
Steatosis, Steatohepatitis and Cancer Immunotherapy: An Intricate Story



Abstract
:1. Introduction
2. Aim of the Review
3. Methods
4. Liver Biopsy Shows That Intrahepatic Fat Accumulation Is Common in Patients Treated with ICIs
5. NAFLD as a Potential Risk Factor for ICI-Induced Hepatotoxicity
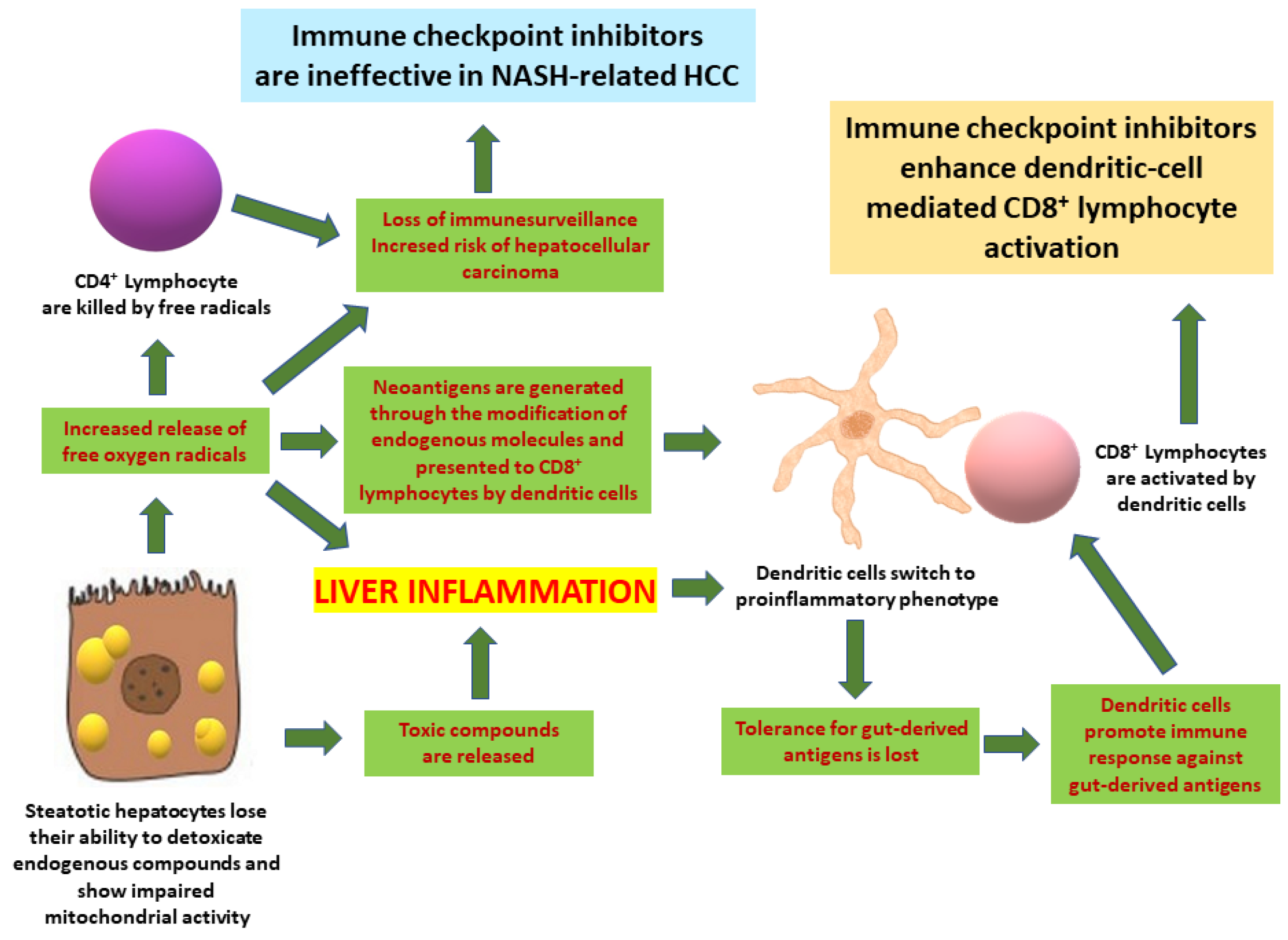
6. Similarities between the Pathogenetic Mechanisms of NASH and Those of ICI-Induced Hepatotoxicity
7. Implications for Hepatocellular Carcinoma
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Geraud, A.; Gougis, P.; Vozy, A.; Anquetil, C.; Allenbach, Y.; Romano, E.; Funck-Brentano, E.; Moslehi, J.J.; Johnson, D.B.; Salem, J.E. Clinical Pharmacology and Interplay of Immune Checkpoint Agents: A Yin-Yang Balance. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 85–112. [Google Scholar] [CrossRef]
- Palmieri, D.J.; Carlino, M.S. Immune Checkpoint Inhibitor Toxicity. Curr. Oncol. Rep. 2018, 20, 72. [Google Scholar] [CrossRef]
- Nguyen, L.S.; Raia, L.; Lebrun-Vignes, B.; Salem, J.E. Graft Versus Host Disease Associated with Immune Checkpoint Inhibitors: A Pharmacovigilance Study and Systematic Literature Review. Front. Pharmacol. 2021, 11, 619649. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.K.; Watson, D.E. Pharmacovigilance assessment of immune-mediated reactions reported for checkpoint inhibitor cancer immunotherapies. Pharmacotherapy 2017, 37, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Spain, L.; Diem, S.; Larkin, J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev. 2016, 44, 51–60. [Google Scholar] [CrossRef]
- Suzman, D.L.; Pelosof, L.; Rosenberg, A.; Avigan, M.I. Hepatotoxicity of immune checkpoint inhibitors: An evolving picture of risk as-sociated with a vital class of immunotherapy agents. Liver Int. 2018, 38, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Li, W.; Hu, J.; Zhu, E.C.; Su, Q. Hepatotoxicity in patients with solid tumors treated with PD-1/PD-L1 inhibitors alone, PD-1/PD-L1 inhibitors plus chemotherapy, or chemotherapy alone: Systematic review and meta-analysis. Eur. J. Clin. Pharmacol. 2020, 76, 1345–1354. [Google Scholar] [CrossRef]
- Wang, P.F.; Chen, Y.; Song, S.Y.; Wang, T.J.; Ji, W.J.; Li, S.W.; Liu, N.; Yan, C.X. Immune-Related Adverse Events Associated with Anti-PD-1/PD-L1 Treatment for Malignancies: A Meta-Analysis. Front. Pharmacol. 2017, 8, 730. [Google Scholar] [CrossRef]
- Peeraphatdit, T.B.; Wang, J.; Odenwald, M.A.; Hu, S.; Hart, J.; Charlton, M.R. Hepatotoxicity from Immune Checkpoint Inhibitors: A Systematic Review and Management Recommendation. Hepatology 2020, 72, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.V.; Dougan, M.; Zubiri, L.; Reynolds, K.L.; Sullivan, R.J.; Misdraji, J. Liver biopsy findings in patients on immune checkpoint inhibitors. Mod. Pathol. 2021, 34, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Jennings, J.J.; Mandaliya, R.; Nakshabandi, A.; Lewis, J.H. Hepatotoxicity induced by immune checkpoint inhibitors: A comprehensive review including current and alternative management strategies. Expert Opin. Drug Metab. Toxicol. 2019, 15, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, S.G.; Moskalenko, M.; Pan, M.; Shah, S.; Sidhu, H.K.; Sicular, S.; Harcharik, S.; Chang, R.; Friedlander, P.; Saenger, Y.M. Elevated rates of transaminitis during ipilimumab therapy for metastatic melanoma. Melanoma. Res. 2013, 23, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.; Srivastava, A.; Misdraji, J. Fibrin Ring Granulomas in Checkpoint Inhibitor-induced Hepatitis. Am. J. Surg. Pathol. 2017, 41, 134–137. [Google Scholar] [CrossRef]
- Ipilimumab. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548335/ (accessed on 10 October 2021).
- Johncilla, M.; Misdraji, J.; Pratt, D.S.; Agoston, A.T.; Lauwers, G.Y.; Srivastava, A.; Doyle, L.A. Ipilimumab-associated Hepatitis: Clinicopathologic Characterization in a Series of 11 Cases. Am. J. Surg. Pathol. 2015, 39, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Ramaiya, N.H.; Krajewski, K.M.; Jagannathan, J.P.; Tirumani, S.H.; Srivastava, A.; Ibrahim, N. Ipilimumab associated hepatitis: Imaging and clinicopathologic findings. Investig. New Drugs 2013, 31, 1071–1077. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Berman, D. Pathologic changes in ipilimumab-related hepatitis in patients with metastatic melanoma. Dig. Dis. Sci. 2012, 57, 2233–2240. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Sugawara, T.; Shinkawa, T.; Kurisu, T.; Kouzen, N.; Tanaka, T.; Fukuta, F.; Yamasaki, K.; Sugita, S.; Matsuo, K.; et al. Fatal fulminant hepatitis induced by combined ipilimumab and nivolumab therapy despite favorable histologic response and confirmed by autopsy in a patient with clear cell renal cell carcinoma. Immunol. Med. 2020, 7, 1–6. [Google Scholar] [CrossRef]
- Ortland, I.; Mirjalili, M.; Kullak-Ublick, G.A.; Peymani, P. Drug-induced liver injury in Switzerland: An analysis of drug-related hepatic disorders in the WHO pharmacovigilance database VigiBaseÔ from 2010 to 2020. Swiss. Med. Wkly. 2021, 151, w20503. [Google Scholar] [CrossRef]
- Tanaka, R.; Fujisawa, Y.; Sae, I.; Maruyama, H.; Ito, S.; Hasegawa, N.; Sekine, I.; Fujimoto, M. Severe hepatitis arising from ipilimumab administration, following melanoma treatment with nivolumab. Jpn. J. Clin. Oncol. 2017, 47, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Yildirim, S.; Deniz, K.; Doğan, E.; Başkol, M.; Gürsoy, Ş.; Özkan, M. Ipilimumab-associated cholestatic hepatitis: A case report and literature review. Melanoma. Res. 2017, 27, 380–382. [Google Scholar] [CrossRef]
- Zen, Y.; Yeh, M.M. Hepatotoxicity of immune checkpoint inhibitors: A histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug-induced liver injury. Mod. Pathol. 2018, 31, 965–973. [Google Scholar] [CrossRef] [Green Version]
- LIBTAYO. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/libtayo-epar-product-information_en.pdf (accessed on 10 October 2021).
- LIBTAYO. Full Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761097s007lbl.pdf (accessed on 10 October 2021).
- Cemiplimab. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548288/ (accessed on 10 October 2021).
- Keytruda. Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125514s096lbl.pdf (accessed on 10 October 2021).
- Keytruda. Summary of Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/keytruda-epar-product-information_en.pdf (accessed on 10 October 2021).
- Pembrolizumab. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548050// (accessed on 10 October 2021).
- Kwan, J.M.; Cheng, R.; Feldman, L.E. Hepatotoxicity and Recurrent NSTEMI While on Pembrolizumab for Metastatic Giant Cell Bone Tumor. Am. J. Med. Sci. 2019, 357, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Cheung, V.; Gupta, T.; Payne, M.; Middleton, M.R.; Collier, J.D.; Simmons, A.; Klenerman, P.; Brain, O.; Cobbold, J.F. Immunotherapy-related hepatitis: Real-world experience from a tertiary centre. Frontline Gastroenterol. 2019, 10, 364–371. [Google Scholar] [CrossRef]
- Kurokawa, K.; Hara, M.; Iwakami, S.I.; Genda, T.; Iwakami, N.; Miyashita, Y.; Fujioka, M.; Sasaki, S.; Takahashi, K. Cholestatic Liver Injury Induced by Pembrolizumab in a Patient with Lung Adenocarcinoma. Intern. Med. 2019, 58, 3283–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koya, Y.; Shibata, M.; Shinohara, N.; Nebuya, S.; Oe, S.; Honma, Y.; Senju, M.; Sato, N.; Harada, M. Secondary sclerosing cholangitis with hemobilia induced by pembrolizumab: Case report and review of published work. Hepatol. Res. 2019, 49, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Onoyama, T.; Takeda, Y.; Yamashita, T.; Hamamoto, W.; Sakamoto, Y.; Koda, H.; Kawata, S.; Matsumoto, K.; Isomoto, H. Programmed cell death-1 inhibitor-related sclerosing cholangitis: A systematic review. World J. Gastroenterol. 2020, 26, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hart, J.; Ding, X.; Zhang, X.; Feely, M.; Yassan, L.; Alpert, L.; Soldevila-Pico, C.; Zhang, X.; Liu, X.; et al. Histologic patterns of liver injury induced by anti-PD-1 therapy. Gastroenterol. Rep. 2019, 8, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opdivo. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/opdivo-epar-product-information_en.pdf (accessed on 10 October 2021).
- Opdivo. Full Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125554s090lbl.pdf (accessed on 10 October 2021).
- Anugwom, C.; Leventhal, T. Nivolumab-Induced Autoimmune-Like Cholestatic Hepatitis in a Liver Transplant Recipient. ACG Case Rep. J. 2020, 7, e00416. [Google Scholar] [CrossRef] [PubMed]
- Imafuku, K.; Yoshino, K.; Yamaguchi, K.; Tsuboi, S.; Ohara, K.; Hata, H. Successful treatment of sudden hepatitis induced by long-term nivolumab administration. Case Rep. Oncol. 2017, 10, 368–371. [Google Scholar] [CrossRef]
- McClure, T.; Cui, W.; Asadi, K.; John, T.; Testro, A. Case of nivolumab-induced sclerosing cholangitis: Lessons from long-term follow-up. BMJ Open Gastroenterol. 2020, 7, e000487. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Ito, T.; Ishigami, M.; Ishizu, Y.; Kuzuya, T.; Honda, T.; Kawashima, H.; Inukai, Y.; Toyoda, H.; Yokota, K.; et al. Real world data of liver injury induced by immune checkpoint inhibitors in Japanese patients with advanced malignancies. J. Gastroenterol. 2020, 55, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Zarrabi, K.; Wu, S. Risk of Liver Toxicity with Nivolumab Immunotherapy in Cancer Patients. Oncology 2018, 94, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Tecentriq- Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761034s033s034s035s036s037s038lbl.pdf (accessed on 10 October 2021).
- Tecentriq- Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/tecentriq-epar-product-information_en.pdf (accessed on 10 October 2021).
- Honma, Y.; Shibata, M.; Gohda, T.; Matsumiya, H.; Kumamoto, K.; Miyama, A.; Morino, K.; Koya, Y.; Taira, A.; Shinohara, S.; et al. Rapid Progression of Liver Fibrosis Induced by Acute Liver Injury Due to Immune-related Adverse Events of Atezolizumab. Intern. Med. 2021, 60, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Khadilkar, A.; Patel Krishen, A. Rare Case of Atezolizumab-Induced Hepatotoxicity. Am. J. Gastr. 2020, 115, S1406. [Google Scholar] [CrossRef]
- Imfinzi. Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761069s029lbl.pdf (accessed on 10 October 2021).
- Imfinzi. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/imfinzi-epar-product-information_en.pdf (accessed on 10 October 2021).
- Kelly, R.J.; Lee, J.; Bang, Y.J.; Almhanna, K.; Blum-Murphy, M.; Catenacci, D.V.T.; Chung, H.C.; Wainberg, Z.A.; Gibson, M.K.; Lee, K.W.; et al. Safety and Efficacy of Durvalumab and Tremelimumab Alone or in Combination in Patients with Advanced Gastric and Gastroesophageal Junction Adenocarcinoma. Clin. Cancer Res. 2020, 26, 846–854. [Google Scholar] [CrossRef] [Green Version]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. 2012; Durvalumab. [Updated 2017 Jul 20]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548150/#_NBK548150_pubdet_ (accessed on 10 October 2021).
- Nakamura, M.; Otsuka, T.; Hayashi, R.; Horita, T.; Ota, M.; Sakurai, N.; Takano, H.; Hayashi, T.; Kumagai, M.; Yamada, S.; et al. Durvalumab-induced Immune-related Hepatitis in a Patient with Non-small Cell Lung Cancer. Intern. Med. 2020, 59, 2711–2717. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.E.; Frei, B.L. Drugs that act on the immune system: Cytokines and monoclonal antibodies in Side Effects of Drugs Annual. In A Worldwide Yearly Survey of New Data in Adverse Drug Reactions; Sidhartha, D., Ray, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 41. [Google Scholar]
- Bavencio-Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761049s006lbl.pdf (accessed on 10 October 2021).
- Bavencio-Summary of Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/bavencio-epar-product-information_en.pdf (accessed on 10 October 2021).
- Kitagataya, T.; Suda, G.; Nagashima, K.; Katsurada, T.; Yamamoto, K.; Kimura, M.; Maehara, O.; Yamada, R.; Shigesawa, T.; Suzuki, K.; et al. Prevalence, clinical course, and predictive factors of immune checkpoint inhibitor monotherapy-associated hepatitis in Japan. J. Gastroenterol. Hepatol. 2020, 35, 1782–1788. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. 2012; Avelumab. [Updated 2017 Jul 20]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548530/ (accessed on 10 October 2021).
- Ramadori, G.; Saile, B. Inflammation, damage repair, immune cells, and liver fibrosis: Specific or nonspecific, this is the question. Gastroenterology 2004, 127, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Ju, C. Hepatic macrophages in drug-induced liver injury. Liv. Res. 2019, 3, 170–175. [Google Scholar] [CrossRef]
- Cataldi, M.; Citro, V.; Resnati, C.; Manco, F.; Tarantino, G. New Avenues for Treatment and Prevention of Drug-Induced Steatosis and Steatohepatitis: Much More Than Antioxidants. Adv. Ther. 2021, 38, 2094–2113. [Google Scholar] [CrossRef]
- Tarantino, G.; Conca, P.; Basile, V.; Gentile, A.; Capone, D.; Polichetti, G.; Leo, E. A prospective study of acute drug-induced liver injury in patients suffering from non-alcoholic fatty liver disease. Hepatol. Res. 2007, 37, 410–415. [Google Scholar] [CrossRef]
- Grieco, A.; Forgione, A.; Miele, L.; Vero, V.; Greco, A.V.; Gasbarrini, A.; Gasbarrini, G. Fatty liver and drugs. Eur. Rev. Med. Pharmacol. Sci. 2005, 9, 261–263. [Google Scholar] [PubMed]
- Schaffner, F.; Thaler, H. Nonalcoholic fatty liver disease. Prog. Liver Dis. 1986, 8, 283–298. [Google Scholar]
- Ahmed, A.; Wong, R.J.; Harrison, S.A. Nonalcoholic Fatty Liver Disease Review: Diagnosis, Treatment, and Outcomes. Clin. Gastroenterol. Hepatol. 2015, 13, 2062–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, D.E.; Brunt, E.M.; Wilson, L.A.; Behling, C.; Guy, C.; Contos, M.; Cummings, O.; Yeh, M.; Gill, R.; Chalasani, N.; et al. Association of Histologic Disease Activity with Progression of Nonalcoholic Fatty Liver Disease. JAMA Netw. Open 2019, 2, e1912565. [Google Scholar] [CrossRef] [Green Version]
- Farrell, G.C. Drugs and steatohepatitis. Semin. Liver Dis. 2002, 22, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.K.; Kuwajima, V.; Nadelson, J.; Atiq, O.; Sanyal, A.J. Drug-induced fatty liver disease: An overview of pathogenesis and management. Ann. Hepatol. 2015, 14, 789–806. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Effenberger, M. From NAFLD to MAFLD: When pathophysiology succeeds. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 387–388. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatology 2021, 73, 194–1198. [Google Scholar] [CrossRef]
- Tarantino, G.; Citro, V.; Capone, D. Nonalcoholic Fatty Liver Disease: A Challenge from Mechanisms to Therapy. J. Clin. Med. 2019, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Sawada, K.; Hayashi, H.; Nakajima, S.; Hasebe, T.; Fujiya, M.; Okumura, T. Non-alcoholic fatty liver disease is a potential risk factor for liver injury caused by immune checkpoint inhibitor. J. Gastroenterol. Hepatol. 2020, 35, 1042–1048. [Google Scholar] [CrossRef]
- Hamid, O.; Ahmed, E.; Abdul, M.; Imad, A. Presence of NAFLD in Patients Receiving Immune Checkpoint Inhibitors Significantly Increases the Prevalence of Immune Medicated Hepatotoxicity. Am. J. Gastr. 2021, 116, S546. [Google Scholar] [CrossRef]
- Nati, M.; Chung, K.J.; Chavakis, T. The Role of Innate Immune Cells in Nonalcoholic Fatty Liver Disease. J. Innate. Immun. 2021, 5, 1–11, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhong, S.; Qu, H.; Xie, Y.; Cao, Z.; Li, Q.; Yang, P.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. Chronic inflammation aggravates metabolic disorders of hepatic fatty acids in high-fat diet-induced obese mice. Sci. Rep. 2015, 5, 10222. [Google Scholar] [CrossRef] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leist, M.; Gantner, F.; Bohlinger, I.; Germann, P.G.; Tiegs, G.; Wendel, A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J. Immunol. 1994, 153, 1778–1788. [Google Scholar] [PubMed]
- Lukacs-Kornek, V.; Schuppan, D. Dendritic cells in liver injury and fibrosis: Shortcomings and promises. J. Hepatol. 2013, 59, 1124–1126. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Sánchez, N.; Córdova-Gallardo, J.; Barranco-Fragoso, B.; Eslam, M. Hepatic Dendritic Cells in the Development and Progression of Metabolic Steatohepatitis. Front. Immunol. 2021, 12, 641240. [Google Scholar] [CrossRef]
- Harley, I.T.; Stankiewicz, T.E.; Giles, D.A.; Softic, S.; Flick, L.M.; Cappelletti, M.; Sheridan, R.; Xanthakos, S.A.; Steinbrecher, K.A.; Sartor, R.B.; et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014, 59, 1830–1839. [Google Scholar] [CrossRef]
- He, B.; Wu, L.; Xie, W.; Shao, Y.; Jiang, J.; Zhao, Z.; Yan, M.; Chen, Z.; Cui, D. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol. 2017, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Ilan, Y.; Shailubhai, K.; Sanyal, A. Immunotherapy with oral administration of humanized anti-CD3 monoclonal antibody: A novel gut-immune system-based therapy for metaflammation and NASH. Clin. Exp. Immunol. 2018, 193, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Lalazar, G.; Mizrahi, M.; Turgeman, I.; Adar, T.; Ben Ya’acov, A.; Shabat, Y.; Nimer, A.; Hemed, N.; Zolotarovya, L.; Lichtenstein, Y.; et al. Oral Administration of OKT3 MAb to Patients with NASH, Promotes Regulatory T-cell Induction, and Alleviates Insulin Resistance: Results of a Phase IIa Blinded Placebo-Controlled Trial. J. Clin. Immunol. 2015, 35, 399–407. [Google Scholar] [CrossRef]
- Sutti, S.; Bruzzì, S.; Heymann, F.; Liepelt, A.; Krenkel, O.; Toscani, A.; Ramavath, N.N.; Cotella, D.; Albano, E.; Tacke, F. CX3CR1 Mediates the Development of Monocyte-Derived Dendritic Cells during Hepatic Inflammation. Cells 2019, 8, 1099. [Google Scholar] [CrossRef] [Green Version]
- Alissafi, T.; Hatzioannou, A.; Legaki, A.I.; Varveri, A.; Verginis, P. Balancing cancer immunotherapy and immune-related adverse events: The emerging role of regulatory T cells. J. Autoimmun. 2019, 104, 102310. [Google Scholar] [CrossRef]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Birebent, B.; Lorho, R.; Lechartier, H.; de Guibert, S.; Alizadeh, M.; Vu, N.; Beauplet, A.; Robillard, N.; Semana, G. Suppressive properties of human CD4+CD25+ regulatory T cells are dependent on CTLA-4 expression. Eur. J. Immunol. 2004, 34, 3485–3496. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Gianchecchi, E.; Fierabracci, A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018, 9, 2374. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Park, J.S.; Jeong, Y.H.; Son, J.; Ban, Y.H.; Lee, B.H.; Chen, L.; Chang, J.; Chung, D.H.; Choi, I.; et al. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J. Immunol. 2015, 194, 5801–5811. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Lau, R.; Yu, D.; Zhu, W.; Korman, A.; Weber, J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int. Immunol. 2009, 21, 1065–1077. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The Role of PD-1/PD-L1 Axis in Treg Development and Function: Implications for Cancer Immunotherapy. Onco. Targets Ther. 2019, 12, 8437–8445. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef]
- Desai, A.; Sandhu, S.; Lai, J.P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- FDA Grants Accelerated Approval to Nivolumab for HCC Previously Treated with Sorafenib. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-hcc-previously-treated-sorafenib (accessed on 10 October 2021).
- FDA Grants Accelerated Approval to Pembrolizumab for Hepatocellular Carcinoma. Available online: https://www.fda.gov/drugs/fda-grants-accelerated-approval-pembrolizumab-hepatocellular-carcinoma (accessed on 10 January 2021).
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Cui, T.M.; Liu, Y.; Wang, J.B.; Liu, L.X. Adverse Effects of Immune-Checkpoint Inhibitors in Hepatocellular Carcinoma. Onco. Targets Ther. 2020, 13, 11725–11740. [Google Scholar] [CrossRef]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491.e1. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef] [Green Version]
- Asfari, M.M.; Talal Sarmini, M.; Alomari, M.; Lopez, R.; Dasarathy, S.; McCullough, A.J. The association of nonalcoholic steatohepatitis and hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2020, 32, 1566–1570. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Min, H.K.; Mirshahi, F.; Verdianelli, A.; Pacana, T.; Patel, V.; Park, C.G.; Choi, A.; Lee, J.H.; Park, C.B.; Ren, S.; et al. Activation of the GP130-STAT3 axis and its potential implications in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G794–G803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zeng, M.; Yan, K.; Yang, Y.; Li, H.; Xu, X. IL-17 promotes hepatocellular carcinoma through inhibiting apoptosis induced by IFN-γ. Biochem. Biophys. Res. Commun. 2020, 522, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.L.; Teijeiro, A.; Burén, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perna, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016, 30, 161–175. [Google Scholar] [CrossRef]
- Morzyglod, L.; Caüzac, M.; Popineau, L.; Denechaud, P.D.; Fajas, L.; Ragazzon, B.; Fauveau, V.; Planchais, J.; Vasseur-Cognet, M.; Fartoux, L.; et al. Growth factor receptor binding protein 14 inhibition triggers insulin-induced mouse hepatocyte proliferation and is associated with hepatocellular carcinoma. Hepatology 2017, 65, 1352–1368. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Eso, Y.; Taura, K.; Seno, H. Does immune checkpoint inhibitor exhibit limited efficacy against non-viral hepatocellular carcinoma? A review of clinical trials. Hepatol. Res. 2021, 9, 13712. [Google Scholar] [CrossRef]
- ESMO Guidelines Committee. Updated Treatment Recommendations for Hepatocellular Carcinoma (HCC) from the ESMO Clinical Practice Guidelines Published: 05 March 2021. Available online: https://www.esmo.org/guidelines/gastrointestinal-cancers/hepatocellular-carcinoma/eupdate-hepatocellular-carcinoma-treatment-recommendations (accessed on 10 November 2021).
- Hilmi, M.; Neuzillet, C.; Calderaro, J.; Lafdil, F.; Pawlotsky, J.M.; Rousseau, B. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: Current knowledge and future research directions. J. Immunother. Cancer 2019, 7, 333. [Google Scholar] [CrossRef]

{kind=link}
| Mechanism of Action | Approved Clinical Indications | Clinical Presentation (Incidence in Clinical Trials/Time to Onset) | References | |
|---|---|---|---|---|
| Ipilimumab | Anti-CTA-4 IgG1 human mAb | Melanoma, Renal Cell Carcinoma, CRC, HCC, NSCLC | -Transaminase elevation (34%/3–9 weeks) -Acute hepatitis (1–2%/3.8 months) -Steatohepatitis (NA) -Cholestatic hepatitis (NA) | [14,15,16,17,18,19,20,21,22,23,24] |
| Celiplimab | Anti-PD1, IgG4 human mAb | Cutaneous Squamous Cell Carcinoma, Basal Cell Carcinoma, NSCLC | -Acute hepatitis (2%) | [25,26,27] |
| Pembrolizumab | Anti-PD1, IgG4 humanized mAb | Melanoma, NSCLC, SCLC, HNSCC, Classical Hodgkin Lymphoma, Primary Mediastinal Large B-Cell Lymphoma, Urothelial Carcinoma, Microsatellite Instability-High or Mismatch Repair Deficient Cancer, Microsatellite Instability-High or Mismatch Repair Deficient CRC, Gastric Cancer, Esophageal Cancer, Cervical Cancer, HCC, Renal Cell Carcinoma, Tumor Mutational Burden-High, Cancer Cutaneous Squamous Cell Carcinoma, Triple-Negative Breast Cancer | -Transaminase elevation (27%) -Acute hepatitis (0.7%/3.8 months) -Steatohepatitis (NA) -Cholestatic hepatitis (NA) -Sclerosing cholangitis (NA) | [21,28,29,30,31,32,33,34,35] |
| Nivolumab | Anti-PD1, IgG4 human mAb | Melanoma, NSCLC, Malignant Pleural Mesothelioma, Classical Hodgkin Lymphoma, Urothelial Carcinoma, CRC, Esophageal Squamous Cell Carcinoma | -Transaminase elevation (monotherapy: 7.3%/2.3 months; in combination with ipilimumab: 29.5%/1.5 months) -Acute Hepatitis (monotherapy: 1.8%/3.3 months; in combination with ipilimumab: 7–13%-2.1 months) -Steatohepatitis (NA) -Cholestatic hepatitis (NA) -Sclerosing cholangitis (NA) | [20,21,35,36,37,38,39,40,41,42,43] |
| Atezolizumab | Anti PDL-1 IgG1 human mAb | Urothelial Carcinoma, NSCLC, Triple-Negative Breast Cancer, SCLC, HCC, Melanoma | -Transaminase elevation (common) -Acute hepatitis (1.8%/1.5 months) -Rapid progression of liver fibrosis (NA) | [44,45,46,47] |
| Durvalumab | Anti PDL-1 IgG1 human mAb | NSCLC, SCLC | -Transaminase elevation (8.1%) -Acute hepatitis (0.8%) | [35,48,49,50,51,52,53] |
| Avelumab | Anti PDL-1 IgG1 human mAb | Merkel carcinoma, Urothelial carcinoma, Renal carcinoma | -Transaminase elevation (common) Acute hepatitis (monotherapy: 0.9%/2.5 months; in combination with axitinib: 7%/2.8 months) | [35,54,55,56,57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cataldi, M.; Manco, F.; Tarantino, G. Steatosis, Steatohepatitis and Cancer Immunotherapy: An Intricate Story. Int. J. Mol. Sci. 2021, 22, 12947. https://doi.org/10.3390/ijms222312947
Cataldi M, Manco F, Tarantino G. Steatosis, Steatohepatitis and Cancer Immunotherapy: An Intricate Story. International Journal of Molecular Sciences. 2021; 22(23):12947. https://doi.org/10.3390/ijms222312947
Chicago/Turabian StyleCataldi, Mauro, Federica Manco, and Giovanni Tarantino. 2021. "Steatosis, Steatohepatitis and Cancer Immunotherapy: An Intricate Story" International Journal of Molecular Sciences 22, no. 23: 12947. https://doi.org/10.3390/ijms222312947
APA StyleCataldi, M., Manco, F., & Tarantino, G. (2021). Steatosis, Steatohepatitis and Cancer Immunotherapy: An Intricate Story. International Journal of Molecular Sciences, 22(23), 12947. https://doi.org/10.3390/ijms222312947