
Crosstalk between Sodium–Glucose Cotransporter Inhibitors and Sodium–Hydrogen Exchanger 1 and 3 in Cardiometabolic Diseases

 , , , ,
, , , , 
 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Pathophysiology of Diabetic Cardiomyopathy
2.1. The Role of Fibrogenesis in DCM
2.2. The Role of Endothelial Dysfunction in the Development of DCM
2.3. The Role of Metabolic Disturbances in the Development of DCM
3. Characteristics of NHE & SGLT Membrane Transporters
3.1. NHE Overview
Activity and Regulation of NHE1 and NHE3
3.2. SGLT Receptors Overview
Activity and Regulation of SGLT
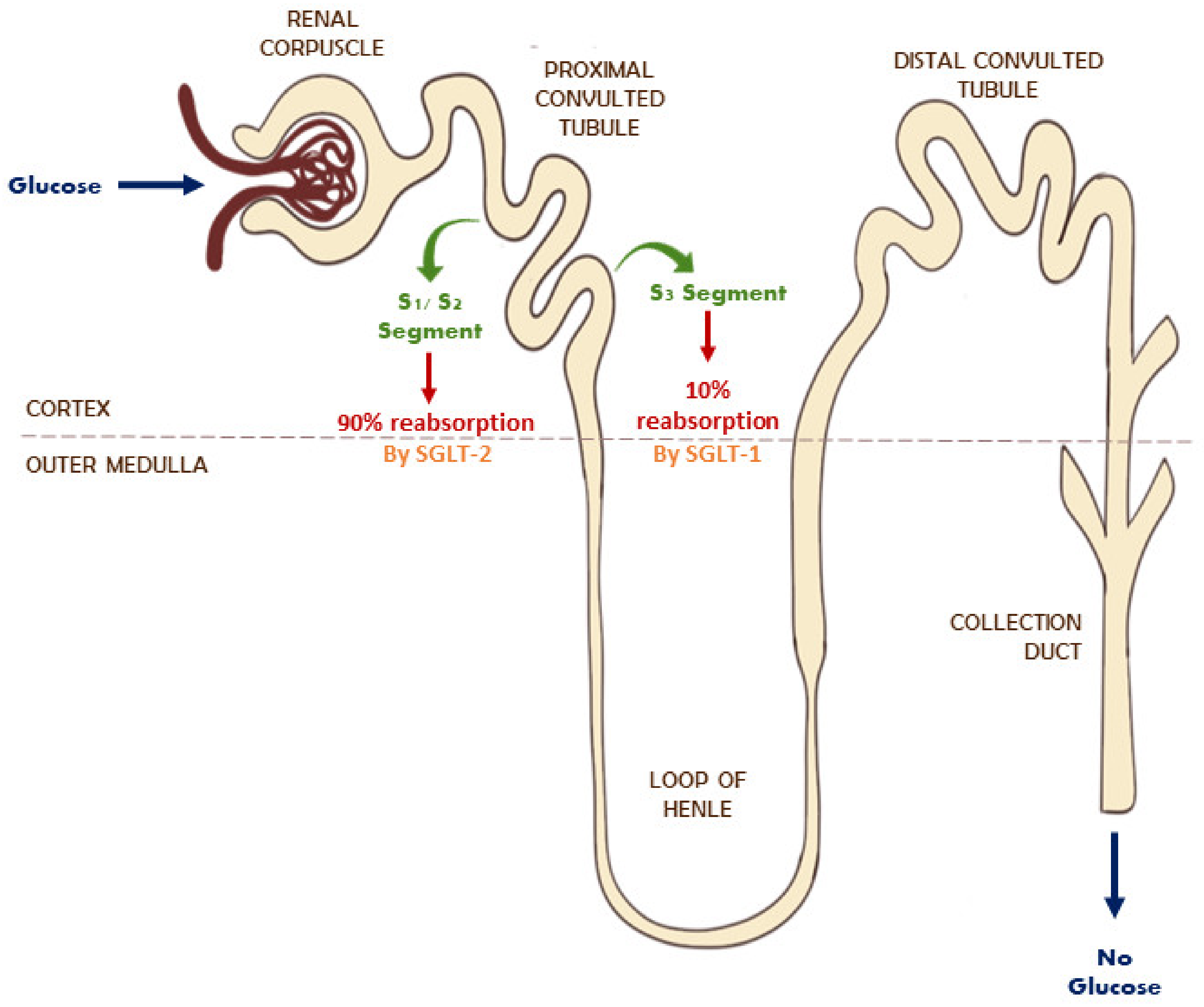
4. Role of SGLT and NHE1 and NHE3 in Diabetes
5. Role of SGLT & NHE1 and NHE3 in Cardiovascular Diseases
5.1. Ischemia-Reperfusion Injury, Cardiac Remodelling, and Hypertrophy
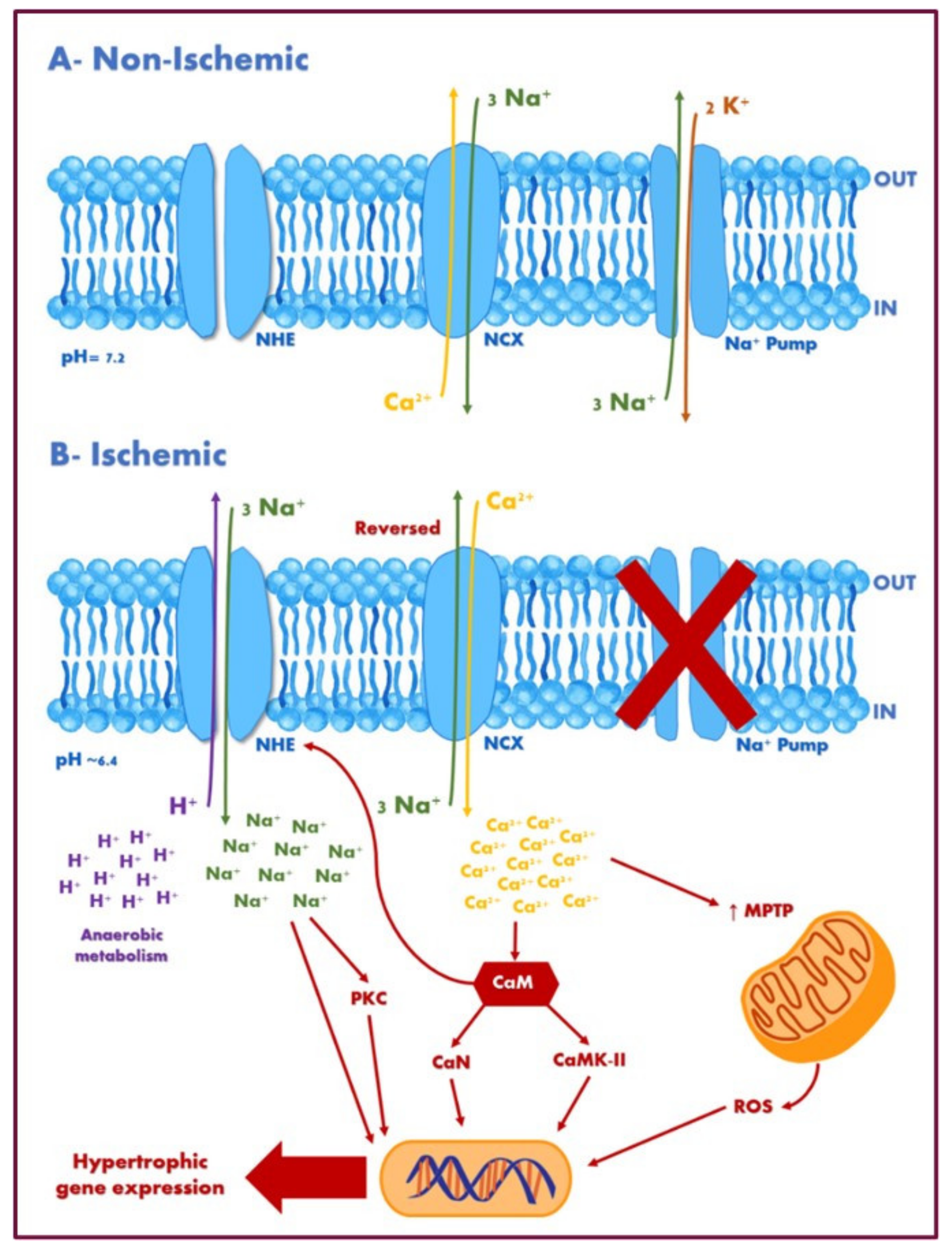
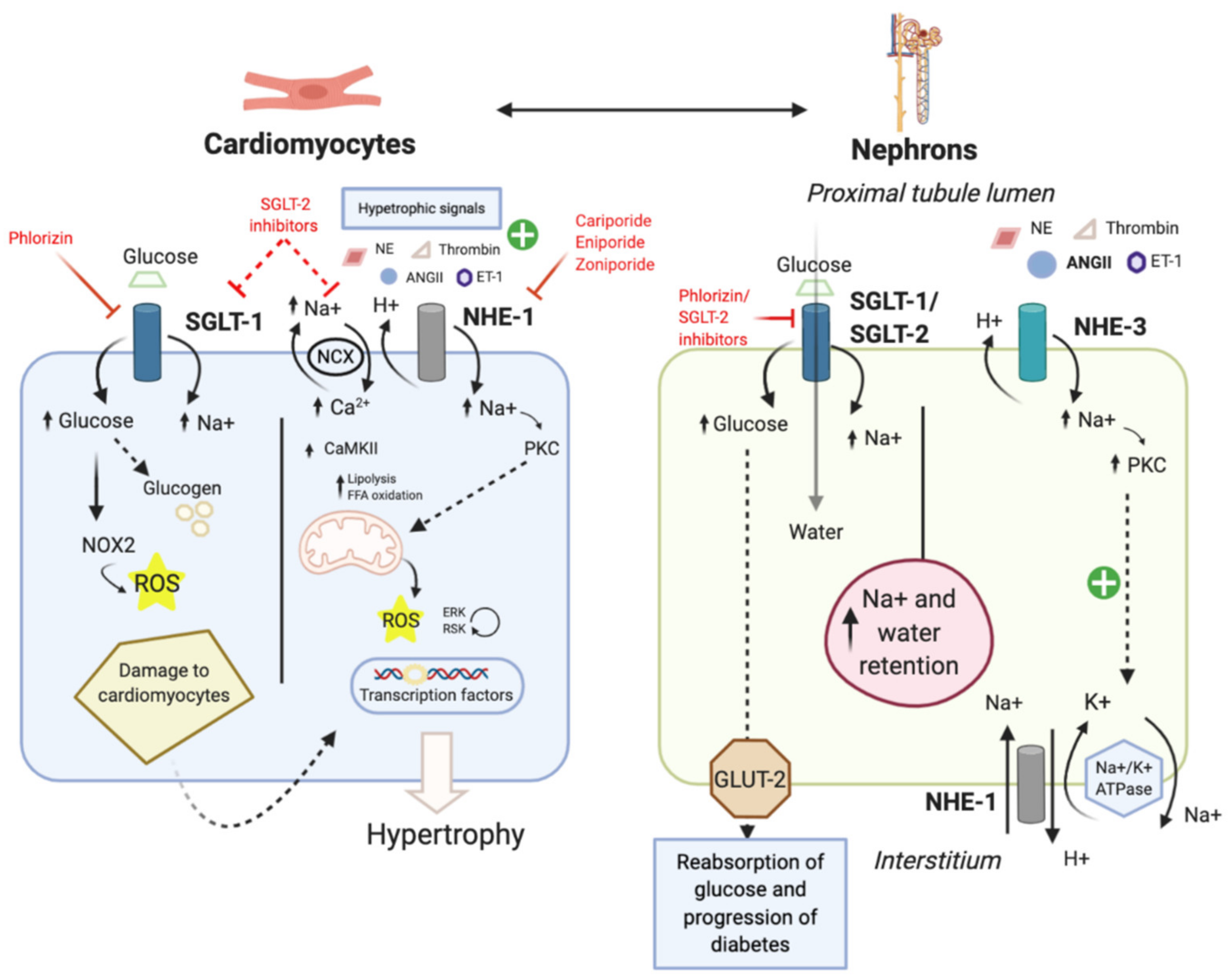
5.2. Diabetic Cardiomyopathy
5.3. Hypertension
6. Available Inhibitors and Their Clinical Outcomes
6.1. Clinical Evaluation of NHE1 Inhibitors
6.2. Clinical Evaluation of SGLT Inhibitors
7. Future Directions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahanovitz, L.; Sluss, P.M.; Russell, S.J. Type 1 diabetes-a clinical perspective. Point Care 2017, 16, 37–40. [Google Scholar] [CrossRef]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes. Diabetes Care 2017, 40, S11–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon, B.M.; Maddox, T.M. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 2015, 6, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Martín-Timón, I.; Sevillano-Collantes, C.; Segura-Galindo, A.; Del Cañizo-Gómez, F.J. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J. Diabetes 2014, 5, 444–470. [Google Scholar] [CrossRef] [PubMed]
- Nichols, G.A.; Gullion, C.M.; Koro, C.E.; Ephross, S.A.; Brown, J.B. The incidence of congestive heart failure in type 2 diabetes: An update. Diabetes Care 2004, 27, 1879–1884. [Google Scholar] [CrossRef] [Green Version]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.W.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Katzmarzyk, P.T.; Horswell, R.; Wang, Y.; Johnson, J.; Hu, G. HbA1c and heart failure risk among diabetic patients. J. Clin. Endocrinol. Metab. 2014, 99, E263–E267. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; DeMarco, V.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Jia, G.; Hill Michael, A.; Sowers James, R. Diabetic cardiomyopathy. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Bain, S.; Hicks, D.; Jones, P.N.; Patel, D.C.; Evans, M.; Fernando, K.; James, J.; Milne, N.; Viljoen, A.; et al. SGLT2 inhibitors: Cardiovascular benefits beyond HbA1c—translating evidence into practice. Diabetes Ther. 2019, 10, 1595–1622. [Google Scholar] [CrossRef] [Green Version]
- Seferović, P.M.; Paulus, W.J. Clinical diabetic cardiomyopathy: A two-faced disease with restrictive and dilated phenotypes. Eur. Heart J. 2015, 36, 1718–1727. [Google Scholar] [CrossRef]
- Aronow, W.S.; Ahn, C. Incidence of heart failure in 2737 older persons with and without diabetes mellitus. Chest 1999, 115, 867–868. [Google Scholar] [CrossRef]
- Herum, K.M.; Lunde, I.G.; McCulloch, A.D.; Christensen, G. The soft- and hard-heartedness of cardiac fibroblasts: Mechanotransduction signaling pathways in fibrosis of the heart. J. Clin. Med. 2017, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and molecular differences between HFpEF and HFrEF: A step ahead in an improved pathological understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac fibrosis. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boer, R.A.; De Keulenaer, G.; Bauersachs, J.; Brutsaert, D.; Cleland, J.G.; Diez, J.; Du, X.J.; Ford, P.; Heinzel, F.R.; Lipson, K.E.; et al. Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the committee of translational research of the heart failure association (HFA) of the European society of cardiology. Eur. J. Heart Fail. 2019, 21, 272–285. [Google Scholar] [CrossRef] [Green Version]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; He, L.-J.; Sun, S.-R. Role of endothelial cells in renal fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; de Kreutzenberg, S.; Fadini, G.P. Endothelial dysfunction in diabetes. Role Reparatory Mech. 2011, 34, S285–S290. [Google Scholar]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vanhoutte, P.M. Reactive oxygen-derived free radicals are key to the endothelial dysfunction of diabetes. J. Diabetes 2009, 1, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.K.; Warnholtz, A.; et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, E14–E22. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Verbeuren, T.J.; Van de Voorde, J.; Lameire, N.H.; Vanhoutte, P.M. Endothelial dysfunction in diabetes. Br. J. Pharmacol. 2000, 130, 963–974. [Google Scholar] [CrossRef] [Green Version]
- Avogaro, A.; Fadini, G.P.; Gallo, A.; Pagnin, E.; De Kreutzenberg, S. Endothelial dysfunction in type 2 diabetes mellitus. Nutr. Metab. Cardiovasc. Dis. 2006, 16, S39–S45. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic reticulum stress: A Critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef] [Green Version]
- Aviello, G.; Knaus, U.G. NADPH oxidases and ROS signaling in the gastrointestinal tract. Mucosal Immunol. 2018, 11, 1011–1023. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, L.; Nyström, T. Antidiabetic agents and endothelial dysfunction–beyond glucose control. Basic Clin. Pharmacol. Toxicol. 2015, 117, 15–25. [Google Scholar] [CrossRef]
- Batzias, K.; Antonopoulos, A.; Oikonomou, E.; Siasos, G.; Bletsa, E.; Stampouloglou, P.K.; Mistakidi, C.-V.; Noutsou, M.; Katsiki, N.; Karopoulos, P.; et al. Effects of newer antidiabetic drugs on endothelial function and arterial stiffness: A systematic review and meta-analysis. J. Diabetes Res. 2018, 2018, 1232583. [Google Scholar] [CrossRef] [PubMed]
- Khemais-Benkhiat, S.; Belcastro, E.; Idris-Khodja, N.; Park, S.-H.; Amoura, L.; Abbas, M.; Auger, C.; Kessler, L.; Mayoux, E.; Toti, F.; et al. Angiotensin II-induced redox-sensitive SGLT1 and 2 expression promotes high glucose-induced endothelial cell senescence. J. Cell. Mol. Med. 2019, 24, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Ganbaatar, B.; Fukuda, D.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; Sata, M. P3111 Empagliflozin, a SGLT2 inhibitor, attenuates endothelial dysfunction and atherogenesis by inhibiting inflammatory responses in the vasculature and adipose tissue in diabetic apolipoprotein E-deficient. Eur. Heart J. 2019, 40, ehz745.0186. [Google Scholar] [CrossRef]
- Tanaka, A.; Shimabukuro, M.; Machii, N.; Teragawa, H.; Okada, Y.; Shima, K.R.; Takamura, T.; Taguchi, I.; Hisauchi, I.; Toyoda, S.; et al. Effect of empagliflozin on endothelial function in patients with type 2 diabetes and cardiovascular disease: Results from the multicenter, randomized, placebo-controlled, double-blind EMBLEM trial. Diabetes Care 2019, 42, e159–e161. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.M.; Battson, M.L.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Weir, T.L.; Gentile, C.L. SGLT2 inhibition via dapagliflozin improves generalized vascular dysfunction and alters the gut microbiota in type 2 diabetic mice. Cardiovasc. Diabetol. 2018, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Gaspari, T.; Spizzo, I.; Liu, H.; Hu, Y.; Simpson, R.W.; Widdop, R.; Dear, A.E. Dapagliflozin attenuates human vascular endothelial cell activation and induces vasorelaxation: A potential mechanism for inhibition of atherogenesis. Diabetes Vasc. Dis. Res. 2017, 15, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Stanley, W.C.; Chandler, M.P. Energy metabolism in the normal and failing heart: Potential for therapeutic interventions. Hear. Fail. Rev. 2002, 7, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Antrás, J.; Picatoste, B.; Ramírez, E.; Egido, J.; Tuñón, J.; Lorenzo, Ó. Targeting metabolic disturbance in the diabetic heart. Cardiovasc. Diabetol. 2015, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.; Young, L.H. AMPK: Energy sensor and survival mechanism in the ischemic heart. Trends Endocrinol. Metab. 2015, 26, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Packer, M. Activation and inhibition of sodium-hydrogen exchanger is a mechanism that links the pathophysiology and treatment of diabetes mellitus with that of heart failure. Circulation 2017, 136, 1548–1559. [Google Scholar] [CrossRef]
- Parker, M.D.; Myers, E.J.; Schelling, J.R. Na+–H+ exchanger-1 (NHE1) regulation in kidney proximal tubule. Cell. Mol. Life Sci. 2015, 72, 2061–2074. [Google Scholar] [CrossRef] [Green Version]
- Das, A.K.; Das, J.; Chandrasekar, S. Specific heart muscle disease in diabetes mellitus—a functional structural correlation. Int. J. Cardiol. 1987, 17, 299–302. [Google Scholar] [CrossRef]
- Dominguez Rieg, J.A.; de la Mora Chavez, S.; Rieg, T. Novel developments in differentiating the role of renal and intestinal sodium hydrogen exchanger 3. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R1186–R1191. [Google Scholar] [CrossRef]
- Vallés, P.G.; Bocanegra, V.; Gil Lorenzo, A.; Costantino, V.V. Physiological functions and regulation of the Na+/H+ Exchanger [NHE1] in renal tubule epithelial cells. Kidney Blood Press. Res. 2015, 40, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Putney, L.K.; Denker, S.P.; Barber, D.L. The changing face of the Na+/H+ exchanger, NHE1: Structure, regulation, and cellular actions. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 527–552. [Google Scholar] [CrossRef]
- Lara, R.; Seckl, M.J.; Pardo, O. The p90 RSK family members: Common functions and isoform specificity. Cancer Res. 2013, 73, 5301–5308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, E.; Abe, J.; Gallis, B.; Aebersold, R.; Spring, D.J.; Krebs, E.G.; Berk, B.C. p90RSK is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J. Biol. Chem. 1999, 274, 20206–20214. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Harding, P.; LaPointe, M.C. PKA, Rap1, ERK1/2, and p90RSK mediate PGE2 and EP4 signaling in neonatal ventricular myocytes. Am. J. Physiol.-Heart Circ. Physiol. 2010, 298, H136–H143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeishi, Y.; Abe, J.-I.; Lee, J.-D.; Kawakatsu, H.; Walsh, R.A.; Berk, B.C. Differential regulation of p90 ribosomal S6 kinase and big mitogen-activated protein kinase 1 by ischemia/reperfusion and oxidative stress in perfused guinea pig hearts. Circ. Res. 1999, 85, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Takeishi, Y.; Huang, Q.; Abe, J.-I.; Che, W.; Lee, J.-D.; Kawakatsu, H.; Hoit, B.D.; Berk, B.; Walsh, R.A. Activation of mitogen-activated protein kinases and p90 ribosomal S6 kinase in failing human hearts with dilated cardiomyopathy. Cardiovasc. Res. 2002, 53, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N.; Chakraborty, A.; Pasek, D.A.; Molkentin, J.D.; Meissner, G. Dysfunctional ryanodine receptor and cardiac hypertrophy: Role of signaling molecules. Am. J. Physiol. Circ. Physiol. 2011, 300, H2187–H2195. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Rajapurohitam, V.; Kilić, A.; Zeidan, A.; Choi, A.; Karmazyn, M. Anti-hypertrophic effect of NHE-1 inhibition involves GSK-3beta-dependent attenuation of mitochondrial dysfunction. J. Mol. Cell Cardiol. 2009, 46, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; Moe, O.; Yamaji, Y.; Cano, A.; Miller, R.T.; Alpern, R.J. Role of protein kinase C and transcription factor AP-1 in the acid-induced increase in Na/H antiporter activity. Proc. Natl. Acad. Sci. USA 1992, 89, 5236–5240. [Google Scholar] [CrossRef] [Green Version]
- Muthusamy, S.; Cheng, M.; Jeong, J.-J.; Kumar, A.; Dudeja, P.K.; Malakooti, J. Extracellular acidosis stimulates NHE2 expression through activation of transcription factor Egr-1 in the intestinal epithelial cells. PLoS ONE 2013, 8, e82023. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K. Akt signaling and growth of the heart. Circulaton 2006, 113, 2032–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiojima, I.; Yefremashvili, M.; Luo, Z.; Kureishi, Y.; Takahashi, A.; Tao, J.; Rosenzweig, A.; Kahn, C.R.; Abel, E.D.; Walsh, K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J. Biol. Chem. 2002, 277, 37670–37677. [Google Scholar] [CrossRef] [Green Version]
- Kemi, O.J.; Ceci, M.; Wisløff, U.; Grimaldi, S.; Gallo, P.; Smith, G.L.; Condorelli, G.; Ellingsen, O. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J. Cell. Physiol. 2008, 214, 316–321. [Google Scholar] [CrossRef]
- Clement, D.L.; Mally, S.; Stock, C.; Lethan, M.; Satir, P.; Schwab, A.; Pedersen, S.F.; Christensen, S.T. PDGFRα signaling in the primary cilium regulates NHE1-dependent fibroblast migration via coordinated differential activity of MEK1/2-ERK1/2-p90RSK and AKT signaling pathways. J. Cell Sci. 2013, 126, 953–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotte, A.; Pasham, V.; Eichenmüller, M.; Bhandaru, M.; Föller, M.; Lang, F. Upregulation of Na+/H+ exchanger by the AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 2010, 398, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Singer, W.D.; Cano, A.; Miller, R.T. G alpha q and G alpha 13 regulate NHE-1 and intracellular calcium in epithelial cells. Am. J. Physiol. Physiol. 1995, 268, C101–C110. [Google Scholar] [CrossRef]
- Avkiran, M.; Haworth, R.S. Regulatory effects of G protein-coupled receptors on cardiac sarcolemmal Na+/H+ exchanger activity: Signalling and significance. Cardiovasc. Res. 2003, 57, 942–952. [Google Scholar] [CrossRef] [Green Version]
- Lambert, R.; Srodulski, S.; Peng, X.; Margulies, K.B.; Despa, F.; Despa, S. Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+–glucose cotransport. J. Am. Heart Assoc. 2015, 4, e002183. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, S.F.; Counillon, L. The SLC9A-C mammalian Na+/H+ Exchanger family: Molecules, mechanisms, and physiology. Physiol. Rev. 2019, 99, 2015–2113. [Google Scholar] [CrossRef]
- Dynia, D.W.; Steinmetz, A.G.; Kocinsky, H.S. NHE3 function and phosphorylation are regulated by a calyculin A-sensitive phosphatase. Am. J. Physiol. Physiol. 2010, 298, F745–F753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- No, Y.R.; He, P.; Yoo, B.; Yun, C.C. Regulation of NHE3 by lysophosphatidic acid is mediated by phosphorylation of NHE3 by RSK2. Am. J. Physiol. Physiol. 2015, 309, C14–C21. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.M.; Loo, D.D.F.; Hirayama, B.A. Biology of Human Sodium Glucose Transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwagi, Y.; Nagoshi, T.; Yoshino, T.; Tanaka, T.D.; Ito, K.; Harada, T.; Takahashi, H.; Ikegami, M.; Anzawa, R.; Yoshimura, M. Expression of SGLT1 in human hearts and impairment of cardiac glucose uptake by phlorizin during ischemia-reperfusion injury in mice. PLoS ONE 2015, 10, e0130605. [Google Scholar] [CrossRef]
- Di Franco, A.; Cantini, G.; Tani, A.; Coppini, R.; Zecchi-Orlandini, S.; Raimondi, L.; Luconi, M.; Mannucci, E. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: A new potential pharmacological target. Int. J. Cardiol. 2017, 243, 86–90. [Google Scholar] [CrossRef]
- Poulsen, S.B.; Fenton, R.A.; Rieg, T. Sodium-glucose cotransport. Curr. Opin. Nephrol. Hypertens. 2015, 24, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Chao, E.C. SGLT-2 inhibitors: A new mechanism for glycemic control. Clin. Diabetes 2014, 32, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Novikov, A.; Vallon, V. Sodium glucose cotransporter 2 inhibition in the diabetic kidney: An update. Curr. Opin. Nephrol. Hypertens. 2016, 25, 50–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannan, K.M.; Little, P.J. Mechanisms regulating the vascular smooth muscle Na/H exchanger (NHE-1) in diabetes. Biochem. Cell Biol. 1998, 76, 751–759. [Google Scholar] [CrossRef]
- Girardi, A.C.C.; Sole, F.D. Deciphering the mechanisms of the Na+/H+ exchanger-3 regulation in organ dysfunction. Am. J. Physiol.-Cell Physiol. 2012, 302, C1569–C1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Knepper, M.A.; Verbalis, J.G.; Ecelbarger, C.A. Increased renal ENaC subunit and sodium transporter abundances in streptozotocin-induced type 1 diabetes. Am. J. Physiol.-Ren. Physiol. 2003, 285, F1125–F1137. [Google Scholar] [CrossRef] [Green Version]
- Klisic, J.; Nief, V.; Reyes, L.; Ambühl, P.M. Acute and chronic regulation of the renal Na+/H+ exchanger NHE3 in rats with STZ-induced diabetes mellitus. Nephron Physiol. 2006, 102, p27–p35. [Google Scholar] [CrossRef] [PubMed]
- Rasch, R. Tubular lesions in streptozotocin-diabetic rats. Diabetologia 1984, 27, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Stevens, V.A.; Saad, S.; Poronnik, P.; Fenton-Lee, C.A.; Polhill, T.S.; Pollock, C.A. The role of SGK-1 in angiotensin II-mediated sodium reabsorption in human proximal tubular cells. Nephrol. Dial. Transplant. 2008, 23, 1834–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, T.F.; Boini, K.M.; Völkl, H.; Bhandaru, M.; Bareiss, P.M.; Just, L.; Vallon, V.; Amann, K.; Kuhl, D.; Feng, Y.; et al. SGK1-sensitive renal tubular glucose reabsorption in diabetes. Am. J. Physiol. Physiol. 2009, 296, F859–F866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuganezawa, H.; Preisig, P.A.; Alpern, R.J. Dominant negative c-Src inhibits angiotensin II induced activation of NHE3 in OKP cells. Kidney Int. 1998, 54, 394–398. [Google Scholar] [CrossRef] [Green Version]
- Du Cheyron, D.; Chalumeau, C.; Defontaine, N.; Klein, C.; Kellermann, O.; Paillard, M.; Poggioli, J. Angiotensin II stimulates NHE3 activity by exocytic insertion of the transporter: Role of PI 3-kinase. Kidney Int. 2003, 64, 939–949. [Google Scholar] [CrossRef] [Green Version]
- He, P.; Klein, J.; Yun, C.C. Activation of Na+/H+ exchanger NHE3 by angiotensin II is mediated by inositol 1,4,5-triphosphate (IP3) receptor-binding protein released with IP3 (IRBIT) and Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 2010, 285, 27869–27878. [Google Scholar] [CrossRef] [Green Version]
- Tojo, A.; Onozato, M.L.; Ha, H.; Kurihara, H.; Sakai, T.; Goto, A.; Fujita, T.; Endou, H. Reduced albumin reabsorption in the proximal tubule of early-stage diabetic rats. Histochem. Cell Biol. 2001, 116, 269–276. [Google Scholar] [CrossRef]
- Drumm, K.; Lee, E.; Stanners, S.; Gassner, B.; Gekle, M.; Poronnik, P.; Pollock, C. Albumin and glucose effects on cell growth parameters, albumin uptake and Na+/H+-exchanger isoform 3 in OK cells. Cell. Physiol. Biochem. 2003, 13, 199–206. [Google Scholar] [CrossRef]
- Uthman, L.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.T.; Kuschma, M.C.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Direct cardiac actions of sodium glucose cotransporter 2 inhibitors target pathogenic mechanisms underlying heart failure in diabetic patients. Front. Physiol. 2018, 9, 1575. [Google Scholar] [CrossRef]
- Wakabayashi, S.; Hisamitsu, T.; Nakamura, T.Y. Regulation of the cardiac Na+/H+ exchanger in health and disease. J. Mol. Cell. Cardiol. 2013, 61, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Odunewu-Aderibigbe, A.; Fliegel, L. The Na+/H+ exchanger and pH regulation in the heart. IUBMB Life 2014, 66, 679–685. [Google Scholar] [CrossRef]
- Karmazyn, M.; Kilić, A.; Javadov, S. The role of NHE-1 in myocardial hypertrophy and remodelling. J. Mol. Cell. Cardiol. 2008, 44, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Cingolani Horacio, E.; Ennis Irene, L. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 2007, 115, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Imahashi, K.; Mraiche, F.; Steenbergen, C.; Murphy, E.; Fliegel, L. Overexpression of the Na+/H+ exchanger and ischemia-reperfusion injury in the myocardium. Am. J. Physiol. Circ. Physiol. 2007, 292, H2237–H2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Meyer, J.W.; Ashraf, M.; Shull, G.E. Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ. Res. 2003, 93, 776–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ropero, Á.; Vargas-Delgado, A.P.; Santos-Gallego, C.G.; Badimon, J.J. Inhibition of sodium glucose cotransporters improves cardiac performance. Int. J. Mol. Sci. 2019, 20, 3289. [Google Scholar] [CrossRef] [Green Version]
- Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Lee, S.J.; Wang, D.; Huang, X.Y.N.; Ahmad, F. Transgenic knockdown of cardiac sodium/glucose cotransporter 1 (SGLT1) attenuates PRKAG2 cardiomyopathy, whereas transgenic overexpression of cardiac SGLT1 causes pathologic hypertrophy and dysfunction in mice. J. Am. Heart Assoc. 2014, 3, e000899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Agrawal, V.; Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Sincoular, A.; Jakubiak, M.; Music, M.L.; Kutschke, W.J.; Huang, X.N.; et al. Cardiac sodium-dependent glucose cotransporter 1 is a novel mediator of ischaemia/reperfusion injury. Cardiovasc. Res. 2019, 115, 1646–1658. [Google Scholar] [CrossRef] [PubMed]
- Balteau, M.; Tajeddine, N.; de Meester, C.; Ginion, A.; Rosiers, C.D.; Brady, N.R.; Sommereyns, C.; Horman, S.; Vanoverschelde, J.-L.; Gailly, P.; et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc. Res. 2011, 92, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Lee, T.W.; Park, G.T.; Kim, J.H.; Lee, H.-C.; Han, J.-H.; Yoon, A.; Yoon, D.; Kim, S.; Jung, S.M.; et al. Sodium/glucose Co-transporter 2 inhibitor, empagliflozin, alleviated transient expression of SGLT2 after myocardial infarction. Korean Circ. J. 2021, 51, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Jia, X.; Bajaj, M.; Birnbaum, Y. Dapagliflozin Attenuates Na+/H+ exchanger-1 in cardiofibroblasts via AMPK activation. Cardiovasc. Drugs Ther. 2018, 32, 553–558. [Google Scholar] [CrossRef]
- Karmazyn, M. Role of NHE-1 in cardiac hypertrophy and heart failure. In The Sodium-Hydrogen Exchanger: From Molecule to its Role in Disease; Karmazyn, M., Avkiran, M., Fliegel, L., Eds.; Springer: Boston, MA, USA, 2003; pp. 211–219. [Google Scholar]
- Mraiche, F.; Oka, T.; Gan, X.T.; Karmazyn, M.; Fliegel, L. Activated NHE1 is required to induce early cardiac hypertrophy in mice. Basic Res. Cardiol. 2011, 106, 603–616. [Google Scholar] [CrossRef]
- Allen, D.G.; Xiao, X.-H. Role of the cardiac Na+/H+ exchanger during ischemia and reperfusion. Cardiovasc. Res. 2003, 57, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Doliba, N.M.; Babsky, A.M.; Osbakken, M.D. The role of sodium in diabetic cardiomyopathy. Front. Physiol. 2018, 9, 1473. [Google Scholar] [CrossRef]
- Silva dos Santos, D.; Polidoro, J.Z.; Borges-Júnior, F.A.; Girardi, A.C.C. Cardioprotection conferred by sodium-glucose cotransporter 2 inhibitors: A renal proximal tubule perspective. Am. J. Physiol.-Cell Physiol. 2019, 318, C328–C336. [Google Scholar] [CrossRef]
- Zhou, Q.; Kesteven, S.; Wu, J.; Aidery, P.; Gawaz, M.; Gramlich, M.; Feneley, M.P.; Harvey, R. Pressure overload by transverse aortic constriction induces maladaptive hypertrophy in a titin-truncated mouse model. BioMed Res. Int. 2015, 2015, 163564. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 276–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, A.; Nagoshi, T.; Kashiwagi, Y.; Kimura, H.; Tanaka, Y.; Oi, Y.; Ito, K.; Yoshino, T.; Tanaka, T.D.; Yoshimura, M. Cardiac ischemia–reperfusion injury under insulin-resistant conditions: SGLT1 but not SGLT2 plays a compensatory protective role in diet-induced obesity. Cardiovasc. Diabetol. 2019, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Sharma, K. Sodium–glucose transport: Role in diabetes mellitus and potential clinical implications. Curr. Opin. Nephrol. Hypertens. 2010, 19, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Li, X.C.; Zheng, X.; Chen, X.; Zhao, C.; Zhu, D.; Zhang, J.; Zhuo, J.L. Genetic and genomic evidence for an important role of the Na+/H+ exchanger 3 in blood pressure regulation and angiotensin II-induced hypertension. Physiol. Genom. 2019, 51, 97–108. [Google Scholar] [CrossRef]
- He, P.; Zhao, L.; No, Y.R.; Karvar, S.; Yun, C.C. The NHERF1 PDZ1 domain and IRBIT interact and mediate the activation of Na+/H+ exchanger 3 by ANG II. Am. J. Physiol. Physiol. 2016, 311, F343–F351. [Google Scholar] [CrossRef] [Green Version]
- Pao, A.C.; Bhargava, A.; Di Sole, F.; Quigley, R.; Shao, X.; Wang, J.; Thomas, S.; Zhang, J.; Shi, M.; Funder, J.W.; et al. Expression and role of serum and glucocorticoid-regulated kinase 2 in the regulation of Na+/H+ exchanger 3 in the mammalian kidney. Am. J. Physiol. Physiol. 2010, 299, F1496–F1506. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Zhang, H.; Lang, F.; Yun, C.C. Acute activation of NHE3 by dexamethasone correlates with activation of SGK1 and requires a functional glucocorticoid receptor. Am. J. Physiol. Physiol. 2007, 292, C396–C404. [Google Scholar] [CrossRef] [Green Version]
- Fuster, D.G.; Bobulescu, I.A.; Zhang, J.; Wade, J.; Moe, O.W. Characterization of the regulation of renal Na+/H+ exchanger NHE3 by insulin. Am. J. Physiol. Physiol. 2007, 292, F577–F585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.C.; Shull, G.E.; Miguel-Qin, E.; Chen, F.; Zhuo, J.L. Role of the Na+ /H+ exchanger 3 in angiotensin II-induced hypertension in NHE3-deficient mice with transgenic rescue of NHE3 in small intestines. Physiol. Rep. 2015, 3, e12605. [Google Scholar] [CrossRef] [PubMed]
- Lima, N.K.; Abbasi, F.; Lamendola, C.; Reaven, G.M. Prevalence of insulin resistance and related risk factors for cardiovascular disease in patients with essential hypertension. Am. J. Hypertens. 2009, 22, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Linz, B.; Hohl, M.; Reil, J.C.; Böhm, M.; Linz, D. Inhibition of NHE3-mediated sodium absorption in the gut reduced cardiac end-organ damage without deteriorating renal function in obese spontaneously hypertensive rats. J. Cardiovasc. Pharmacol. 2016, 67, 225–231. [Google Scholar] [CrossRef]
- Linz, D.; Wirth, K.; Linz, W.; Heuer, H.O.O.; Frick, W.; Hofmeister, A.; Heinelt, U.; Arndt, P.; Schwahn, U.; Böhm, M.; et al. Antihypertensive and laxative effects by pharmacological inhibition of sodium-proton-exchanger subtype 3–mediated sodium absorption in the gut. Hypertension 2012, 60, 1560–1567. [Google Scholar] [CrossRef]
- Kuro-O, M.; Hanaoka, K.; Hiroi, Y.; Noguchi, T.; Fujimori, Y.; Takewaki, S.-I.; Hayasaka, M.; Katoh, H.; Miyagishi, A.; Nagai, R.; et al. Salt-sensitive hypertension in transgenic mice overexpressing Na+-proton exchanger. Circ. Res. 1995, 76, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.N.; Adragna, N.C.; Adarichev, V.A.; Hamet, P. Genetic and biochemical determinants of abnormal monovalent ion transport in primary hypertension. Am. J. Physiol. Content 1999, 276, C511–C536. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Damkier, H.H.; Aalkjaer, C. NHE1 knockout reduces blood pressure and arterial media/lumen ratio with no effect on resting pH(i) in the vascular wall. J. Physiol. 2012, 590, 1895–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobulescu, I.A.; Di Sole, F.; Moe, O.W. Na+/H+ exchangers: Physiology and link to hypertension and organ ischemia. Curr. Opin. Nephrol. Hypertens. 2005, 14, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef] [Green Version]
- Cianciolo, G.; De De Pascalis, A.; Gasperoni, L.; Tondolo, F.; Zappulo, F.; Capelli, I.; Cappuccilli, M.; La La Manna, G. The off-target effects, electrolyte and mineral disorders of SGLT2i. Molecules 2020, 25, 2757. [Google Scholar] [CrossRef]
- Bautista, R.; Manning, R.; Martínez, F.; Avila-Casado, M.D.C.; Soto, V.; Medina, A.; Escalante, B. Angiotensin II-dependent increased expression of Na+-glucose cotransporter in hypertension. Am. J. Physiol. Physiol. 2004, 286, F127–F133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Pardo, H.; Bautista, R.; Vargas-Robles, H.; Rios, A.; Sanchez, D.; Escalante, B. Role of sodium/glucose cotransporter inhibition on a rat model of angiotensin II–dependent kidney damage. BMC Nephrol. 2019, 20, 292. [Google Scholar] [CrossRef]
- Sanidas, E.A.; Papadopoulos, D.P.; Hatziagelaki, E.; Grassos, C.; Velliou, M.; Barbetseas, J. Sodium-glucose cotransporter 2 (SGLT2) inhibitors across the spectrum of hypertension. Am. J. Hypertens. 2019, 33, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Trimarco, B.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Baker, W.L.; Smyth, L.R.; Riche, D.M.; Bourret, E.M.; Chamberlin, K.W.; White, W.B. Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: A systematic review and meta-analysis. J. Am. Soc. Hypertens. 2014, 8, 262–275.e9. [Google Scholar] [CrossRef]
- Filippatos, T.; Tsimihodimos, V.; Elisaf, M.S. Mechanisms of blood pressure reduction with sodium-glucose co-transporter 2 (SGLT2) inhibitors. Expert Opin. Pharmacother. 2016, 17, 1581–1583. [Google Scholar] [CrossRef]
- Gorboulev, V.; Schürmann, A.; Vallon, V.; Kipp, H.; Jaschke, A.; Klessen, D.; Friedrich, A.; Scherneck, S.; Rieg, T.; Cunard, R.; et al. Na+-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 2011, 61, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Tavakkolizadeh, A.; Berger, U.V.; Shen, K.R.; Levitsky, L.L.; Zinner, M.J.; Hediger, M.A.; Ashley, S.W.; Whang, E.E.; Rhoads, D.B. Diurnal rhythmicity in intestinal SGLT-1 function, V max, and mRNA expression topography. Am. J. Physiol. Liver Physiol. 2001, 280, G209–G215. [Google Scholar] [CrossRef] [Green Version]
- Mate, A.; Barfull, A.; Hermosa, Á.M.; Gómez-Amores, L.; Vázquez, C.M.; Planas, J.M. Regulation of sodium-glucose cotransporter SGLT1 in the intestine of hypertensive rats. Am. J. Physiol. Integr. Comp. Physiol. 2006, 291, R760–R767. [Google Scholar] [CrossRef]
- Zeymer, U.; Suryapranata, H.; Monassier, J.P.; Opolski, G.; Davies, J.; Rasmanis, G.; Linssen, G.; Tebbe, U.; Schröder, R.; Tiemann, R.; et al. The Na+/H+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J. Am. Coll. Cardiol. 2001, 38, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- Klein, H.H.; Pich, S.; Bohle, R.M.; Lindert-Heimberg, S.; Nebendahl, K. Na+ /H+ exchange inhibitor cariporide attenuates cell injury predominantly during ischemia and not at onset of reperfusion in porcine hearts with low residual blood flow. Circulation 2000, 102, 1977–1982. [Google Scholar] [CrossRef] [Green Version]
- Rupprecht, H.-J.; Dahl, J.V.; Terres, W.; Seyfarth, K.M.; Richardt, G.; Schultheiß, H.-P.; Buerke, M.; Sheehan, F.H.; Drexler, H. Cardioprotective effects of the Na+/H+ exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct ptca. Circulation 2000, 101, 2902–2908. [Google Scholar] [CrossRef] [Green Version]
- Chaitman, B.R. A review of the GUARDIAN trial results: Clinical Implications and the Significance of Elevated perioperative CK-MB on 6-month survival. J. Card. Surg. 2003, 18, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Mentzer, R.M.; Bartels, C.; Bolli, R.; Boyce, S.; Buckberg, G.D.; Chaitman, B.; Haverich, A.; Knight, J.; Menasché, P.; Myers, M.L.; et al. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: Results of the expedition study. Ann. Thorac. Surg. 2008, 85, 1261–1270. [Google Scholar] [CrossRef]
- Raskin, P. Sodium-glucose cotransporter inhibition: Therapeutic potential for the treatment of type 2 diabetes mellitus. Diabetes/Metab. Res. Rev. 2013, 29, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Dominguez Rieg, J.A.; Rieg, T. What does sodium-glucose co-transporter 1 inhibition add: Prospects for dual inhibition. Diabetes Obes. Metab. 2019, 21, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chasis, H.; Jolliffe, N.; Smith, H.W. THE action of phlorizin on the excretion of glucose, xylose, sucrose, creatinine and urea by maN. J. Clin. Investig. 1933, 12, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, L.; Shulman, G.I.; Zawalich, W.; DeFronzo, R.A. Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J. Clin. Investig. 1987, 80, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, L.; Smith, D.; Shulman, G.; Papachristou, D.; DeFronzo, R.A. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J. Clin. Investig. 1987, 79, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Crespy, V.; Aprikian, O.; Morand, C.; Besson, C.; Manach, C.; Demigné, C.; Rémésy, C. Bioavailability of phloretin and phloridzin in rats. J. Nutr. 2001, 131, 3227–3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oku, A.; Ueta, K.; Arakawa, K.; Ishihara, T.; Nawano, M.; Kuronuma, Y.; Matsumoto, M.; Saito, A.; Tsujihara, K.; Anai, M.; et al. T-1095, an inhibitor of renal Na+-glucose cotransporters, may provide a novel approach to treating diabetes. Diabetes 1999, 48, 1794–1800. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Steg, P.G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; Metra, M.; et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in patients with diabetes and chronic kidney disease. N. Engl. J. Med. 2021, 384, 129–139. [Google Scholar] [CrossRef]
- Rosenstock, J.; Cefalu, W.T.; Lapuerta, P.; Zambrowicz, B.; Ogbaa, I.; Banks, P.; Sands, A. Greater dose-ranging effects on A1C levels than on glucosuria with LX4211, a dual inhibitor of SGLT1 and SGLT2, in patients with type 2 diabetes on metformin monotherapy. Diabetes Care 2014, 38, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Powell, D.; Doree, D.; Jeter-Jones, S.; Ding, Z.-M.; Zambrowicz, B.; Sands, A. Sotagliflozin improves glycemic control in nonobese diabetes-prone mice with type 1 diabetes. Diabetes Metab. Syndr. Obesity Targets Ther. 2015, 8, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Sands, A.T.; Zambrowicz, B.P.; Rosenstock, J.; Lapuerta, P.; Bode, B.W.; Garg, S.K.; Buse, J.B.; Banks, P.; Heptulla, R.; Rendell, M.; et al. Sotagliflozin, a dual SGLT1 and SGLT2 inhibitor, as adjunct therapy to insulin in type 1 diabetes. Diabetes Care 2015, 38, 1181–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, S.K.; Henry, R.R.; Banks, P.; Buse, J.; Davies, M.; Fulcher, G.R.; Pozzilli, P.; Gesty-Palmer, D.; Lapuerta, P.; Simó, R.; et al. Effects of sotagliflozin added to insulin in patients with type 1 diabetes. N. Engl. J. Med. 2017, 377, 2337–2348. [Google Scholar] [CrossRef]
- Polidori, D.; Sha, S.; Mudaliar, S.; Ciaraldi, T.P.; Ghosh, A.; Vaccaro, N.; Farrell, K.; Rothenberg, P.; Henry, R.R. Canagliflozin lowers postprandial glucose and insulin by delaying intestinal glucose absorption in addition to increasing urinary glucose excretion: Results of a randomized, placebo-controlled study. Diabetes Care 2013, 36, 2154–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, T.D.; Campos, L.C.G.; Carraro-Lacroix, L.; Girardi, A.C.C.; Malnic, G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J. Am. Soc. Nephrol. 2014, 25, 2028. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.; Rao, V.S.; Ivey-Miranda, J.; Fleming, J.; Mahoney, D.; Maulion, C.; Suda, N.; Siwakoti, K.; Ahmad, T.; Jacoby, D.; et al. Empagliflozin in heart failure: Diuretic and cardiorenal effects. Circulation 2020, 142, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Schumacher, C.A.; Wust, R.C.; Fiolet, J.W.T.; Stienen, G.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, K.M.; Lau, Y.M.; Dhandhania, V.; Cai, Z.J.; Lee, Y.K.; Lai, W.H.; Tse, H.F.; Siu, C.W. Empagliflozin ammeliorates high glucose induced-cardiac dysfuntion in human iPSC-derived cardiomyocytes. Sci. Rep. 2018, 8, 14872. [Google Scholar] [CrossRef]
- American Diabetes Association. 9. pharmacologic approaches to glycemic treatment: Standards of medical care in diabetes—2021. Diabetes Care 2021, 44, S111–S124. [Google Scholar] [CrossRef]
- Packer, M. SGLT2 inhibitors produce cardiorenal benefits by promoting adaptive cellular reprogramming to induce a state of fasting mimicry: A paradigm shift in understanding their mechanism of action. Diabetes Care 2020, 43, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, K.R.; Brosius, F.C., III; Cavender, M.A.; Fioretto, P.; Fowler, K.J.; Heerspink, H.J.; Manley, T.; McGuire, D.K.; Molitch, M.E.; Mottl, A.K.; et al. SGLT2 inhibition for CKD and cardiovascular disease in type 2 diabetes: Report of a scientific workshop sponsored by the national kidney foundation. Am. J. Kidney Dis. 2021, 77, 94–109. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Shamasi, A.-A.; Elkaffash, R.; Mohamed, M.; Rayan, M.; Al-Khater, D.; Gadeau, A.-P.; Ahmed, R.; Hasan, A.; Eldassouki, H.; Yalcin, H.C.; et al. Crosstalk between Sodium–Glucose Cotransporter Inhibitors and Sodium–Hydrogen Exchanger 1 and 3 in Cardiometabolic Diseases. Int. J. Mol. Sci. 2021, 22, 12677. https://doi.org/10.3390/ijms222312677
Al-Shamasi A-A, Elkaffash R, Mohamed M, Rayan M, Al-Khater D, Gadeau A-P, Ahmed R, Hasan A, Eldassouki H, Yalcin HC, et al. Crosstalk between Sodium–Glucose Cotransporter Inhibitors and Sodium–Hydrogen Exchanger 1 and 3 in Cardiometabolic Diseases. International Journal of Molecular Sciences. 2021; 22(23):12677. https://doi.org/10.3390/ijms222312677
Chicago/Turabian StyleAl-Shamasi, Al-Anood, Rozina Elkaffash, Meram Mohamed, Menatallah Rayan, Dhabya Al-Khater, Alain-Pierre Gadeau, Rashid Ahmed, Anwarul Hasan, Hussein Eldassouki, Huseyin Cagatay Yalcin, and et al. 2021. "Crosstalk between Sodium–Glucose Cotransporter Inhibitors and Sodium–Hydrogen Exchanger 1 and 3 in Cardiometabolic Diseases" International Journal of Molecular Sciences 22, no. 23: 12677. https://doi.org/10.3390/ijms222312677
APA StyleAl-Shamasi, A.-A., Elkaffash, R., Mohamed, M., Rayan, M., Al-Khater, D., Gadeau, A.-P., Ahmed, R., Hasan, A., Eldassouki, H., Yalcin, H. C., Abdul-Ghani, M., & Mraiche, F. (2021). Crosstalk between Sodium–Glucose Cotransporter Inhibitors and Sodium–Hydrogen Exchanger 1 and 3 in Cardiometabolic Diseases. International Journal of Molecular Sciences, 22(23), 12677. https://doi.org/10.3390/ijms222312677