
Natural History of a Mouse Model Overexpressing the Dp71 Dystrophin Isoform

 , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
2. Results
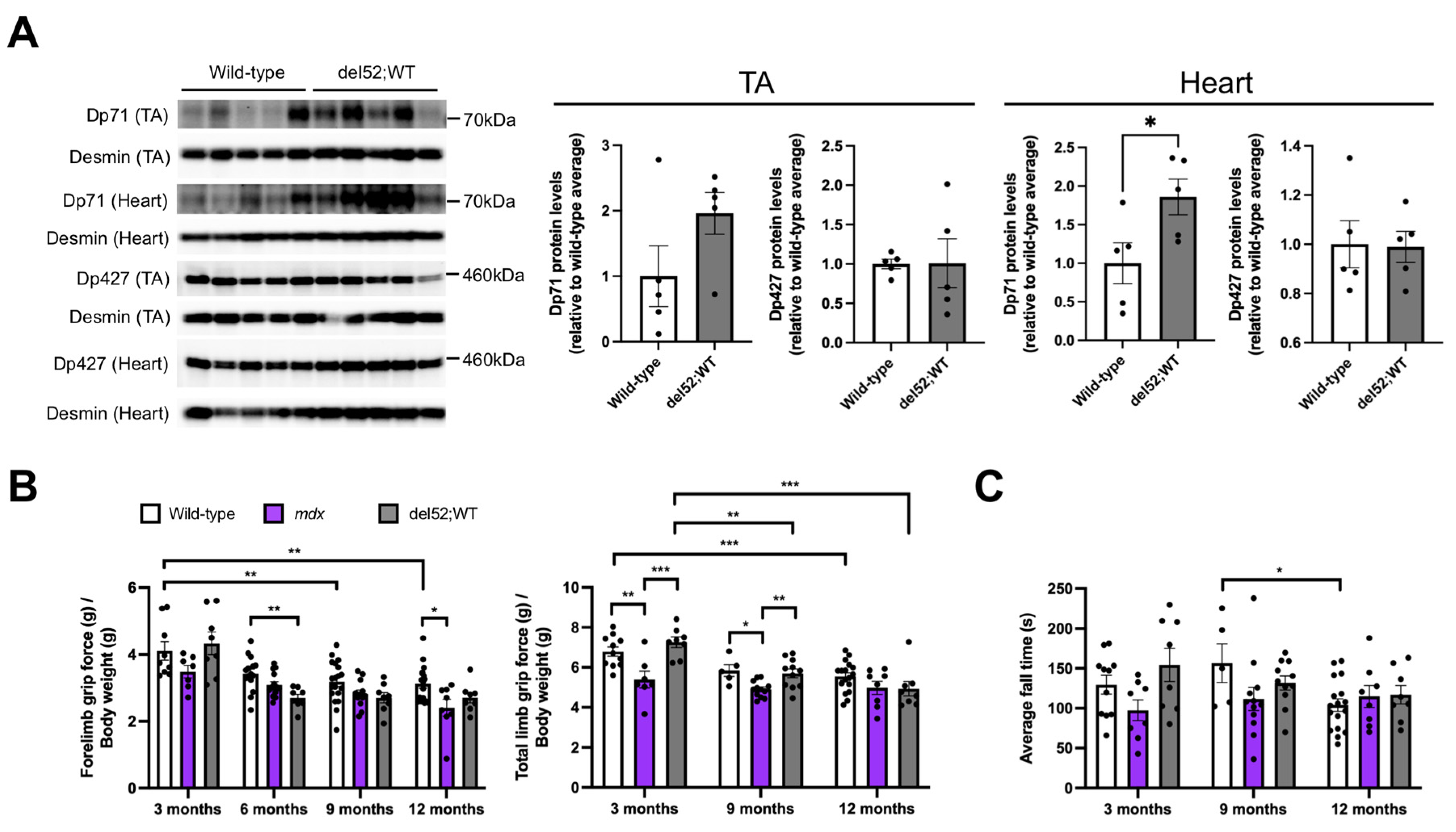
2.1. del52;WT Mice Have Significantly Elevated Dp71 Levels in the Heart but Not Skeletal Muscle, and Have Mostly Normal Skeletal Muscle Function
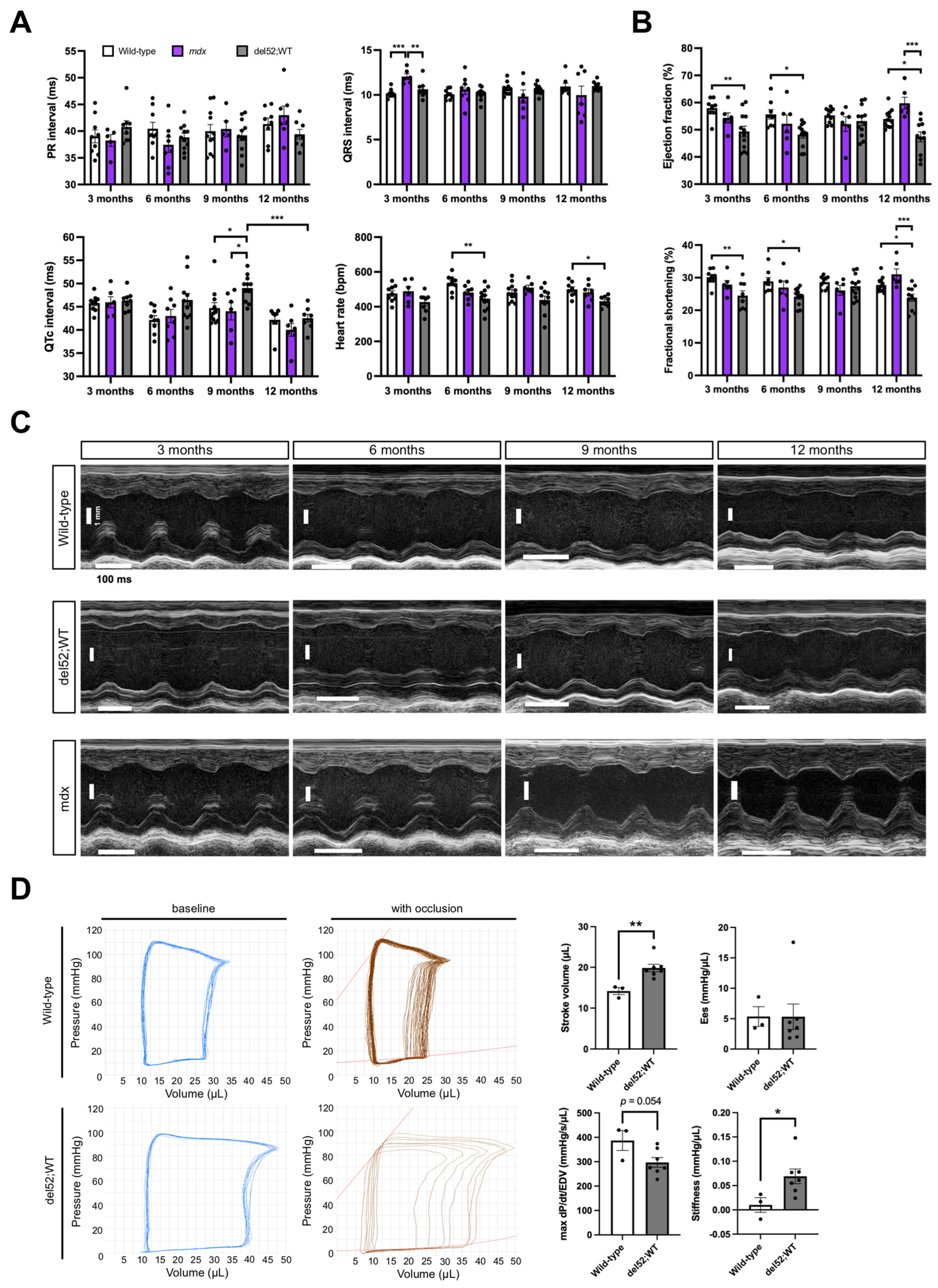
2.2. Cardiac Function in del52;WT Mice Shows Signs of Impairment
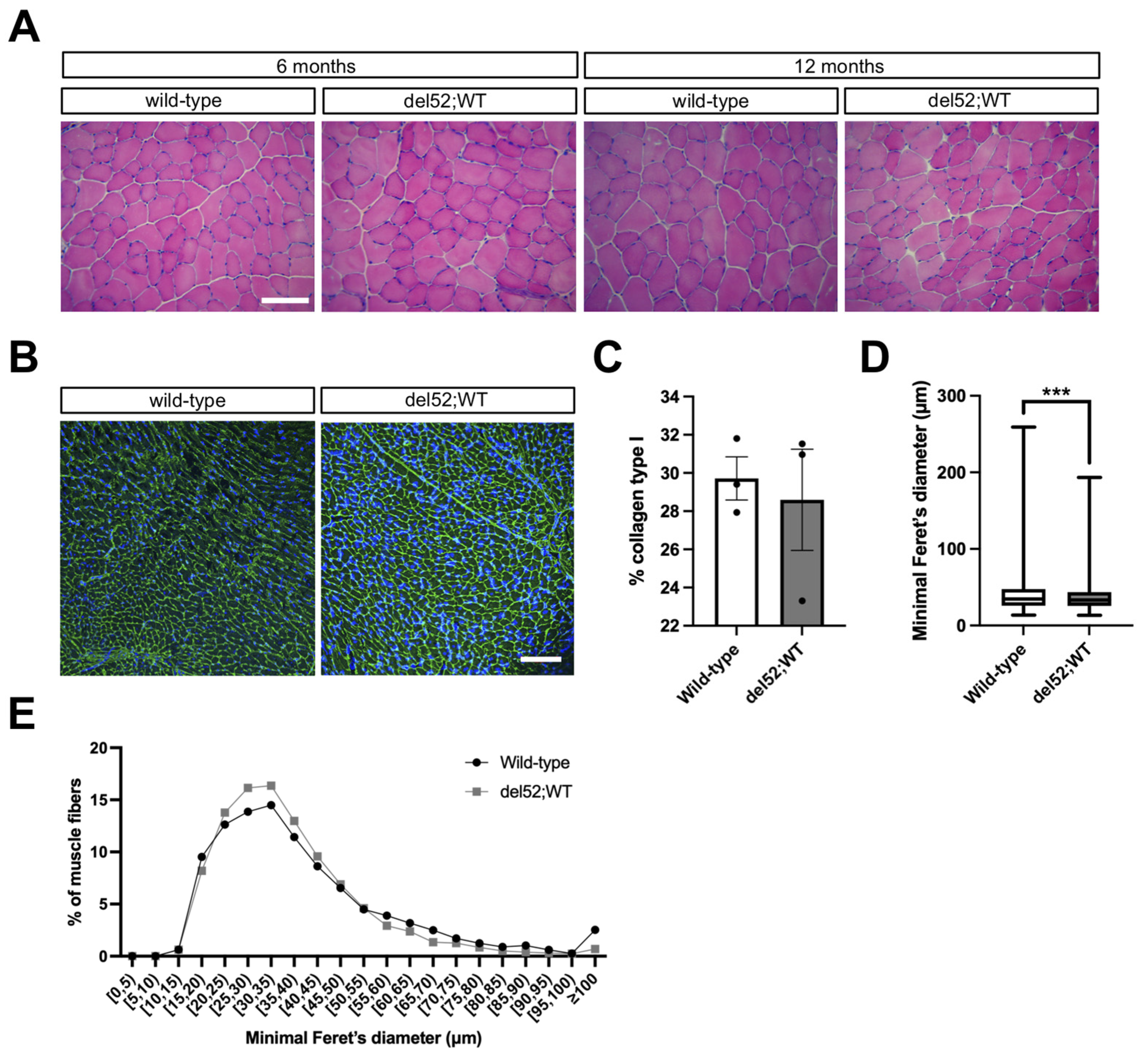
2.3. del52;WT Mice Have Histologically Normal Skeletal and Cardiac Muscle
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Muscle Function Tests
4.3. Electrocardiographic (ECG) Recording
4.4. Echocardiographic Imaging
4.5. Pressure-Volume Loop Measurements
4.6. Western Blot
4.7. Histology
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ervasti, J.M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzur, A.Y.; Kinali, M.; Muntoni, F. Update on the management of Duchenne muscular dystrophy. Arch. Dis. Child. 2008, 93, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Barnea, E.; Zuk, D.; Simantov, R.; Nudel, U.; Yaffe, D. Specificity of expression of the muscle and brain dystrophin gene promoters in muscle and brain cells. Neuron 1990, 5, 881–888. [Google Scholar] [CrossRef]
- Gorecki, D.; Monaco, A.; Derry, J.M.J.; Walker, A.P.; Barnard, E.A.; Barnard, P.J. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum. Mol. Genet. 1992, 1, 505–510. [Google Scholar] [CrossRef]
- D’Souza, V.N.; Man, N.T.; Morris, G.E.; Karges, W.; Pillers, D.-A.M.; Ray, P.N. A novel dystrophin isoform is required for normal retinal electrophysiology. Hum. Mol. Genet. 1995, 4, 837–842. [Google Scholar] [CrossRef]
- Lidov, H.G.W.; Selig, S.; Kunkel, L.M. Dp140: A novel 140 kDa CNS transcript from the dystrophin locus. Hum. Mol. Genet. 1995, 4, 329–335. [Google Scholar] [CrossRef]
- Byers, T.J.; Lidov, H.G.W.; Kunkel, L.M. An alternative dystrophin transcript specific to peripheral nerve. Nat. Genet. 1993, 4, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Lederfein, D.; Levy, Z.; Augier, N.; Mornet, D.; Morris, G.; Fuchs, O.; Yaffe, D.; Nudel, U. A 71-kilodalton protein is a major product of the Duchenne muscular dystrophy gene in brain and other nonmuscle tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 5346–5350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, T.; Niba, E.T.E.; Rani, A.Q.M.; Onishi, Y.; Koizumi, M.; Awano, H.; Matsumoto, M.; Nagai, M.; Yoshida, S.; Sakakibara, S.; et al. Detection of Dystrophin Dp71 in Human Skeletal Muscle Using an Automated Capillary Western Assay System. Int. J. Mol. Sci. 2018, 19, 1546. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, D.; Sunada, Y.; Campbell, K.; Yaffe, D.; Nudel, U. Exogenous Dp71 restores the levels of dystrophin associated proteins but does not alleviate muscle damage in mdx mice. Nat. Genet. 1994, 8, 340–344. [Google Scholar] [CrossRef]
- Urasawa, N.; Wada, M.R.; Machida, N.; Yuasa, K.; Shimatsu, Y.; Wakao, Y.; Yuasa, S.; Sano, T.; Nonaka, I.; Nakamura, A.; et al. Selective Vacuolar Degeneration in Dystrophin-Deficient Canine Purkinje Fibers Despite Preservation of Dystrophin-Associated Proteins With Overexpression of Dp71. Circulation 2008, 117, 2437–2448. [Google Scholar] [CrossRef] [PubMed]
- Leibovitz, S.; Meshorer, A.; Fridman, Y.; Wieneke, S.; Jockusch, H.; Yaffe, D.; Nudel, U. Exogenous Dp71 is a dominant negative competitor of dystrophin in skeletal muscle. Neuromuscul. Disord. 2002, 12, 836–844. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. Genotype–Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry. J. Pers. Med. 2020, 10, 241. [Google Scholar] [CrossRef]
- Tandon, A.; Jefferies, J.L.; Villa, C.R.; Hor, K.N.; Wong, B.L.; Ware, S.M.; Gao, Z.; Towbin, J.A.; Mazur, W.; Fleck, R.J.; et al. Dystrophin Genotype–Cardiac Phenotype Correlations in Duchenne and Becker Muscular Dystrophies Using Cardiac Magnetic Resonance Imaging. Am. J. Cardiol. 2015, 115, 967–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.; Towbin, J.A. Genetic Predictors and Remodeling of Dilated Cardiomyopathy in Muscular Dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef] [Green Version]
- Ashwath, M.L.; Jacobs, I.B.; Crowe, C.A.; Ashwath, R.; Super, D.M.; Bahler, R.C. Left Ventricular Dysfunction in Duchenne Muscular Dystrophy and Genotype. Am. J. Cardiol. 2014, 114, 284–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, Q.; Yokota, T. Antisense oligonucleotides for the treatment of cardiomyopathy in Duchenne muscular dystrophy. Am. J. Transl. Res. 2019, 11, 1202–1218. [Google Scholar]
- Veltrop, M.; Van Vliet, L.; Hulsker, M.; Claassens, J.; Brouwers, C.; Breukel, C.; Van Der Kaa, J.; Linssen, M.M.; Dunnen, J.T.D.; Verbeek, S.; et al. A dystrophic Duchenne mouse model for testing human antisense oligonucleotides. PLoS ONE 2018, 13, e0193289. [Google Scholar] [CrossRef]
- Yavas, A.; Weij, R.; Van Putten, M.; Kourkouta, E.; Beekman, C.; Puoliväli, J.; Bragge, T.; Ahtoniemi, T.; Knijnenburg, J.; Hoogenboom, M.E.; et al. Detailed genetic and functional analysis of the hDMDdel52/mdx mouse model. PLoS ONE 2020, 15, e0244215. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-H.; Ms, C.H.H.; Zhang, K.; Duan, D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve 2010, 43, 283–286. [Google Scholar] [CrossRef] [Green Version]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The Molecular Basis of Muscular Dystrophy in the mdx Mouse: A Point Mutation. Sci. 1989, 244, 1578–1580. [Google Scholar] [CrossRef]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar]
- Tamiyakul, H.; Kemter, E.; Kösters, M.; Ebner, S.; Blutke, A.; Klymiuk, N.; Flenkenthaler, F.; Wolf, E.; Arnold, G.J.; Fröhlich, T. Progressive Proteome Changes in the Myocardium of a Pig Model for Duchenne Muscular Dystrophy. iScience 2020, 23. [Google Scholar] [CrossRef]
- Lederfein, D.; Yaffe, D.; Nudel, U. A housekeeping type promoter, located in the 3’ region of the Duchenne muscular dystrophy gene, controls the expression of Dp71, a major product of the gene. Hum. Mol. Genet. 1993, 2, 1883–1888. [Google Scholar] [CrossRef]
- Richard, C.A.; Howard, P.L.; D’Souza, V.N.; Klamut, H.J.; Ray, P.N. Cloning and characterization of alternatively spliced isoforms of Dp71. Hum. Mol. Genet. 1995, 4, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.A.; Sunada, Y.; Campbell, K.; Chamberlain, J.S. Dp71 can restore the dystrophin-associated glycoprotein complex in muscle but fails to prevent dystrophy. Nat. Genet. 1994, 8, 333–339. [Google Scholar] [CrossRef]
- de Leon, M.B.; Montañez, C.; Gómez, P.; Morales-Lázaro, S.L.; Tapia-Ramírez, V.; Valadez-Graham, V.; Recillas-Targa, F.; Yaffe, D.; Nudel, U.; Cisneros, B. Dystrophin Dp71 Expression Is Down-regulated during Myogenesis. J. Biol. Chem. 2005, 280, 5290–5299. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2019, 57, 1748–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, C.; Loch, D.; Laws, N.; Trebbin, A.; Hoey, A.J. Timeline of cardiac dystrophy in 3-18-month-old MDX mice. Muscle Nerve 2010, 42, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Sarig, R.; Mezger-Lallemand, V.; Gitelman, I.; Davis, C.; Fuchs, O.; Yaffe, D.; Nudel, U. Targeted inactivation of Dp71, the major non-muscle product of the DMD gene: Differential activity of the Dp71 promoter during development. Hum. Mol. Genet. 1999, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Aragón, J.; González-Reyes, M.; Romo-Yañez, J.; Vacca, O.; Aguilar-González, G.; Rendón, A.; Vaillend, C.; Montañez, C. Dystrophin Dp71 Isoforms Are Differentially Expressed in the Mouse Brain and Retina: Report of New Alternative Splicing and a Novel Nomenclature for Dp71 Isoforms. Mol. Neurobiol. 2018, 55, 1376–1386. [Google Scholar] [CrossRef]
- Dalloz, C.; Sarig, R.; Fort, P.; Yaffe, D.; Bordais, A.; Pannicke, T.; Grosche, J.; Mornet, D.; Reichenbach, A.; Sahel, J.-A.; et al. Targeted inactivation of dystrophin gene product Dp71: Phenotypic impact in mouse retina. Hum. Mol. Genet. 2003, 12, 1543–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aartsma-Rus, A.; Van Putten, M. Assessing Functional Performance in the Mdx Mouse Model. J. Vis. Exp. 2014, 85, e51303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhabyeyev, P.; McLean, B.; Chen, X.; Vanhaesebroeck, B.; Oudit, G.Y. Inhibition of PI3Kinase-α is pro-arrhythmic and associated with enhanced late Na+ current, contractility, and Ca2+ release in murine hearts. J. Mol. Cell. Cardiol. 2019, 132, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhabyeyev, P.; Chen, X.; Wang, F.; Paul, M.; Fan, D.; McLean, B.A.; Basu, R.; Zhang, P.; Shah, S.; et al. PI3Kα-regulated gelsolin activity is a critical determinant of cardiac cytoskeletal remodeling and heart disease. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Maruyama, R.; Echigoya, Y.; Caluseriu, O.; Aoki, Y.; Takeda, S.; Yokota, T. Systemic Delivery of Morpholinos to Skip Multiple Exons in a Dog Model of Duchenne Muscular Dystrophy. Adv. Struct. Saf. Stud. 2017, 1565, 201–213. [Google Scholar] [CrossRef]
- Desgeorges, T.; Liot, S.; Lyon, S.; Bouvière, J.; Kemmel, A.; Trignol, A.; Rousseau, D.; Chapuis, B.; Gondin, J.; Mounier, R.; et al. Open-CSAM, a new tool for semi-automated analysis of myofiber cross-sectional area in regenerating adult skeletal muscle. Skelet. Muscle 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Parameter | 3 Months | 6 Months | 9 Months | 12 Months | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | mdx | del52;WT | WT | mdx | del52;WT | WT | mdx | del52;WT | WT | mdx | del52;WT | |
| Number | 8 | 4–5 | 12 | 8 | 5–6 | 11 | 10 | 3–6 | 11–12 | 12 | 6 | 10–11 |
| Heart rate | 443.0 (31.3) | 425.3 (32.8) | 426.6 (48.7) | 464.6 (32.7) | 450.5 (50.1) | 418.4 (36.8) b | 423.8 (45.6) | 528.7 (23.9) aa | 430.4 (38.9) cc | 427.1 (62.6) | 531.5 (63.4) aa | 393.3 (55.0) ccc |
| LVIDd | 3.9 (0.2) | 4.1 (0.2) | 4.2 (0.4) | 4.3 (0.4) | 4.0 (0.4) | 4.3 (0.3) | 4.3 (0.3) | 3.3 (0.4) aa | 4.5 (0.4) cc | 4.3 (0.2) | 3.5 (0.5) aa | 4.6 (0.4) cc |
| LVIDs | 2.8 (0.2) | 3.0 (0.2) | 3.2 (0.5) | 3.0 (0.3) | 2.9 (0.4) | 3.3 (0.3) | 3.1 (0.2) | 2.4 (0.3) aa | 3.3 (0.4) cc | 3.1 (0.2) | 2.4 (0.3) aa | 3.6 (0.4) cc |
| LVPWd | 0.6 (0.1) | 0.5 (0.2) | 0.5 (0.1) bb | 0.7 (0.05) | 0.7 (0.1) | 0.6 (0.1) bb,cc | 0.7 (0.1) | 0.8 (0.2) | 0.5 (0.1) b,cc | 0.6 (0.1) | 0.7 (0.1) | 0.5 (0.1) c |
| LVPWs | 1.0 (0.1) | 0.8 (0.2) | 0.7 (0.1) | 1.1 (0.1) | 0.9 (0.2) a | 0.8 (0.1) bb,c | 1.0 (0.1) | 1.1 (0.1) | 0.8 (0.1) bb,cc | 1.0 (0.2) | 1.1 (0.2) | 0.7 (0.2) bb,cc |
| LV vol s | 29.1 (4.1) | 34.1 (5.6) | 41.2 (15.4) | 36.3 (8.0) | 33.6 (12.1) | 44.1 (9.9) | 37.7 (4.6) | 20.9 (6.2) aa | 43.8 (11.7) cc | 38.4 (4.9) | 20.1 (7.2) aa | 54.0 (16.8) bb,cc |
| LV vol d | 68.1 (8.1) | 75.7 (7.1) | 79.8 (19.4) | 82.0 (16.4) | 68.9 (15.2) | 84.4 (15.6) | 84.7 (12.4) | 46.3 (12.9) aa | 92.2 (17.7) cc | 84.7 (7.0) | 51.6 (18.9) aa | 98.3 (18.4) cc |
| EF | 57.9 (4.0) | 54.3 (5.1) | 49.3 (7.2) bb | 55.7 (5.4) | 52.8 (7.6) | 48.2 (4.9) bb | 55.3 (3.4) | 52.1 (8.4) | 53.2 (5.9) | 53.9 (4.1) | 59.8 (5.7) a | 47.4 (7.1) bb,cc |
| FS | 30.1 (2.7) | 27.9 (3.3) | 24.8 (4.4) bb | 28.9 (3.7) | 27.0 (4.8) | 24.2 (2.9) bb | 28.6 (2.3) | 26.1 (4.8) | 27.4 (3.7) | 27.7 (2.7) | 31.0 (4.2) a | 23.8 (4.2) bb,cc |
| LA | 1.5 (52.4) | 1.6 (0.2) a | 1.6 (0.04) bb | 1.4 (0.1) | 1.7 (0.3) aa | 1.7 (0.1) bb | 1.4 (0.1) | 1.8 (0.7) | 1.9 (0.3) b | 1.6 (0.3) | 1.9 (0.5) | 1.9 (0.2) |
| IVRT | 16.9 (2.0) | 14.7 (2.1) | 16.5 (4.2) | 13.5 (1.0) | 14.1 (1.4) | 14.3 (1.8) | 13.4 (1.6) | 13.7 (2.4) | 13.3 (1.5) | 13.9 (2.0) | 12.6 (5.0) | 16.0 (2.7) |
| IVCT | 16.7 (56.6) | 13.9 (5.4) | 16.1 (5.5) | 13.5 (3.2) | 10.9 (2.7) | 12.5 (2.6) | 11.7 (3.3) | 10.5 (2.7) | 10.9 (2.9) | 12.3 (3.5) | 9.5 (2.4) | 13.8 (2.4) c |
| MV decel | 22.4 (4.9) | 18.3 (2.4) | 22.0 (5.6) | 17.7 (3.0) | 16.9 (5.4) | 24.5 (5.2) b,c | 19.8 (6.0) | 14.5 (7.2) | 22.9 (5.6) | 16.2 (4.6) | 16.5 (7.9) | 19.8 (3.5) |
| MV E | 663.5 (85.5) | 702.6 (70.6) | 681.9 (118.6) | 619.5 (65.7) | 713.8 (118.2) | 697.8 (76.1) | 610.8 (46.7) | 476.8 (196.5) | 727.9 (114.4) cc | 610.5 (36.8) | 541.5 (206.2) | 636.4 (103.0) |
| MV A | 476.0 (43.7) | 444.2 (65.1) | 438.5 (99.6) | 437.9 (92.8) | 513.8 (102.9) | 452.3 (98.1) | 423.2 (66.5) | 368.7 (179.0) | 458.2 (119.9) | 441.6 (102.3) | 412.9 (151.1) | 420.2 (178.2) |
| MV E/A | 1.4 (0.2) | 1.6 (0.2) | 1.6 (0.3) | 1.5 (0.2) | 1.4 (0.2) | 1.6 (0.4) | 1.5 (0.2) | 1.6 (1.2) | 1.7 (0.4) | 1.4 (0.3) | 1.6 (1.0) | 1.7 (0.4) |
| E’ | −24.6 (4.6) | −19.0 (2.8) | −23.6 (3.9) | −23.5 (5.5) | −25.8 (3.6) | −22.7 (3.3) | −23.1 (4.2) | −27.4 (4.5) | −21.8 (3.6) c | −19.1 (4.0) | −24.3 (9.5) | −18.0 (5.2) |
| A’ | −24.3 (5.3) | −26.9 (5.1) | −23.2 (1.8) | −22.9 (4.8) | −32.1 (9.3) a | −21.7 (3.6) cc | −21.1 (5.4) | −26.3 (5.2) | −23.8 (3.2) | −23.5 (5.0) | −27.5 (11.8) | −22.0 (6.9) |
| E’/A’ | 1.0 (0.2) | 0.7 (0.04) a | 1.0 (0.2) c | 1.1 (0.3) | 0.9 (0.3) | 1.1 (0.3) | 1.2 (0.4) | 1.1 (0.3) | 0.9 (0.2) | 0.9 (0.3) | 1.0 (0.7) | 0.9 (0.3) |
| MV E/E’ | −28.2 (8.8) | −37.5 (6.8) | −29.6 (6.7) | −27.9 (7.9) | −29.0 (6.4) | −31.3 (5.9) | −27.2 (5.3) | −17.5 (8.7) | −34.1 (6.3) b,cc | −33.1 (6.1) | −26.8 (16.0) | −38.1 (12.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.R.Q.; Shah, M.N.A.; Woo, S.; Wilton-Clark, H.; Zhabyeyev, P.; Wang, F.; Maruyama, R.; Oudit, G.Y.; Yokota, T. Natural History of a Mouse Model Overexpressing the Dp71 Dystrophin Isoform. Int. J. Mol. Sci. 2021, 22, 12617. https://doi.org/10.3390/ijms222312617
Lim KRQ, Shah MNA, Woo S, Wilton-Clark H, Zhabyeyev P, Wang F, Maruyama R, Oudit GY, Yokota T. Natural History of a Mouse Model Overexpressing the Dp71 Dystrophin Isoform. International Journal of Molecular Sciences. 2021; 22(23):12617. https://doi.org/10.3390/ijms222312617
Chicago/Turabian StyleLim, Kenji Rowel Q., Md Nur Ahad Shah, Stanley Woo, Harry Wilton-Clark, Pavel Zhabyeyev, Faqi Wang, Rika Maruyama, Gavin Y. Oudit, and Toshifumi Yokota. 2021. "Natural History of a Mouse Model Overexpressing the Dp71 Dystrophin Isoform" International Journal of Molecular Sciences 22, no. 23: 12617. https://doi.org/10.3390/ijms222312617
APA StyleLim, K. R. Q., Shah, M. N. A., Woo, S., Wilton-Clark, H., Zhabyeyev, P., Wang, F., Maruyama, R., Oudit, G. Y., & Yokota, T. (2021). Natural History of a Mouse Model Overexpressing the Dp71 Dystrophin Isoform. International Journal of Molecular Sciences, 22(23), 12617. https://doi.org/10.3390/ijms222312617