
Paradigmatic De Novo GRIN1 Variants Recapitulate Pathophysiological Mechanisms Underlying GRIN1-Related Disorder Clinical Spectrum

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. GRIN1 Variants-Associated Clinical Cases
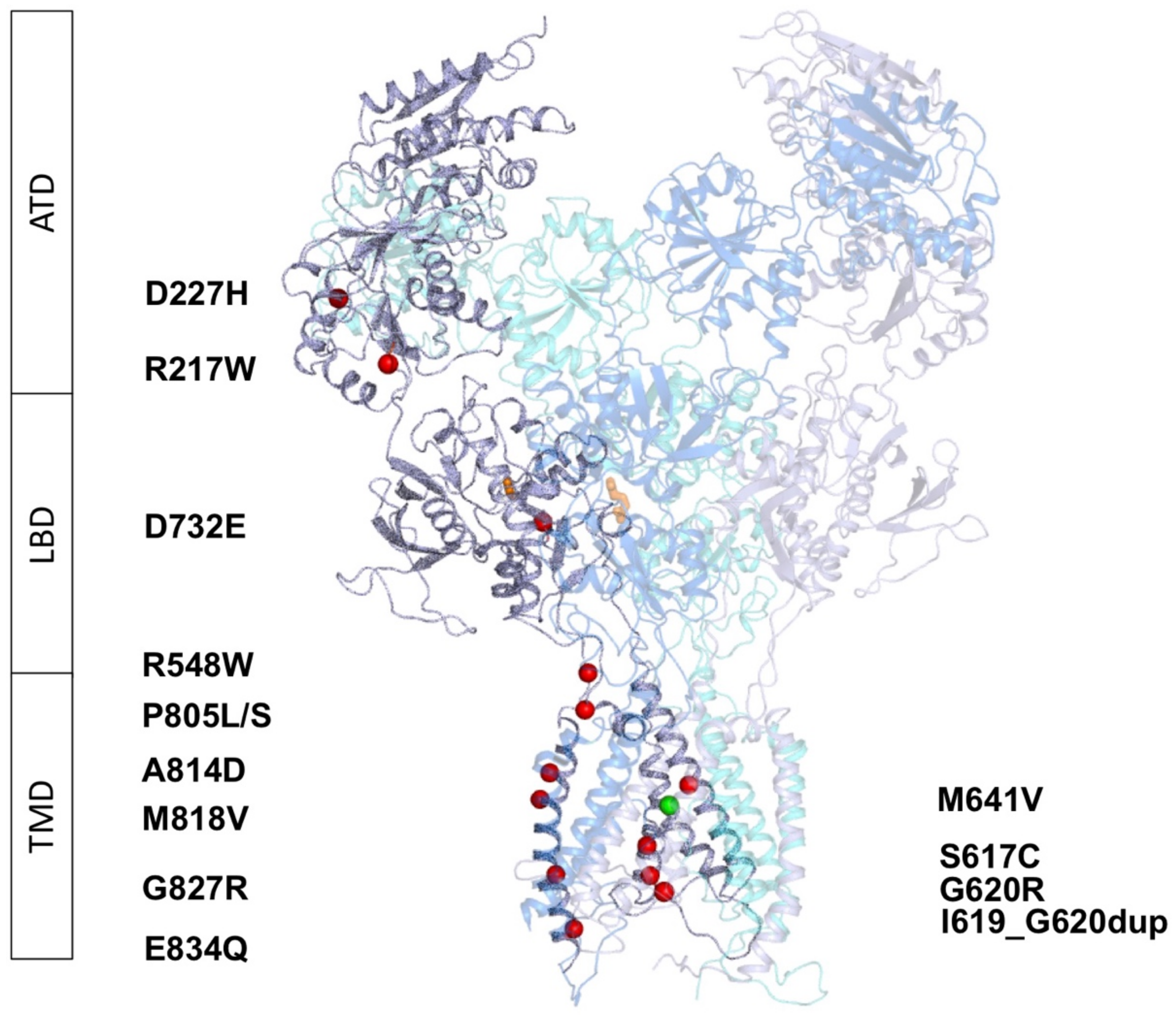
2.2. Molecular Modeling of GRIN1-DNVs in Triheteromeric NMDA Receptors
2.2.1. Variants Affecting GluN1 Subunit Folding and/or NMDAR Oligomerisation
2.2.2. GRIN1-D732E Missense Variant Affects Coagonist Binding
2.2.3. GRIN1-DNVs Affecting NMDAR Channel Properties
2.3. Functional Annotations of GRIN1-DNVs
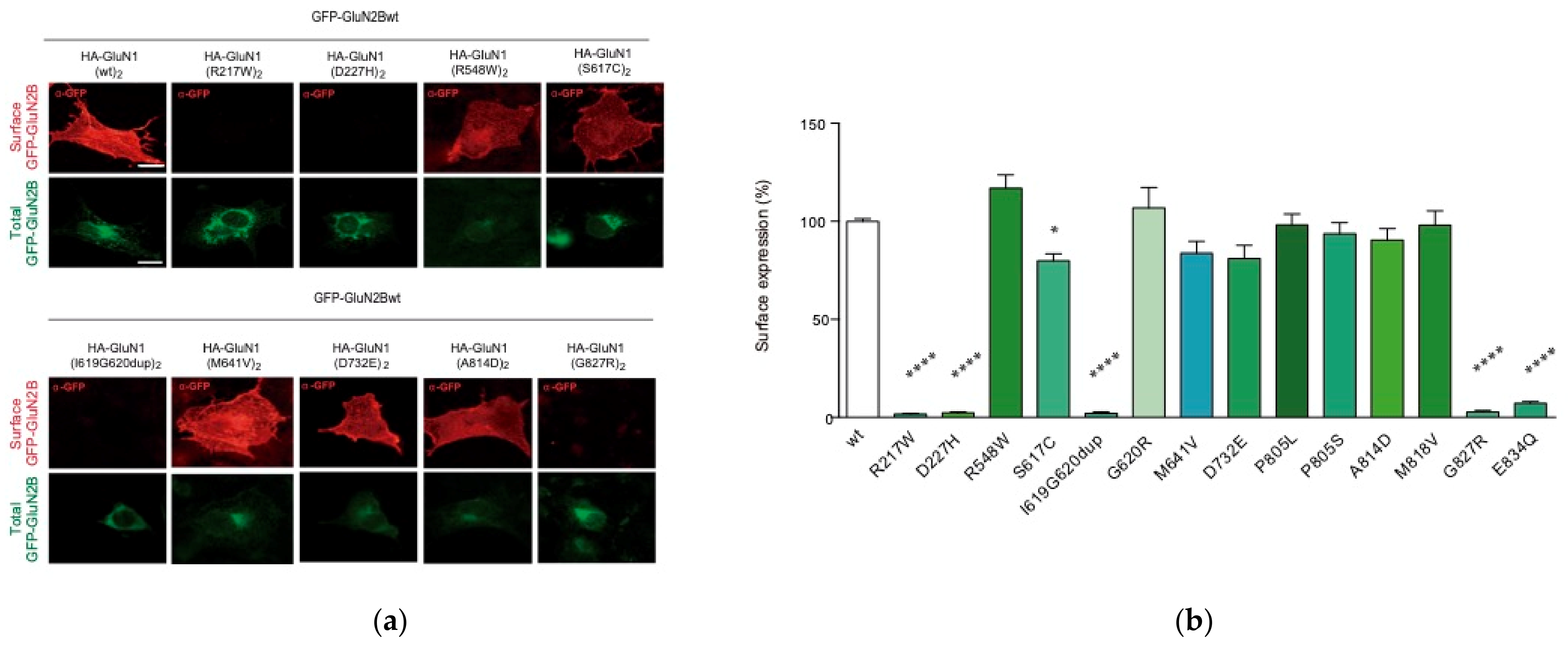
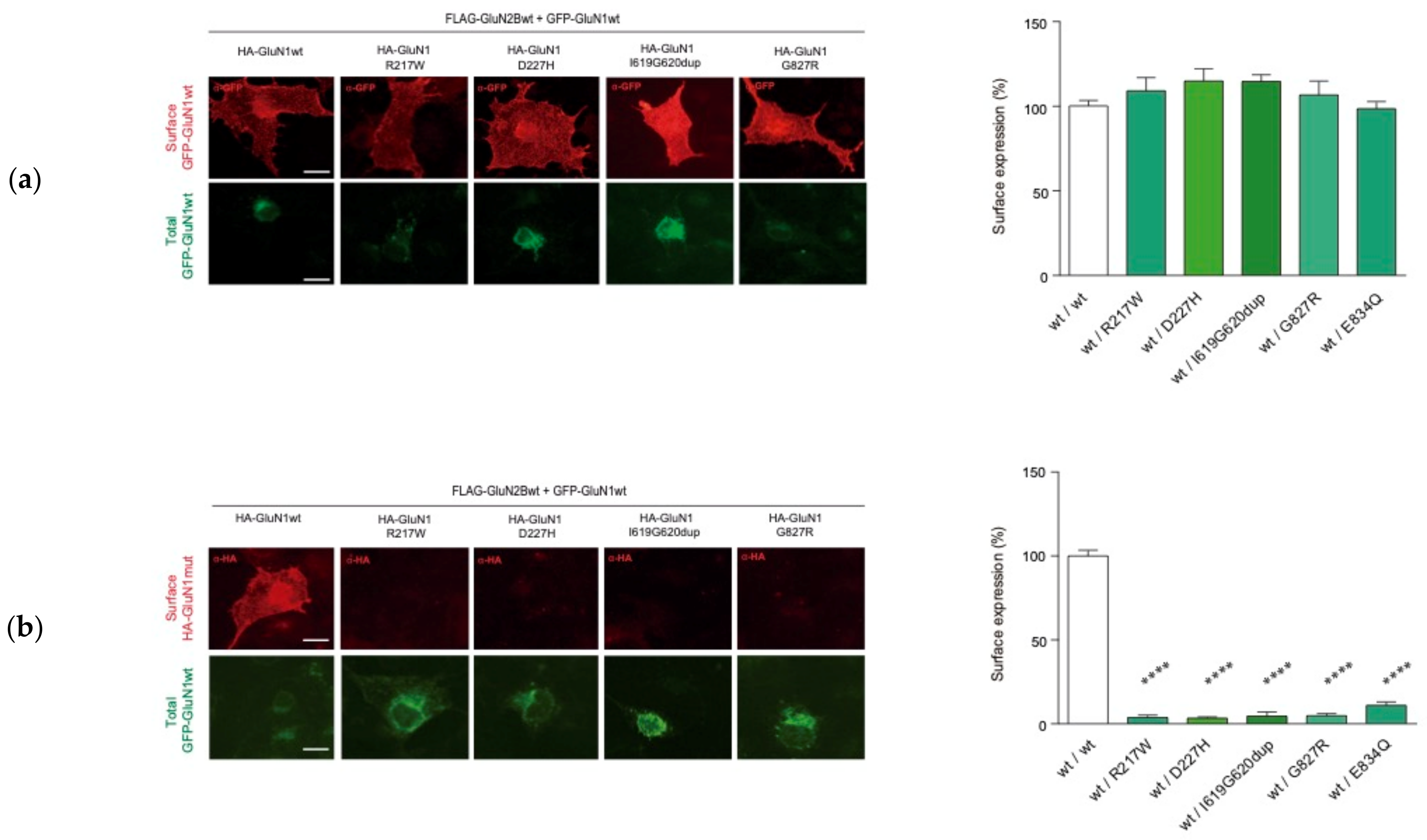
2.3.1. GRIN1 Variants Affecting NMDAR Surface Density
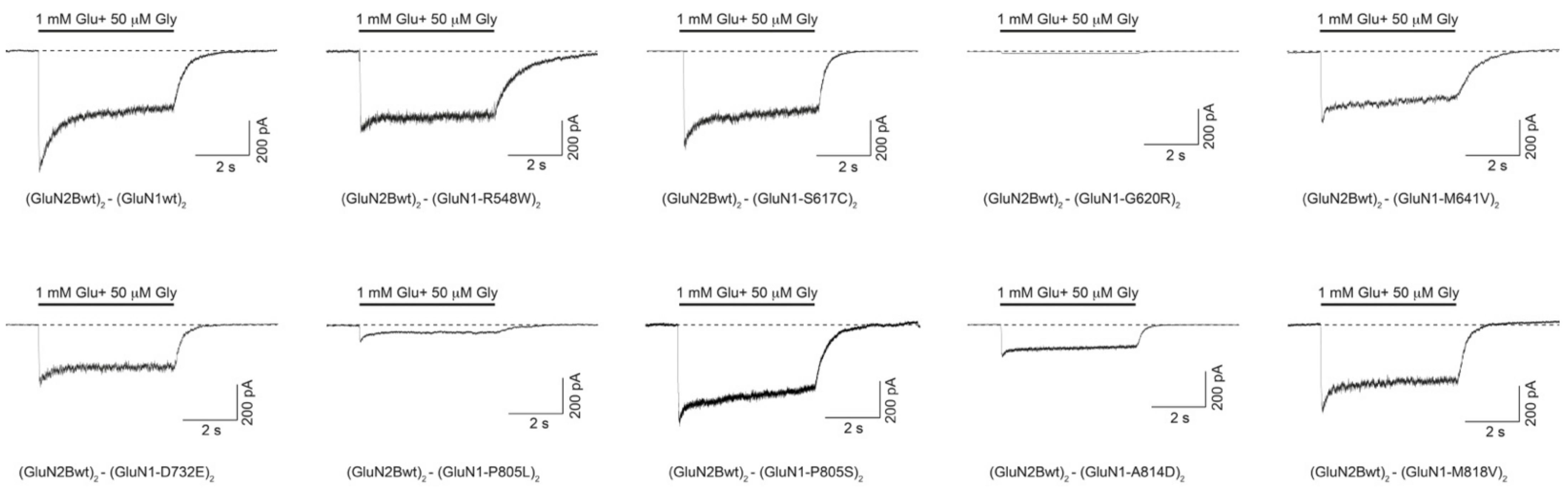
2.3.2. GRIN1 Variants Disturbing NMDAR Channel Gating Properties
2.4. GRIN1-DNV Functional Stratification and GRD Severity
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Plasmids
4.3. Cell Culture and Transfection
4.4. Immunofluorescence Analysis of NMDAR Surface Expression in COS-7 Cells
4.5. Electrophysiological Recordings of NMDAR-Mediated Whole-Cell Currents in HEK-293T Cells
4.6. Statistical Analysis
4.7. Structural Models
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Volk, L.; Chiu, S.-L.; Sharma, K.; Huganir, R.L. Glutamate synapses in human cognitive disorders. Annu. Rev. Neurosci. 2015, 38, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Endele, S.; Rosenberger, G.; Geider, K.; Popp, B.; Tamer, C.; Stefanova, I.; Milh, M.; Kortüm, F.; Fritsch, A.; Pientka, F.K.; et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 2010, 42, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Tarabeux, J.; Kebir, O.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; Piton, A.; Spiegelman, D.; Henrion, É.; Millet, B.; S2D Team; et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 2011, 1, e55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ligt, J.; Willemsen, M.H.; van Bon, B.W.M.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- Lemke, J.R. Predicting incidences of neurodevelopmental disorders. Brain 2020, 143, 1046–1048. [Google Scholar] [CrossRef]
- García-Recio, A.; Santos-Gómez, A.; Soto, D.; Julia-Palacios, N.; García-Cazorla, À.; Altafaj, X.; Olivella, M. GRIN database: A unified and manually curated repertoire of GRIN variants. Hum. Mutat. 2021, 42, 8–18. [Google Scholar] [CrossRef]
- Strehlow, V.; Heyne, H.O.; Vlaskamp, D.R.M.; Marwick, K.F.M.; Rudolf, G.; de Bellescize, J.; Biskup, S.; Brilstra, E.H.; Brouwer, O.F.; Callenbach, P.M.C.; et al. GRIN2A-related disorders: Genotype and functional consequence predict phenotype. Brain 2019, 142, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Platzer, K.; Yuan, H.; Schütz, H.; Winschel, A.; Chen, W.; Hu, C.; Kusumoto, H.; Heyne, H.O.; Helbig, K.L.; Tang, S.; et al. GRIN2B encephalopathy: Novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 2017, 54, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Lemke, J.R.; Geider, K.; Helbig, K.L.; Heyne, H.O.; Schütz, H.; Hentschel, J.; Courage, C.; Depienne, C.; Nava, C.; Heron, D.; et al. Delineating the GRIN1 phenotypic spectrum: A distinct genetic NMDA receptor encephalopathy. Neurology 2016, 86, 2171–2178. [Google Scholar] [CrossRef] [Green Version]
- XiangWei, W.; Jiang, Y.; Yuan, H. De Novo Mutations and Rare Variants Occurring in NMDA Receptors. Curr. Opin. Physiol. 2018, 2, 27–35. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Furukawa, H.; Singh, S.K.; Mancusso, R.; Gouaux, E. Subunit arrangement and function in NMDA receptors. Nature 2005, 438, 185–192. [Google Scholar] [CrossRef]
- Wang, H.; Lv, S.; Stroebel, D.; Zhang, J.; Pan, Y.; Huang, X.; Zhang, X.; Paoletti, P.; Zhu, S. Gating mechanism and a modulatory niche of human GluN1-GluN2A NMDA receptors. Neuron 2021, 109, 2443–2456.e5. [Google Scholar] [CrossRef]
- Chou, T.-H.; Tajima, N.; Romero-Hernandez, A.; Furukawa, H. Structural Basis of Functional Transitions in Mammalian NMDA Receptors. Cell 2020, 182, 357–371.e13. [Google Scholar] [CrossRef]
- Soto, D.; Olivella, M.; Grau, C.; Armstrong, J.; Alcon, C.; Gasull, X.; Santos-Gómez, A.; Locubiche, S.; de Salazar, M.G.; García-Díaz, R.; et al. l-Serine dietary supplementation is associated with clinical improvement of loss-of-function GRIN2B-related pediatric encephalopathy. Sci. Signal. 2019, 12, eaaw0936. [Google Scholar] [CrossRef]
- Mullier, B.; Wolff, C.; Sands, Z.A.; Ghisdal, P.; Muglia, P.; Kaminski, R.M.; André, V.M. GRIN2B gain of function mutations are sensitive to radiprodil, a negative allosteric modulator of GluN2B-containing NMDA receptors. Neuropharmacology 2017, 123, 322–331. [Google Scholar] [CrossRef]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar] [CrossRef]
- Snyder, M.A.; Gao, W.-J. NMDA receptor hypofunction for schizophrenia revisited: Perspectives from epigenetic mechanisms. Schizophr. Res. 2020, 217, 60–70. [Google Scholar] [CrossRef]
- Ossnat Bar-Shira, R.M.G.C. Gene Expression Switching of Receptor Subunits in Human Brain Development. PLoS Comput. Biol. 2015, 11, e1004559. [Google Scholar] [CrossRef]
- Santos-Gómez, A.; Miguez-Cabello, F.; García-Recio, A.; Locubiche-Serra, S.; García-Díaz, R.; Soto-Insuga, V.; Guerrero-López, R.; Juliá-Palacios, N.; Ciruela, F.; García-Cazorla, À.; et al. Disease-associated GRIN protein truncating variants trigger NMDA receptor loss-of-function. Hum. Mol. Genet. 2021, 29, 3859–3871. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Lehner, B.; Lindeboom, R.G.H. To NMD or Not To NMD: Nonsense-Mediated mRNA Decay in Cancer and Other Genetic Diseases. Trends Genet. 2021, 37, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Vicini, S.; Wang, J.F.; Li, J.H.; Zhu, W.J.; Wang, Y.H.; Luo, J.H.; Wolfe, B.B.; Grayson, D.R. Functional and Pharmacological Differences Between RecombinantN-Methyl-d-Aspartate Receptors. J. Neurophysiol. 1998, 79, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Karakas, E.; Furukawa, H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 2014, 344, 992–997. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2014, 47, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SUPERIOR FUNCTIONS | MOTOR FUNCTION | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Clinical Case | Variant | Gender, Age | Intellectual Disability | Mood Disorders | Sleep Disorder | Verbal Communication | ASD Traits | Movement Disorders | Gait | Muscle Tone | Oculogyric Crisis |
| 1 | GRIN1-R548W | F, exitus (10 m.o.) | Severe | No | Yes (melatonin treatment) | No | No | Akinetic rigid syndrome, stereotypies | Non-acquired ambulation | Lower limb spasticity, axial hypotonia | N.A. |
| 2 | GRIN1-S617C | F; 9.5 years-old | Severe | Hyperactivity | Sleeps with the mother | <10 words | No | No | Motor delay, autonomous ambulation aquired at 3 y.o. | Normal | No |
| 3 | GRIN1-I619G620dup | F; 12 years-old | Severe | Irritability | No | No | No | No | Ambulation with double support | Hypotonia | No |
| 4 | GRIN1-G620R | M, 5 years-old | Severe | No | No | No. Language restricted to 5 purposeful sounds, understands easy orders | No | No ambulation. No sitting. Cephalic control | Severe axial hypotonia, hyperlaxity | No | |
| 5 | GRIN1-G620R | F, 5.5 years-old | Severe | No | No | No. Unintelligible jargon, absence of purposeful language | Yes | Hyperkinesia, dyskinetic movements, hands-washing stereotypies | No ambulation. Sitting acquired at 4 y.o., standing with supports | Axial hypotonia, Upper limb hypertonia, hyperlaxity | N.A. |
| 6 | GRIN1-M641V | F, Exitus (20 y.o.) | Severe | Agressivity | No | No | Yes | No | No ambulation | Limbs spaticity axial hypotonia | |
| 7 | GRIN1-D732E | F, 11.5 years-old | Severe | Intolerance, frequent hanger behaviour, hypo- and hypersensitivity | No | Poor language (7 words, gestures, shouts) acquired at 4 years-old. Sporadic use od 2-3 bisyllabic words, and 3 bimodal signs | ASD traits, social interaction interest | Hands and swinging stereotypies; manual hyperkinesia | Walking acquired at 5 y.o., with external hand support, ability to walk and go up and down stairs | Axial hypotonia, lower limbs mild spactic signs, hyperlaxity | No |
| 8 | GRIN1-D732E | F, 18 years-old | Severe | No | No | No | No | Akinetic rigid syndrome, stereotypies | Autonomous ambulation in familiar spaces, unstability in uneven terrains | Upper limbs spasticity (lower limbs, difficult to assess) | No |
| 9 | GRIN1-P805S | F, 11.8 years-old | Severe | Hyperactivity, impulsiveness | No | No. Unintelligible jargon | No | No | Normal ambulation | Axial hypotonia, spasticity | No |
| 10 | GRIN1-P805L | M, 5 years-old | Severe | No | Difficulties falling asleep, multiple awakenings. (melatonin-tryptophane treatment) | No | No. Hand stereotypies | Oculomotor dsirturbances (oculogyric crisis starting at 4 m.o., visual tonic upgaze, horizontal nistagmus); dyskinetic movements | Intermitent cephalic control, no sitting, no ambulation | Spastic tetraparesis, axial hypotonia with limbs spasticity, generalized hypotonia | Yes |
| 11 | GRIN1-A814D | F, 11.8 years-old | Severe | Motor restlessness, paroxysmal agitation episodes | Difficulties falling asleep, awakenings; Resistant to neuroleptics, benzodiazepines, antihistaminic and melatonin | No | Yes | Dystonia and choreoathetosis | No ambulation, no cephalic control | Axial hypotonia | N.A. |
| 12 | GRIN1-M818V | M, 13.8 years-old | Moderate | No | Mild difficulties falling asleep, autolimitated | Yes. Difficulties in pragmatics | Yes | Stereotypies | Ambulation with motor clumsiness | Hypotonia | N.A. |
| 13 | GRIN1-G827R | M, 5.8 years-old | Severe | No | Yes | No | No | Akinetic rigid syndrome, stereotypies. Dystonic movements. Oculogyric crisis | Non-acquired | Lower limbs spasticity, axial hypotonia | Yes |
| 14 | GRIN1-E834Q | M, 14 years-old | Severe | Aggresivity, motor hyperkinesia | No | No | Yes | Stereotypies | Autonomous ambulation | Normal tone | No |
| 15 | GRIN1-E834Q | 10 years-old | Severe | Disruptive, hyperkinetic and restlessness behaviour | No | No | No | Hyperkinesia, akathisia. Motor clumsiness, poor motor coordination, without ataxia or specific movement disorder | Autonomous ambulation acquired at 2.8 y.o. | Normal tone; Low axial tone until 1 y.o. | No |
| EPILEPSY | BRAIN IMAGING | G-I DISTRESS | |||||||||
| Clinical case | Variant | ||||||||||
| 1 | GRIN1-R548W | Neonatal myoclonia, tonic seizures and spasms. AED-controled | Asymmetric ventriculomegaly, thin corpus callosum and pulvinar nuclei | Dysphagia | |||||||
| 2 | GRIN1-S617C | No | Normal | No | |||||||
| 3 | GRIN1-I619G620dup | Absence seizures, onset at 18 m.o. | Prominence and rounding of the LVs frontal horns (Evans’ index= 0.35); Mega cisterna magna with prominence of the IV ventricle median aperture | No | |||||||
| 4 | GRIN1-G620R | No | Macrocephaly; Reduced corpus callosum size, prominent ventricles and enlarged subdural space | Yes. Liquid dysphagia, gastroesophageal reflux, constipation, normal digestive endoscopy | |||||||
| 5 | GRIN1-G620R | Oculocephalic crisis (onset: 5 m.o.) with remission. Valproate treatment stopped (4 y.o.) | Increased extra-axial spaces | No | |||||||
| 6 | GRIN1-M641V | Absences, tonic seizures (period: 13 m.o to 13 y.o.) | Cortico-subcortical atrophy, corpus callosum hypoplasia | N.A. | |||||||
| 7 | GRIN1-D732E | Single seizure episode (4 y.o.) | Normal (3 y.o.) | Oropharingeal dysphagia, constipation, episodic laxative treatment | |||||||
| 8 | GRIN1-D732E | Non-febrile crisis (7 y.o.) | Normal | No | |||||||
| 9 | GRIN1-P805S | No | Normal | No | |||||||
| 10 | GRIN1-P805L | Yes, onset at 2 m.o. Tonic seizures. Clonic seizures and rigidity of lower limbs (frequency: 4-5 times/month). AED: Efficient carbamazepine and levetiracetam treatments | Normal | Yes, severe gastroesophageal reflux, gastric ulcers | |||||||
| 11 | GRIN1-A814D | Epileptic encephalopathy (onset: 6 m.o.) with spasms and tonic seizures | Mild brain atrophy | Dysphagia, gastrostomy | |||||||
| 12 | GRIN1-M818V | Focal epilepsy (onset: 2 y.o.) evolving to myoclonic seizures with reflex component (audiogenic). Topiramate efficacy. Absence of seizures since 3 y.o. | Normal | No | |||||||
| 13 | GRIN1-G827R | Tonic seizures with laugh (onset: 3 m.o.) Absnece seizures with elevated gaze. AED efficacy | Increased extracerebral space with prominent sulci and ventricular size; reduced corpus callosum volume and thickness; decreased hippocampal size | No | |||||||
| 14 | GRIN1-E834Q | No | Normal | No | |||||||
| 15 | GRIN1-E834Q | Focal seizures with secondary generalised seizures (onset: 3 m.o.), evolving to night tonico-clonic seizures. Resistance to ketogenic diet, levociteram, clobazam, TPM combined to valproic acid. Seizures controled under valproic acid and LTG | Increased extra-axial space (diffuse brain atrophy) | No | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos-Gómez, A.; Miguez-Cabello, F.; Juliá-Palacios, N.; García-Navas, D.; Soto-Insuga, V.; García-Peñas, J.J.; Fuentes, P.; Ibáñez-Micó, S.; Cuesta, L.; Cancho, R.; et al. Paradigmatic De Novo GRIN1 Variants Recapitulate Pathophysiological Mechanisms Underlying GRIN1-Related Disorder Clinical Spectrum. Int. J. Mol. Sci. 2021, 22, 12656. https://doi.org/10.3390/ijms222312656
Santos-Gómez A, Miguez-Cabello F, Juliá-Palacios N, García-Navas D, Soto-Insuga V, García-Peñas JJ, Fuentes P, Ibáñez-Micó S, Cuesta L, Cancho R, et al. Paradigmatic De Novo GRIN1 Variants Recapitulate Pathophysiological Mechanisms Underlying GRIN1-Related Disorder Clinical Spectrum. International Journal of Molecular Sciences. 2021; 22(23):12656. https://doi.org/10.3390/ijms222312656
Chicago/Turabian StyleSantos-Gómez, Ana, Federico Miguez-Cabello, Natalia Juliá-Palacios, Deyanira García-Navas, Víctor Soto-Insuga, Juan J. García-Peñas, Patricia Fuentes, Salvador Ibáñez-Micó, Laura Cuesta, Ramón Cancho, and et al. 2021. "Paradigmatic De Novo GRIN1 Variants Recapitulate Pathophysiological Mechanisms Underlying GRIN1-Related Disorder Clinical Spectrum" International Journal of Molecular Sciences 22, no. 23: 12656. https://doi.org/10.3390/ijms222312656
APA StyleSantos-Gómez, A., Miguez-Cabello, F., Juliá-Palacios, N., García-Navas, D., Soto-Insuga, V., García-Peñas, J. J., Fuentes, P., Ibáñez-Micó, S., Cuesta, L., Cancho, R., Andreo-Lillo, P., Gutiérrez-Aguilar, G., Alonso-Luengo, O., Málaga, I., Hedrera-Fernández, A., García-Cazorla, À., Soto, D., Olivella, M., & Altafaj, X. (2021). Paradigmatic De Novo GRIN1 Variants Recapitulate Pathophysiological Mechanisms Underlying GRIN1-Related Disorder Clinical Spectrum. International Journal of Molecular Sciences, 22(23), 12656. https://doi.org/10.3390/ijms222312656