
Primaquine Inhibits the Endosomal Trafficking and Nuclear Localization of EGFR and Induces the Apoptosis of Breast Cancer Cells by Nuclear EGFR/Stat3-Mediated c-Myc Downregulation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
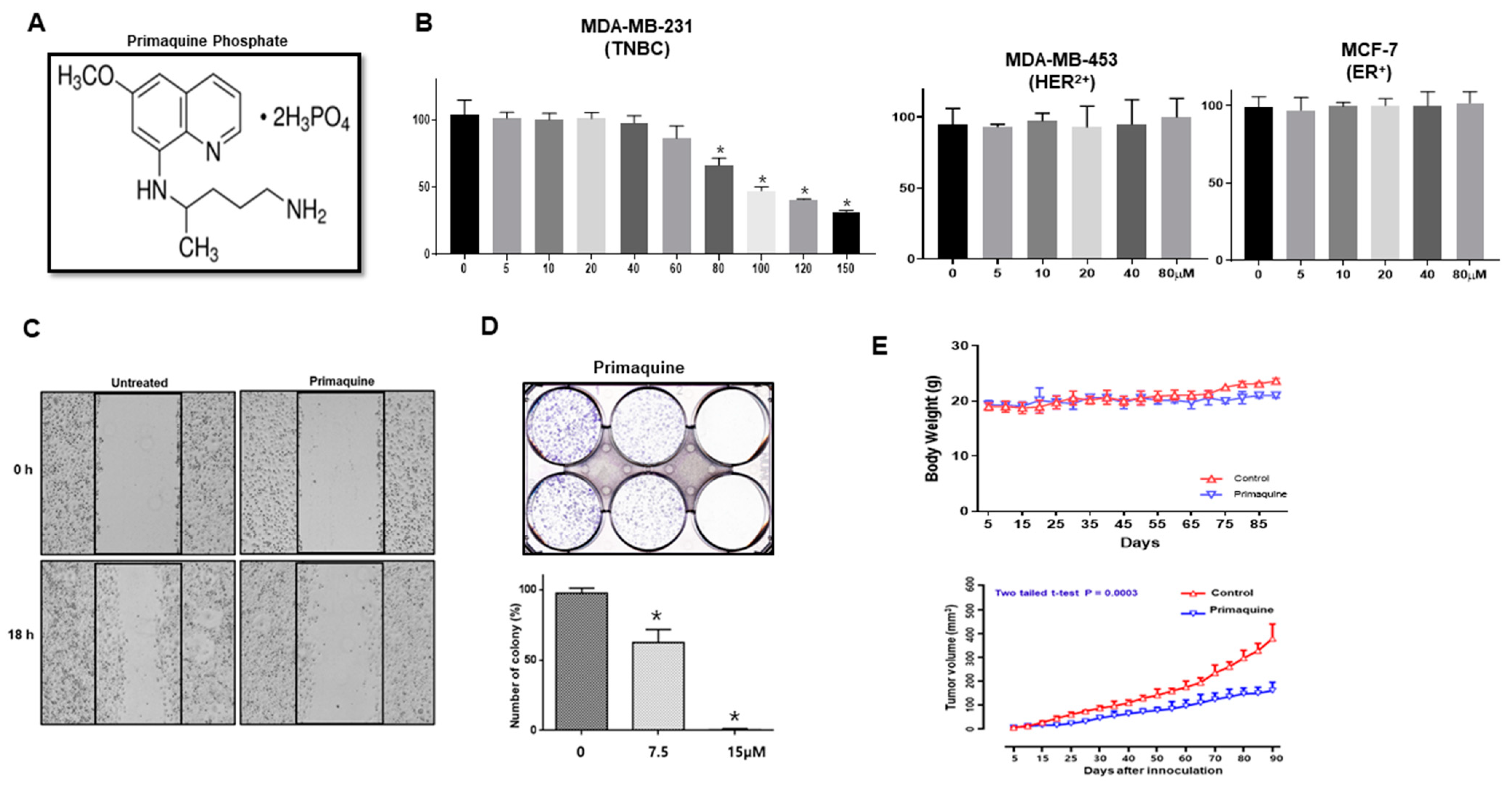
2.1. The Antimalarial Drug Primaquine Decreases Breast Cancer Cell Viability and Inhibits Tumor Growth in a Mouse Xenograft Model
2.2. Primaquine Affects the Endolysosomal System and Impairs the Endocytosis-Mediated Degradation of EGFR
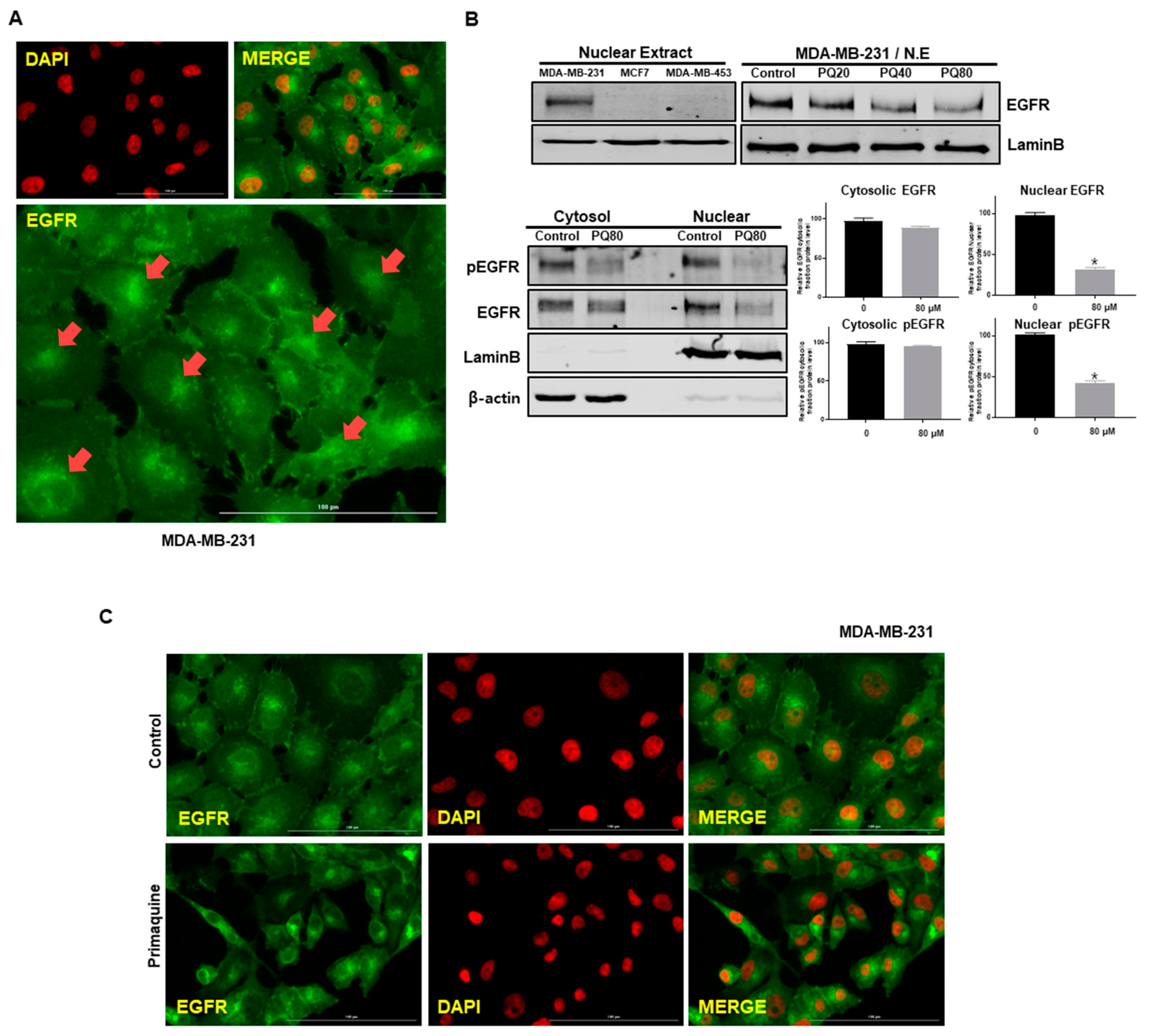
2.3. Primaquine Reduces the Expression of nEGFR in Breast Cancer
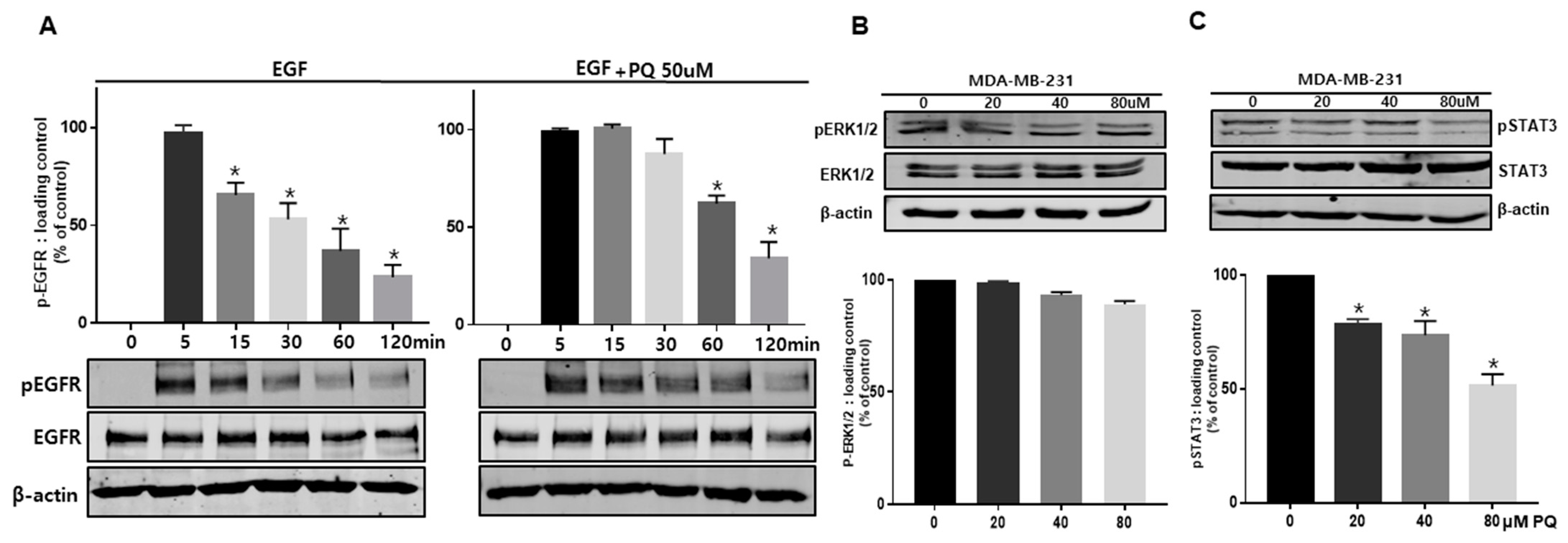
2.4. Primaquine Regulates EGFR Phosphorylation and the EGFR Downstream Signaling Pathway
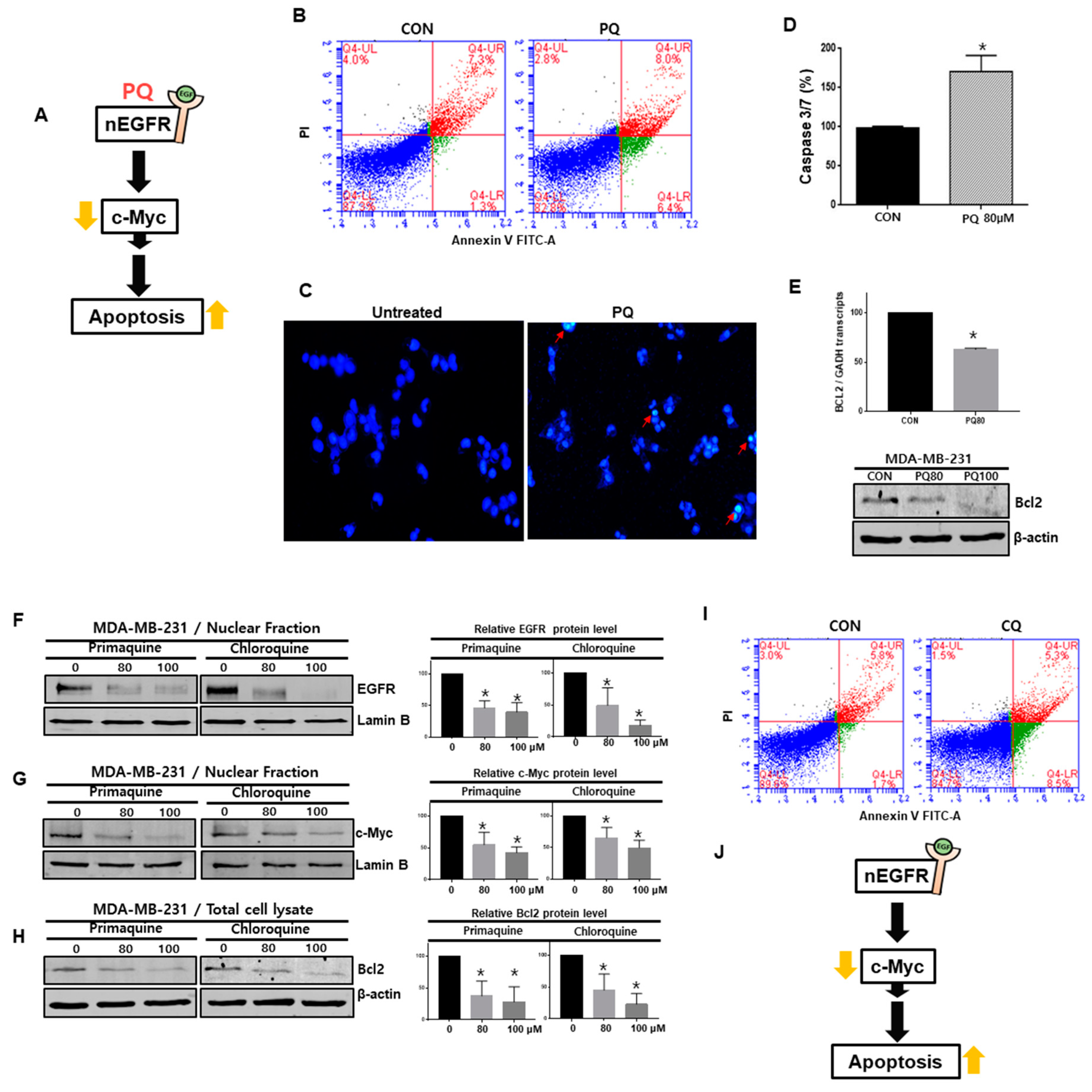
2.5. The nEGFR Protein Physically Interacts with Stat3 and this Interaction Is Inhibited by Primaquine Treatment
2.6. Primaquine Induces the Apoptosis of Breast Cancer Cells through nEGFR/Stat3-Mediated c-Myc and Bcl-2 Downregulation
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Lines and Media
4.3. Cell Proliferation Assay
4.4. Clonogenic and Scratch Assays
4.5. Xenograft Transplantation
4.6. Western Blot Analysis
4.7. EGFR Degradation and Trafficking
4.8. Immunofluorescence
4.9. Immunoprecipitation
4.10. Gene Expression Analysis
4.11. Small Interfering RNA (siRNA)
4.12. Hoechst 33342 Staining and Annexin V/PI Assay
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schneider, B.P.; Winer, E.P.; Foulkes, W.D.; Garber, J.; Perou, C.M.; Richardson, A.; Sledge, G.W.; Carey, L.A. Triple-negative breast cancer: Risk factors to potential targets. Clin. Cancer Res. 2008, 14, 8010–8018. [Google Scholar] [CrossRef] [Green Version]
- Stevens, K.N.; Vachon, C.M.; Couch, F.J. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013, 73, 2025–2030. [Google Scholar] [CrossRef] [Green Version]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, T.M.; Iida, M.; Dunn, E.F.; Luthar, N.; Kostopoulos, K.T.; Corrigan, K.L.; Wleklinski, M.J.; Yang, D.; Wisinski, K.B.; Salgia, R.; et al. Nuclear epidermal growth factor receptor is a functional molecular target in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 1356–1368. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Faivre, S.; Armand, J.P. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs 2000, 60 (Suppl. 1), 15–23. [Google Scholar] [CrossRef]
- Wang, Y.N.; Yamaguchi, H.; Hsu, J.M.; Hung, M.C. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene 2010, 29, 3997–4006. [Google Scholar] [CrossRef] [Green Version]
- Marti, U.; Burwen, S.J.; Wells, A.; Barker, M.E.; Huling, S.; Feren, A.M.; Jones, A.L. Localization of Epidermal Growth-Factor Receptor in Hepatocyte Nuclei. Hepatology 1991, 13, 15–20. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Flanigan, B.G.; Stein, A.P.; Salgia, R.; Rolff, J.; Kimple, R.J.; et al. The receptor tyrosine kinase AXL mediates nuclear translocation of the epidermal growth factor receptor. Sci. Signal. 2017, 10, 1064. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Li, C.; Wheeler, D.L. The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov. Med. 2011, 12, 419–432. [Google Scholar]
- Du, Y.; Shen, J.; Hsu, J.L.; Han, Z.; Hsu, M.C.; Yang, C.C.; Kuo, H.P.; Wang, Y.N.; Yamaguchi, H.; Miller, S.A.; et al. Syntaxin 6-mediated Golgi translocation plays an important role in nuclear functions of EGFR through microtubule-dependent trafficking. Oncogene 2014, 33, 756–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, H.W.; Ali-Seyed, M.; Wu, Y.; Bartholomeusz, G.; Hsu, S.C.; Hung, M.C. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J. Cell Biochem. 2006, 98, 1570–1583. [Google Scholar] [CrossRef]
- Wang, Y.N.; Yamaguchi, H.; Huo, L.; Du, Y.; Lee, H.J.; Lee, H.H.; Wang, H.; Hsu, J.M.; Hung, M.C. The translocon Sec61beta localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus. J. Biol. Chem. 2010, 285, 38720–38729. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Tao, Y.; Jiang, Y.; Xu, Y.; Yan, B.; Chen, X.; Xiao, L.; Cao, Y. Nuclear epidermal growth factor receptor interacts with transcriptional intermediary factor 2 to activate cyclin D1 gene expression triggered by the oncoprotein latent membrane protein 1. Carcinogenesis 2012, 33, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Hanada, N.; Lo, H.W.; Day, C.P.; Pan, Y.; Nakajima, Y.; Hung, M.C. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol. Carcinog. 2006, 45, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.Y.; Tseng, J.T.; Lee, Y.C.; Xia, W.; Wang, Y.N.; Wu, M.L.; Chuang, Y.H.; Lai, C.H.; Chang, W.C. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008, 36, 4337–4351. [Google Scholar] [CrossRef]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol. Cancer Res. 2010, 8, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Chen, Y.J.; Li, L.Y.; Wei, Y.L.; Hsu, S.C.; Tsai, S.L.; Chiu, P.C.; Huang, W.P.; Wang, Y.N.; Chen, C.H.; et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J. Biol. Chem. 2011, 286, 20558–20568. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Carpenter, R.L.; Cao, X.; Lo, H.W. STAT1 gene expression is enhanced by nuclear EGFR and HER2 via cooperation with STAT3. Mol. Carcinog. 2013, 52, 959–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liccardi, G.; Hartley, J.A.; Hochhauser, D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 2011, 71, 1103–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Iida, M.; Dunn, E.F.; Ghia, A.J.; Wheeler, D.L. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009, 28, 3801–3813. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.L.; Iida, M.; Kruser, T.J.; Nechrebecki, M.M.; Dunn, E.F.; Armstrong, E.A.; Huang, S.; Harari, P.M. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol. Ther. 2009, 8, 696–703. [Google Scholar] [CrossRef] [Green Version]
- Iida, M.; Brand, T.M.; Campbell, D.A.; Li, C.; Wheeler, D.L. Yes and Lyn play a role in nuclear translocation of the epidermal growth factor receptor. Oncogene 2013, 32, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Le Roy, C.; Wrana, J.L. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat. Rev. Mol. Cell Biol. 2005, 6, 112–126. [Google Scholar] [CrossRef]
- Jo, U.; Park, K.H.; Whang, Y.M.; Sung, J.S.; Won, N.H.; Park, J.K.; Kim, Y.H. EGFR endocytosis is a novel therapeutic target in lung cancer with wild-type EGFR. Oncotarget 2014, 5, 1265–1278. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Luthar, N.; Starr, M.M.; Huppert, E.J.; Wheeler, D.L. Nuclear EGFR as a molecular target in cancer. Radiother. Oncol. 2013, 108, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cao, Y.; Sun, X.; Feng, Y.; Du, Y.; Liu, F.; Yu, C.; Jin, F. Chloroquine (CQ) exerts anti-breast cancer through modulating microenvironment and inducing apoptosis. Int. Immunopharmacol. 2017, 42, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Pavic, K.; Rubinic, B.; Rajic, Z.; Fontinha, D.; Prudencio, M.; Uzelac, L.; Kralj, M.; Held, J.; Zorc, B. Primaquine homodimers as potential antiplasmodial and anticancer agents. Bioorg. Med. Chem. Lett. 2019, 29, 126614. [Google Scholar] [CrossRef]
- Beus, M.; Rajic, Z.; Maysinger, D.; Mlinaric, Z.; Antunovic, M.; Marijanovic, I.; Fontinha, D.; Prudencio, M.; Held, J.; Olgen, S.; et al. SAHAquines, Novel Hybrids Based on SAHA and Primaquine Motifs, as Potential Cytostatic and Antiplasmodial Agents. Chem. Open 2018, 7, 624–638. [Google Scholar] [CrossRef] [Green Version]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, A.R.; Kim, Y.K.; Yoon, S. Co-treatment with the anti-malarial drugs mefloquine and primaquine highly sensitizes drug-resistant cancer cells by increasing P-gp inhibition. Biochem. Biophys. Res. Commun. 2013, 441, 655–660. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Huang, S.F.; Hung, M.C. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005, 65, 338–348. [Google Scholar] [PubMed]
- Hadzisejdic, I.; Mustac, E.; Jonjic, N.; Petkovic, M.; Grahovac, B. Nuclear EGFR in ductal invasive breast cancer: Correlation with cyclin-D1 and prognosis. Mod. Pathol. 2010, 23, 392–403. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.A.; Lu, H.; Soutto, M.; Capobianco, A.; Rai, P.; Zaika, A.; El-Rifai, W. Exposure of Barrett’s and esophageal adenocarcinoma cells to bile acids activates EGFR-STAT3 signaling axis via induction of APE1. Oncogene 2018, 37, 6011–6024. [Google Scholar] [CrossRef]
- Hoffman, B.; Liebermann, D.A. Apoptotic signaling by c-MYC. Oncogene 2008, 27, 6462–6472. [Google Scholar] [CrossRef] [Green Version]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.S.; Kim, J.H.; Kim, S.L.; Deng, H.Y.; Lee, D.; Kim, C.S.; Yun, B.S.; Lee, D.S. Catechol derived from aronia juice through lactic acid bacteria fermentation inhibits breast cancer stem cell formation via modulation Stat3/IL-6 signaling pathway. Mol. Carcinog. 2018, 57, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, S.L.; Kim, J.H.; Deng, H.Y.; Yun, B.S.; Lee, D.S. Triterpene Acid (3-O-p-Coumaroyltormentic Acid) Isolated From Aronia Extracts Inhibits Breast Cancer Stem Cell Formation through Downregulation of c-Myc Protein. Int. J. Mol. Sci. 2018, 19, 2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.L.; Choi, H.S.; Kim, J.H.; Jeong, D.K.; Kim, K.S.; Lee, D.S. Dihydrotanshinone-Induced NOX5 Activation Inhibits Breast Cancer Stem Cell through the ROS/Stat3 Signaling Pathway. Oxid. Med. Cell Longev. 2019, 2019, 9296439. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, H.S.; Kim, S.L.; Lee, D.S. The PAK1-Stat3 Signaling Pathway Activates IL-6 Gene Transcription and Human Breast Cancer Stem Cell Formation. Cancers 2019, 11, 1527. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Choi, H.-S.; Lee, D.-S. Primaquine Inhibits the Endosomal Trafficking and Nuclear Localization of EGFR and Induces the Apoptosis of Breast Cancer Cells by Nuclear EGFR/Stat3-Mediated c-Myc Downregulation. Int. J. Mol. Sci. 2021, 22, 12961. https://doi.org/10.3390/ijms222312961
Kim J-H, Choi H-S, Lee D-S. Primaquine Inhibits the Endosomal Trafficking and Nuclear Localization of EGFR and Induces the Apoptosis of Breast Cancer Cells by Nuclear EGFR/Stat3-Mediated c-Myc Downregulation. International Journal of Molecular Sciences. 2021; 22(23):12961. https://doi.org/10.3390/ijms222312961
Chicago/Turabian StyleKim, Ji-Hyang, Hack-Sun Choi, and Dong-Sun Lee. 2021. "Primaquine Inhibits the Endosomal Trafficking and Nuclear Localization of EGFR and Induces the Apoptosis of Breast Cancer Cells by Nuclear EGFR/Stat3-Mediated c-Myc Downregulation" International Journal of Molecular Sciences 22, no. 23: 12961. https://doi.org/10.3390/ijms222312961
APA StyleKim, J.-H., Choi, H.-S., & Lee, D.-S. (2021). Primaquine Inhibits the Endosomal Trafficking and Nuclear Localization of EGFR and Induces the Apoptosis of Breast Cancer Cells by Nuclear EGFR/Stat3-Mediated c-Myc Downregulation. International Journal of Molecular Sciences, 22(23), 12961. https://doi.org/10.3390/ijms222312961