmol) was added that brought the pH = 1.0 and the resulting precipitate was filtered and washed with cold water to give a crude product (0.67 g) that was purified by column chromatography (150 mL SiO2) with 20% ethyl acetate/hexanes to 70% ethyl acetate/hexanes to give 82 (0.4606 g, 49%) as white solid, m.p. 213.7–216.2 °C: 1H NMR (400 MHz, CDCl3) δ 10.79 (s, 1H), 7.94 (d, J = 8.0, 1H), 7.71 (d, J = 1.6, 1H), 7.58 (dd, J = 8.0, 1.6, 1H), 3.99 (s, 3H). ES-MS- (M-H)- calcd for C9H7O5 195.0293, found 195.0285.
6.55. 3-Acetoxy-4-(methoxycarbonyl)benzoic Acid (83)
To a solution of 3-hydroxy-4-(methoxycarbonyl)benzoic acid (82) (0.5328 g, 2.716 mmols) in acetic anhydride (20.0 mL) in a 100 mL round bottom flask equipped with a stir bar was added concentrated sulfuric acid (3 drops) and the reaction was stirred in an oil bath at 45 °C for 40 min. The acetic anhydride was removed in vacuo and the crude oil was purified by column chromatography (150 mL SiO2) with 10% ethyl acetate/hexanes to pure ethyl acetate to give 83 (0.5715 g, 88%) as white solid, m.p. 182.6–185.1 °C: 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.4, 1H), 8.03 (dd, J = 8.4, 1.6, 1H), 7.84 (d, J = 1.6, 1H), 3.91 (s, 3H), 2.38 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 169.8, 169.5, 164.1, 150.5, 134.0, 131.9, 127.8, 127.4, 125.6, 52.6, 20.9. ES-MS (M + Na)+ calcd for C11H10O6Na 261.0375, found 261.0366.
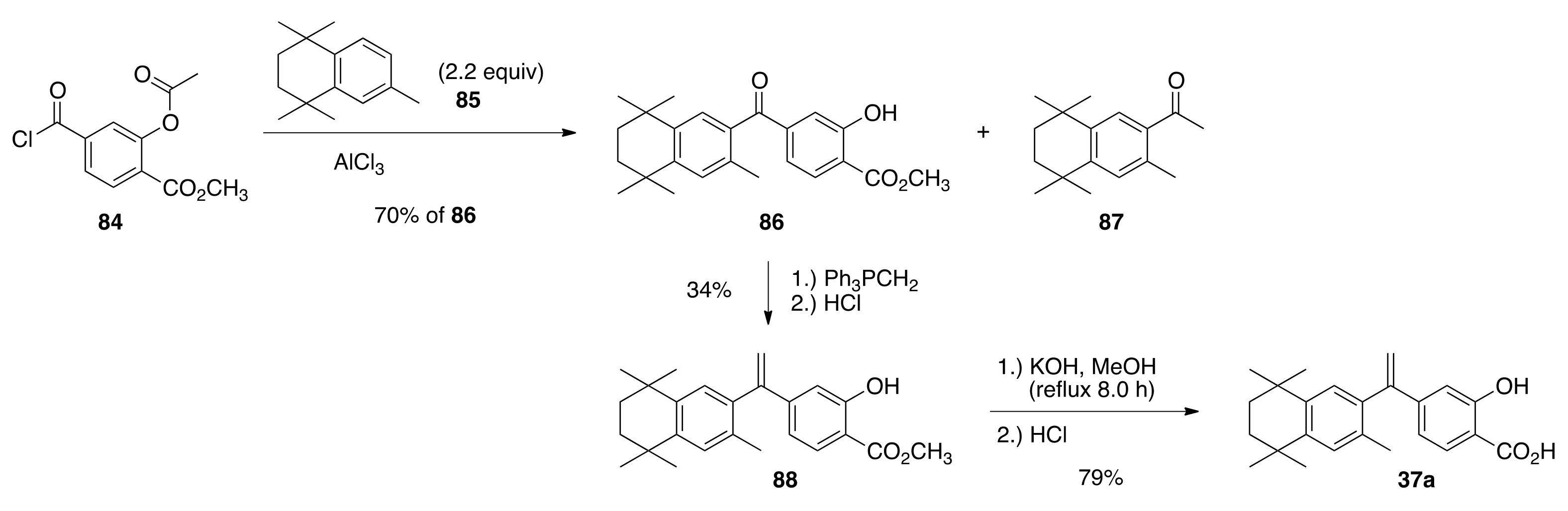
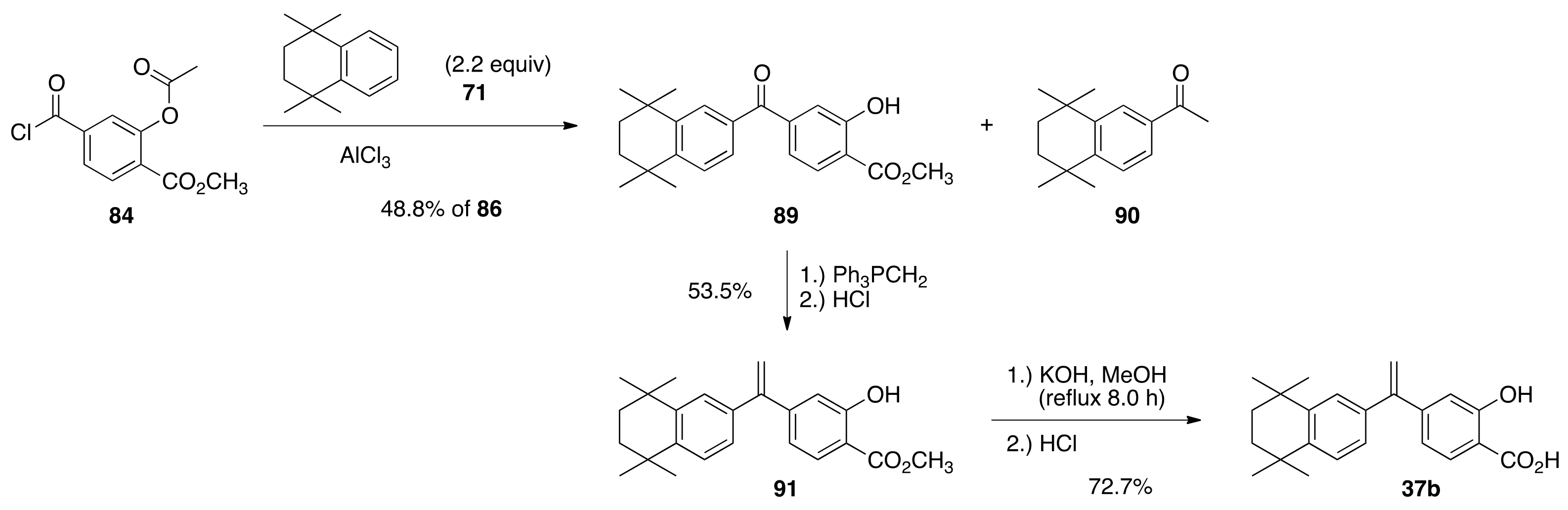
6.56. Methyl 2-hydroxy-4-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalene-2-carbonyl)benzoate (86)
To a 100 mL round bottom flask charged with 3-acetoxy-4-(methoxycarbonyl)benzoic acid (83) (3.236 g, 13.59 mmols) was added thionyl chloride (22 mL, 300 mmols) and a few drops of DMF. A water condenser was added to the flask, and the solution was refluxed in an oil bath for 1.2 h to give methyl 2-acetoxy-4-(chlorocarbonyl)benzoate (84) in quantitative yield after the excess thionyl chloride was removed in vacuo. To the 100 mL round bottom flask containing 84 was added 1,1,4,4,6-pentamethyl-1,2,3,4-tetrahydronaphthalene (85) (6.0508 g, 29.9 mmols) and DCM (30 mL). To the resulting homogeneous solution was slowly added aluminum chloride (3.0 g) at room temperature, with the observed evolution of gas, and the reaction was refluxed for 15 min at 55 °C in an oil bath. The reaction solution was cooled to 0 °C in an ice bath and poured onto 100 mL of an ice water solution. The layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water and then brine, dried over sodium sulfate, filtered, and concentrated in vacuo to give a crude product that was purified by column chromatography (250 mL SiO2) with 1.5% to 5% ethyl acetate/hexanes to give 86 (3.64 g, 70%) as a white solid, m.p. 104.2–106.3 °C: 1H NMR (400 MHz, CDCl3) δ 10.78 (s, 1H), 7.93 (d, J = 8.0, 1H), 7.33 (d, J = 1.2, 1H), 7.31 (dd, J = 8.0, 1.6, 1H), 7.27 (s, 1H), 7.18 (s, 1H), 3.99 (s, 3H), 2.32 (s, 3H), 1.68 (s, 4H), 1.30 (s, 6H), 1.20 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 197.5, 170.0, 161.3, 148.3, 144.5, 141.8, 134.5, 134.4, 129.9, 129.3, 128.3, 119.8, 119.4, 115.2, 52.6, 34.9, 34.8, 34.3, 33.8, 31.7, 31.6, 20.0. ES-MS (M + Na)+ calcd for C24H28O4Na 403.1885, found 403.1888.
6.57. Methyl 2-hydroxy-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoate (88)
To a 100 mL round bottom flask containing a solution of diisopropylamine (5.67 mL, 40.5 mmols) in THF (16.8 mL) was added a 1.6 M solution of n-butyl lithium in hexanes (22.65 mL, 36.24 mmols) at room temperature, and the reaction was stirred for 15 min followed by the addition of methyl triphenylphosphonium bromide (9.7201 g, 27.21 mmols). After stirring this reaction for 1 h, the resulting solution was added to a 100 mL round bottom flask contain a solution of methyl 2-hydroxy-4-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalene-2-carbonyl)benzoate (86) (3.8134 g, 10.02 mmols) in THF (8.86 mL) and the resulting reaction solution was stirred for 1 h, poured into 1 N hydrochloric acid (150 mL, 150 mmols), and extracted with ethyl acetate. The combined organic layers were washed with water and brine, dried over sodium sulfate, filtered, and concentrated in vacuo to give a crude product that was purified by column chromatography (150 mL SiO2) with 1.5% to 5% ethyl acetate/hexanes to give a mixture of spots containing 88 and this mixture was again purified by column chromatography (250 mL SiO2) with 1% to 2% ethyl acetate/hexanes to give pure 88 (1.2997 g, 34%) as white solid, m.p. 103.6–106.6 °C: 1H NMR (400 MHz, CDCl3) δ 10.74 (s, 1H), 7.76 (d, J = 8.4, 1H), 7.11 (s, 1H), 7.06 (s, 1H), 6.88 (dd, J = 8.4, 1.6, 1H), 6.84 (d, J = 2.0, 1H), 5.81 (d, J = 1.2, 1H), 5.33 (d, J = 1.2, 1H), 3.94 (s, 3H), 1.96 (s, 3H), 1.69 (s, 4H), 1.30 (s, 6H), 1.27 (s, 6H). ES-MS (M + Na)+ calcd for C25H30O3Na 401.2093, found 401.2110.
6.58. 2-Hydroxy-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoic acid (37a)
To a 100 mL round bottom flask containing a suspension of methyl 2-hydroxy-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoate (88) (0.3027 g, 0.800 mmols) in methanol (4.0 mL) was added a solution of potassium hydroxide (0.1634 g, 2.9 mmols) in water (0.20 mL), and the flask was fitted with a condenser and refluxed in an oil bath set to 85 °C for 1.2 h. The solution was cooled to room temperature and acidified with 1 N hydrochloric acid (90 mL, 90 mmols), and the resulting precipitate was filtered and dried to give crude 37 (0.2380 g, 81.6%) as a white solid. This crude material was purified by column chromatography (25 mL SiO2) with 40% ethyl acetate/hexanes to pure ethyl acetate to 8% methanol/ethyl acetate to give pure 37a (0.2316 g, 79%) as white solid, m.p. 220.4–224.9 °C: 1H NMR (400 MHz, CDCl3) δ 10.35 (br s, 1H), 7.85 (d, J = 8.4, 1H), 7.11 (s, 1H), 7.07 (s, 1H), 6.92 (dd, J = 8.4, 1.6, 1H), 6.86 (d, J = 1.6, 1H), 5.84 (d, J = 0.8, 1H), 5.36 (d, J = 1.2, 1H), 1.96 (s, 3H), 1.69 (s, 4H), 1.30 (s, 6H), 1.27 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 174.5, 162.1, 149.8, 148.8, 144.4, 142.3, 137.5, 132.6, 130.7, 128.0, 128.0, 118.0, 117.9, 115.7, 109.9, 35.2, 35.1, 33.9, 33.8, 31.9, 31.8, 19.8. ES-MS (M + H)+ calcd for C24H29O3 365.2117, found 365.2122.
6.59. Methyl 2-hydroxy-4-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalene-2-carbonyl)benzoate (89)
To a 100 mL round bottom flask charged with 3-acetoxy-4-(methoxycarbonyl)benzoic acid (83) (4.7646 g, 20.00 mmols) was added thionyl chloride (32 mL, 440 mmols) and a few drops of DMF. A water condenser was added to the flask, and the solution was refluxed in an oil bath for 1.2 h to give methyl 2-acetoxy-4-(chlorocarbonyl)benzoate (84) in quantitative yield after the excess thionyl chloride was removed in vacuo. To the 100 mL round bottom flask containing 84 was added 1,1,4,4-tetramethyl-1,2,3,4-tetrahydronaphthalene (71) (7.4885 g, 39.8 mmols) and DCM (45 mL). To the resulting homogeneous solution was slowly added aluminum chloride (6.7750 g) at room temperature, with the observed evolution of gas, and the reaction was refluxed for 15 min at 55 °C in an oil bath. The reaction solution was cooled to 0 °C in an ice bath and poured onto 100 mL of an ice water solution. The layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water and then brine, dried over sodium sulfate, filtered, and concentrated in vacuo to give a crude product that was purified by column chromatography (250 mL SiO2) with 1.5% to 5% ethyl acetate/hexanes to give 89 (3.5757 g, 48.8%) as white solid, m.p. 76.7–81.3 °C: 1H NMR (400 MHz, CDCl3) δ 10.82 (s, 1H), 7.95 (d, J = 8.4, 1H), 7.81 (d, J = 1.6, 1H), 7.53 (dd, J = 8.4, 1.6, 1H), 7.39 (d, J = 8.0, 1H), 7.32 (d, J = 1.2, 1H), 7.25 (dd, J = 8.0, 1.6, 1H), 3.95 (s, 3H), 1.71 (s, 4H), 1.31 (s, 6H), 1.29 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 195.4, 170.0, 161.1, 150.8, 145.2, 144.5, 133.8, 129.8, 128.8, 127.4, 126.6, 119.8, 118.8, 114.6, 52.6, 34.8, 34.7, 34.6, 34.3, 31.7, 31.5; ES-MS (M + Na)+ calcd for C23H26O4Na 389.1729, found 389.1728.
6.60. Methyl 2-hydroxy-4-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoate (91)
To a 100 mL round bottom flask containing a solution of diisopropylamine (5.07 mL, 36.2 mmols) in THF (15.0 mL) was added a 1.6 M solution of n-butyl lithium in hexanes (20.25 mL, 32.40 mmols) at room temperature, and the reaction was stirred for 15 min followed by the addition of methyl triphenylphosphonium bromide (8.700 g, 24.35 mmols). After stirring this reaction for 1 h, the resulting solution was added to a 100 mL round bottom flask containing a solution of methyl 2-hydroxy-4-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalene-2-carbonyl)benzoate (89) (3.3653 g, 9.18 mmols) in THF (7.92 mL) and the resulting reaction solution was stirred for 1 h, poured into 1 N hydrochloric acid (180 mL, 180 mmols), and extracted with ethyl acetate. The combined organic layers were washed with water and brine, dried over sodium sulfate, filtered, and concentrated in vacuo to give a crude product that was purified by column chromatography (250 mL SiO2) with 1% to 2% ethyl acetate/hexanes to give pure 91 (1.7909 g, 53.5%) as a white solid, m.p. 104.7–112.3 °C: 1H NMR (400 MHz, CDCl3) δ 10.75 (s, 1H), 7.78 (d, J = 8.0, 1H), 7.26 (d, J = 8.0, 1H), 7.25 (dd, J = 7.2, 2.0), 7.05 (dd, J = 8.0, 2.0, 1H); 7.00 (d, J = 1.6, 1H), 6.88 (dd, J = 8.4, 1.6, 1H), 5.51 (d, J = 0.8, 1H), 5.49 (d, J = 1.2, 1H), 3.95 (s, 3H), 1.69 (s, 4H), 1.29 (s, 6H), 1.25 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 170.4, 161.3, 149.2, 149.1, 144.7, 144.6, 137.3, 129.4, 126.3, 125.4, 119.4, 117.1, 115.3, 111.3, 52.2, 35.0, 34.9, 34.2, 34.1, 31.7; ES-MS (M + Na)+ calcd for C24H28O3Na 387.1936, found 387.1939.
6.61. 2-Hydroxy-4-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoic Acid (37b)
To a 100 mL round bottom flask containing a suspension of methyl 2-hydroxy-4-(1-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)benzoate (91) (1.6706 g, 4.58 mmols) in methanol (5.0 mL) was added a solution of potassium hydroxide (0.9389 g, 16.7 mmols) in water (1.16 mL), and the flask was fitted with a condenser and refluxed in an oil bath set to 85 °C for 1.5 h. The solution was cooled to room temperature and acidified with 1 N hydrochloric acid (85 mL, 85 mmols), and the resulting precipitate was filtered and dried to give crude 37b (1.5797 g, 98.3%) as a white solid. This crude material was purified by column chromatography (150 mL SiO2) with 40% ethyl acetate/hexanes to pure ethyl acetate to give pure 37b (1.1678 g, 72.7%) as a white solid, m.p. 183.9–191.5 °C: 1H NMR (400 MHz, CDCl3) δ 10.37 (br s, 1H), 7.88 (d, J = 8.4, 1H), 7.27 (d, J = 8.4, 1H), 7.25 (d, J = 2.4, 1H), 7.06 (dd, J = 8.4, 2.0, 1H), 7.03 (d, J = 1.2, 1H), 6.95 (dd, J = 8.4, 1.2, 1H), 5.54 (d, J = 5.6, 1H), 1.70 (s, 4H), 1.30 (s, 6H), 1.26 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 174.8, 162.0, 150.6, 149.0, 144.8, 144.7, 137.1, 130.5, 126.4, 126.3, 125.4, 119.8, 117.3, 115.7, 110.2, 35.0, 34.9, 34.2, 34.1, 31.8; ES-MS (M + H)+ calcd for C23H27O3 351.1960, found 351.1964.


 ,
, 






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
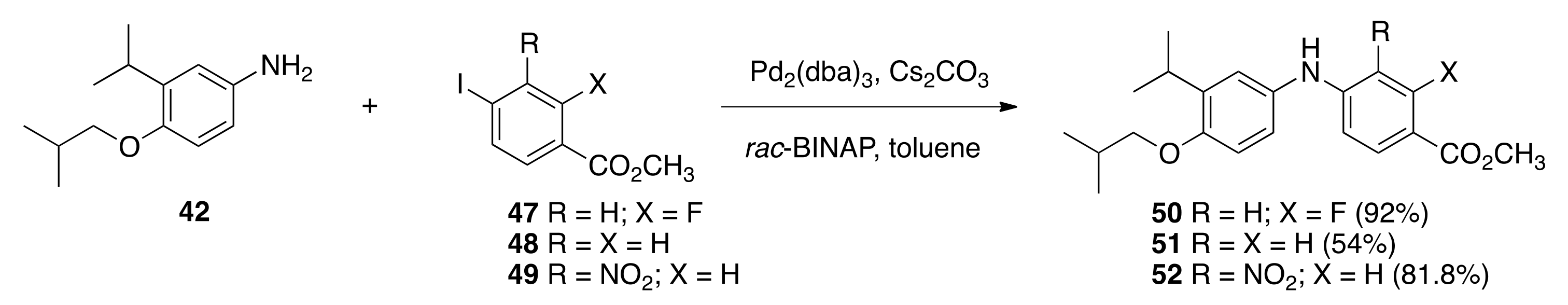{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
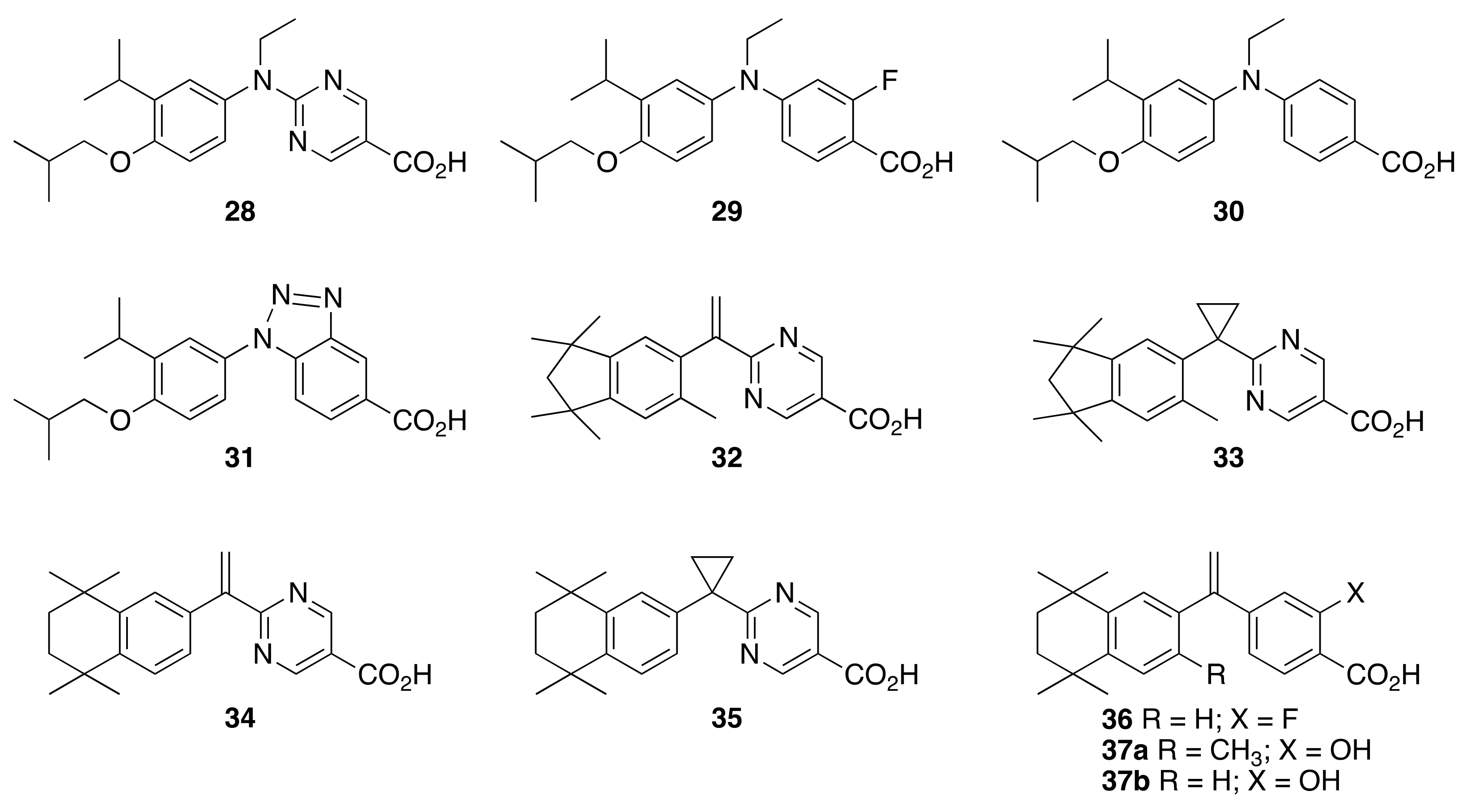
 1
1 25
25 26
26 27
27 28
28 29
29 30
30 31
31 32
32 33
33 34
34 35
35 36
36 37a
37a 37b
37b