
Investigation of the Antihypertrophic and Antifibrotic Effects of Losartan in a Rat Model of Radiation-Induced Heart Disease

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
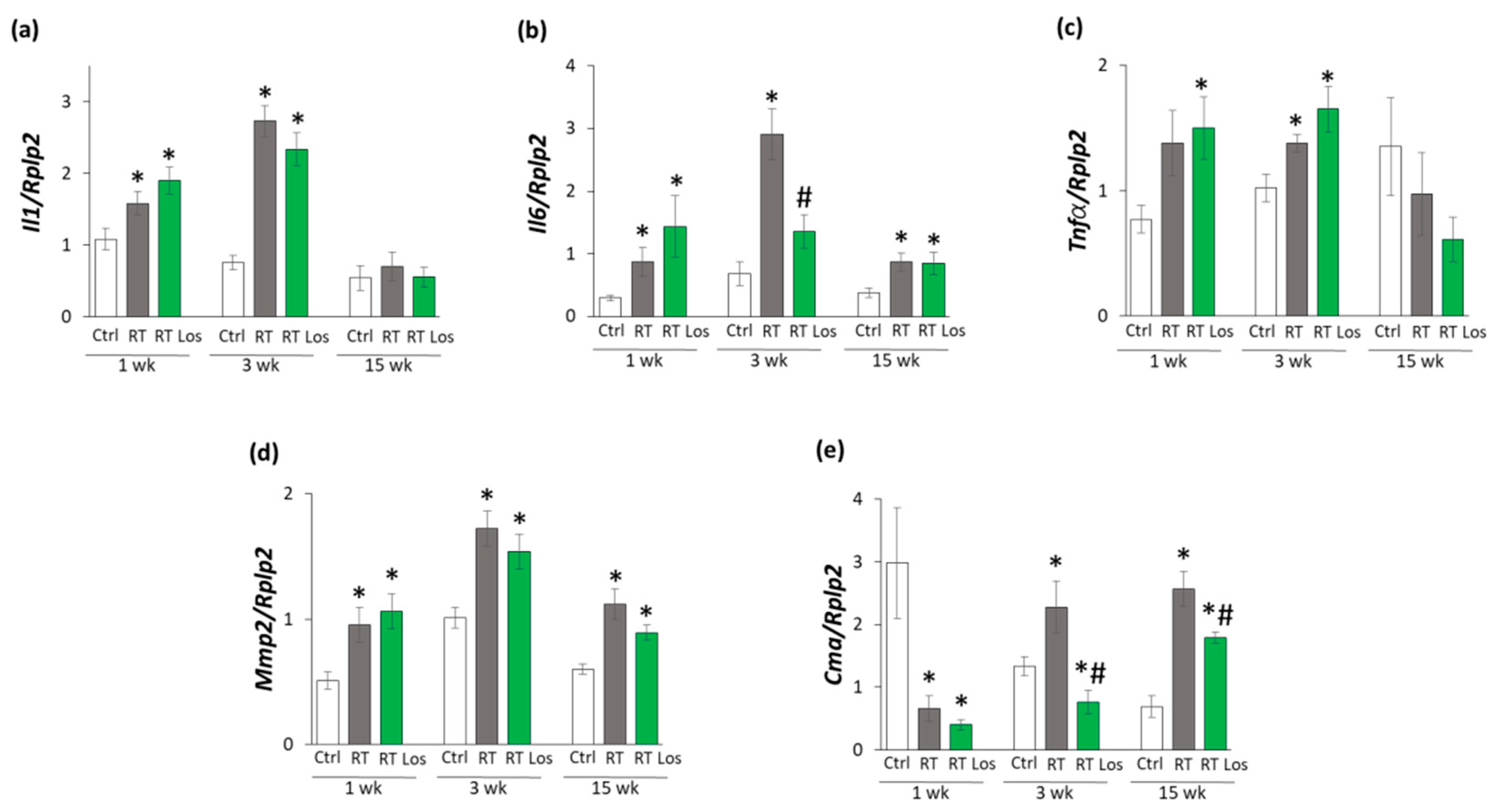
2.1. Systemic Effects of the Radiotherapy in Our RIHD Model
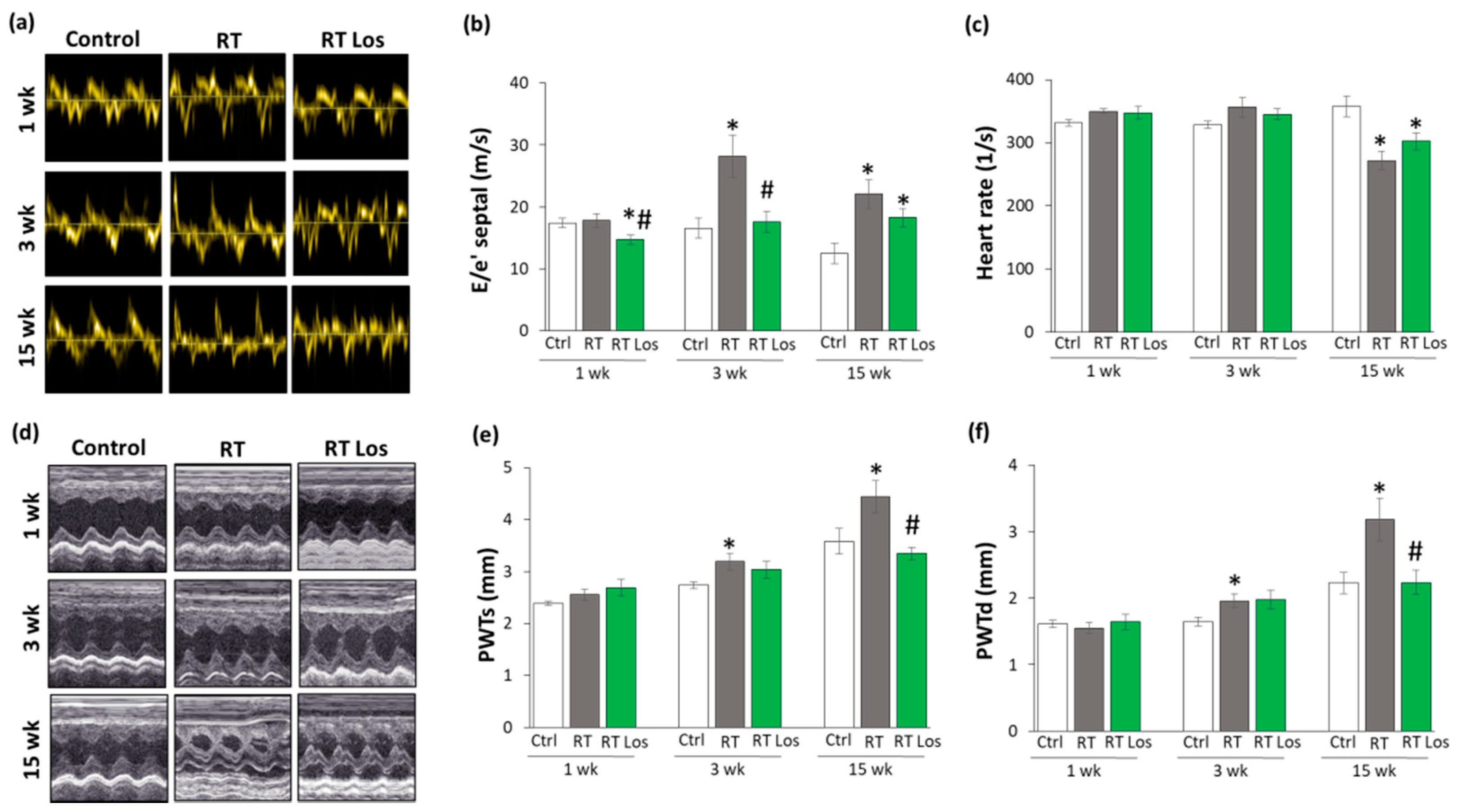
2.2. Post-RT Diastolic Dysfunction Was Alleviated by Losartan at Weeks 1 and 3 but Not at Week 15
2.3. RT-Related Echocardiographic Signs of Left Ventricular Hypertrophy (LVH) Were Alleviated by Losartan at Week 15
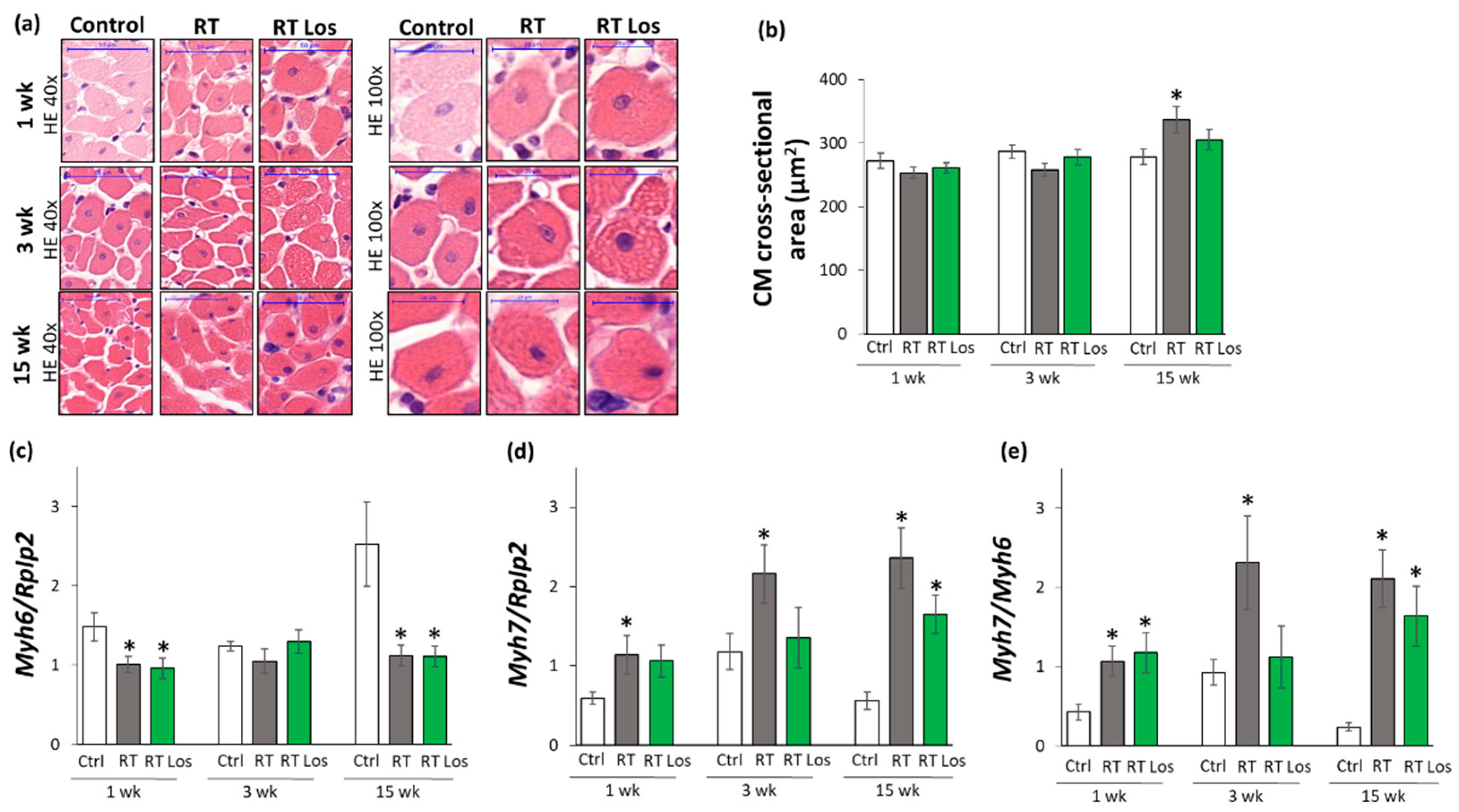
2.4. Cardiomyocyte Hypertrophy and the Overexpression of LVH Markers Were Reduced in the Losartan-Treated Animals 3 and 15 Weeks after RT
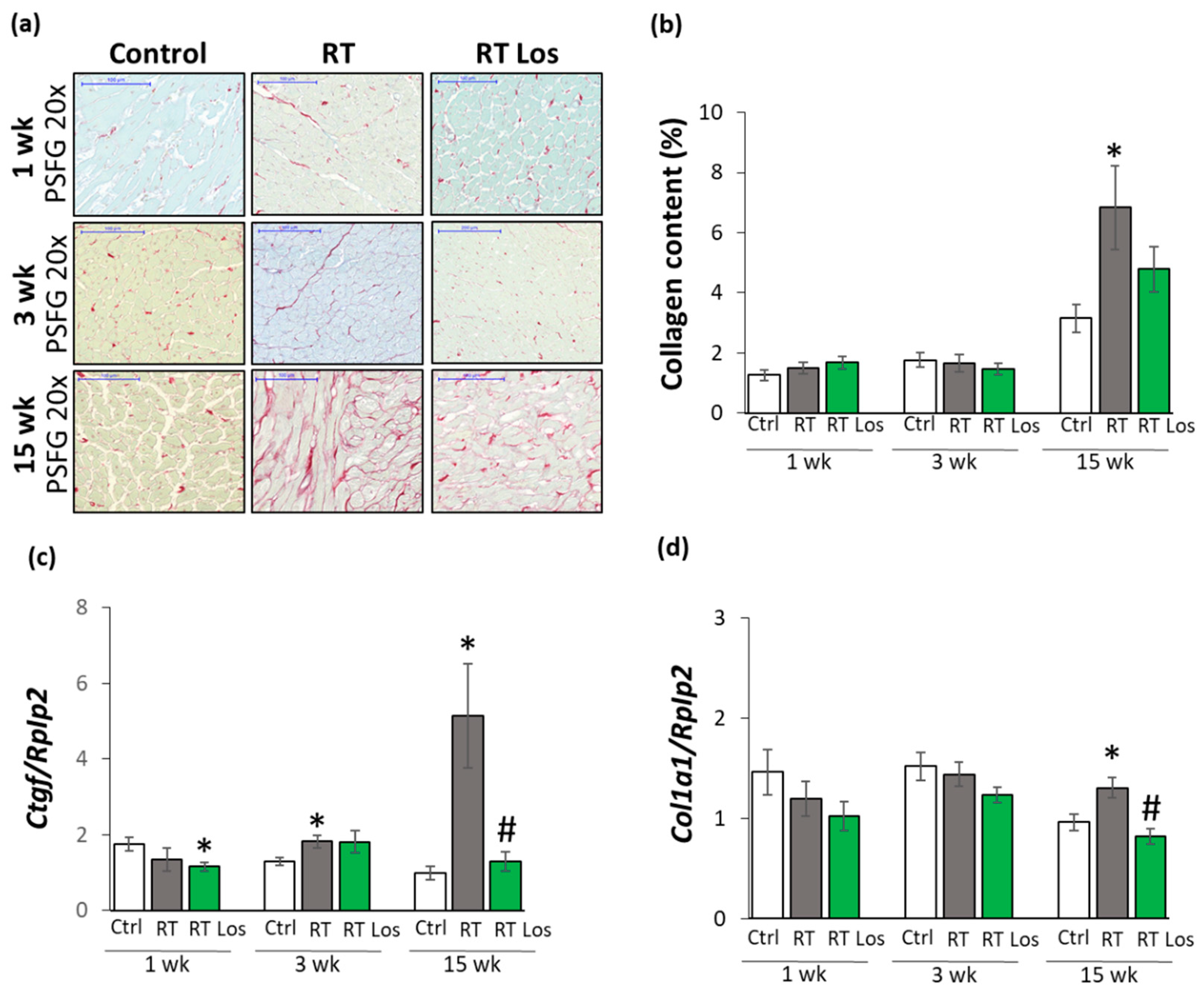
2.5. Interstitial Fibrosis Was Reduced in the Losartan-Treated Animals 15 Weeks after RT
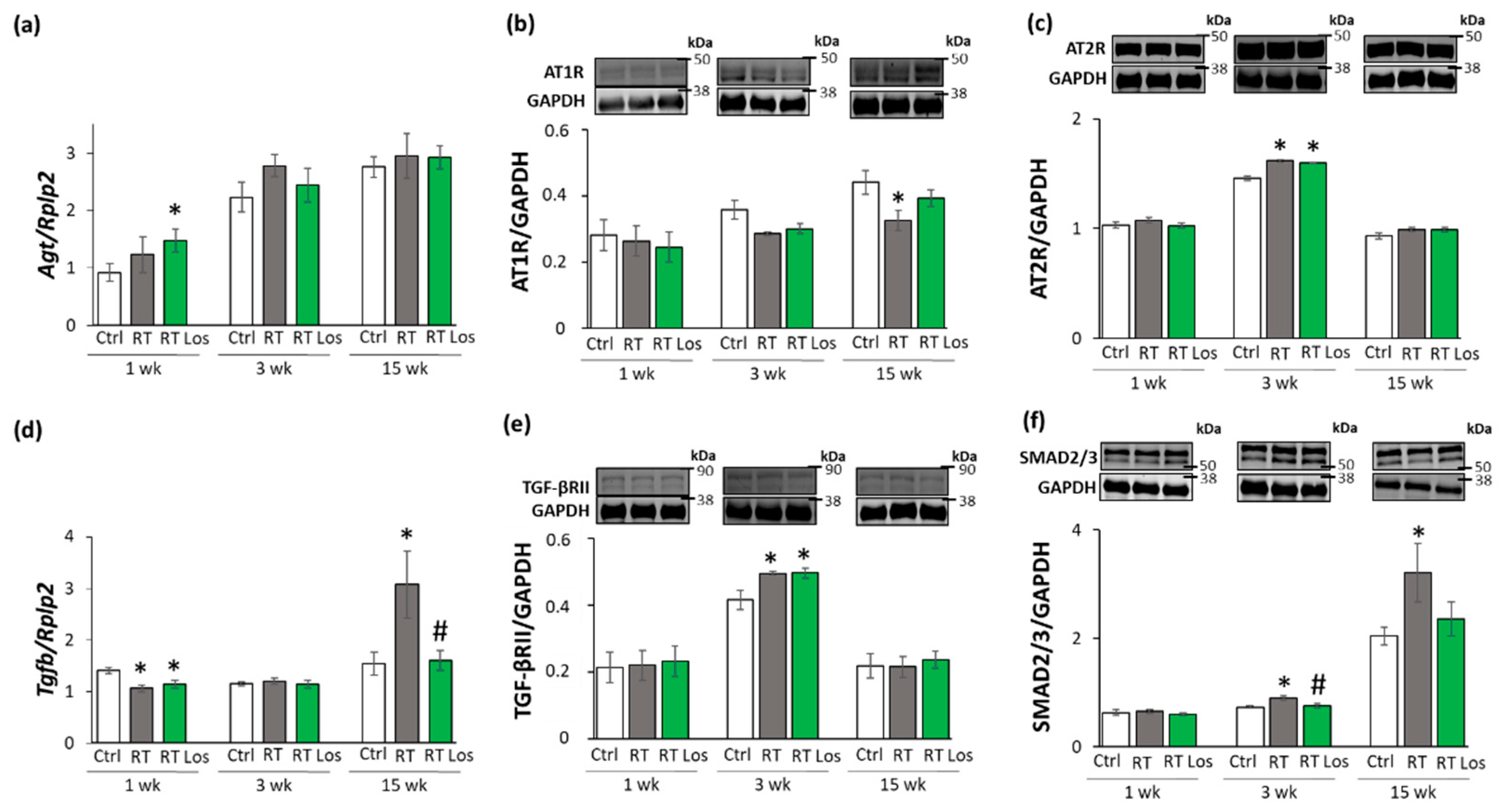
2.6. Losartan Reduced the Chymase Overexpression at Weeks 3 and 15 after RT
2.7. Losartan Alleviated the Cardiac Fibrosis via Inhibiting the TGF-β-Mediated SMAD-Dependent Pathway in Our RIHD Model at Weeks 3 and 15
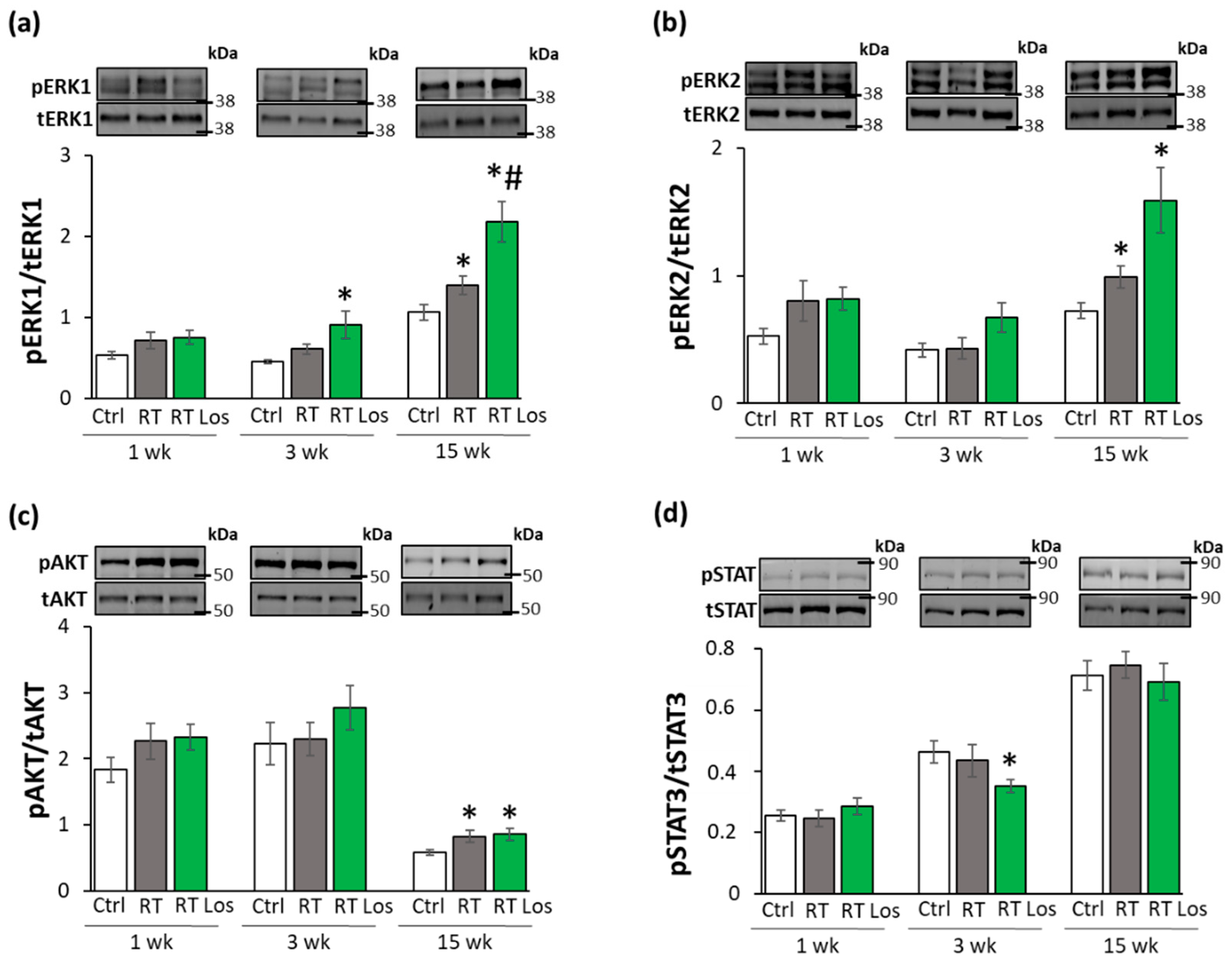
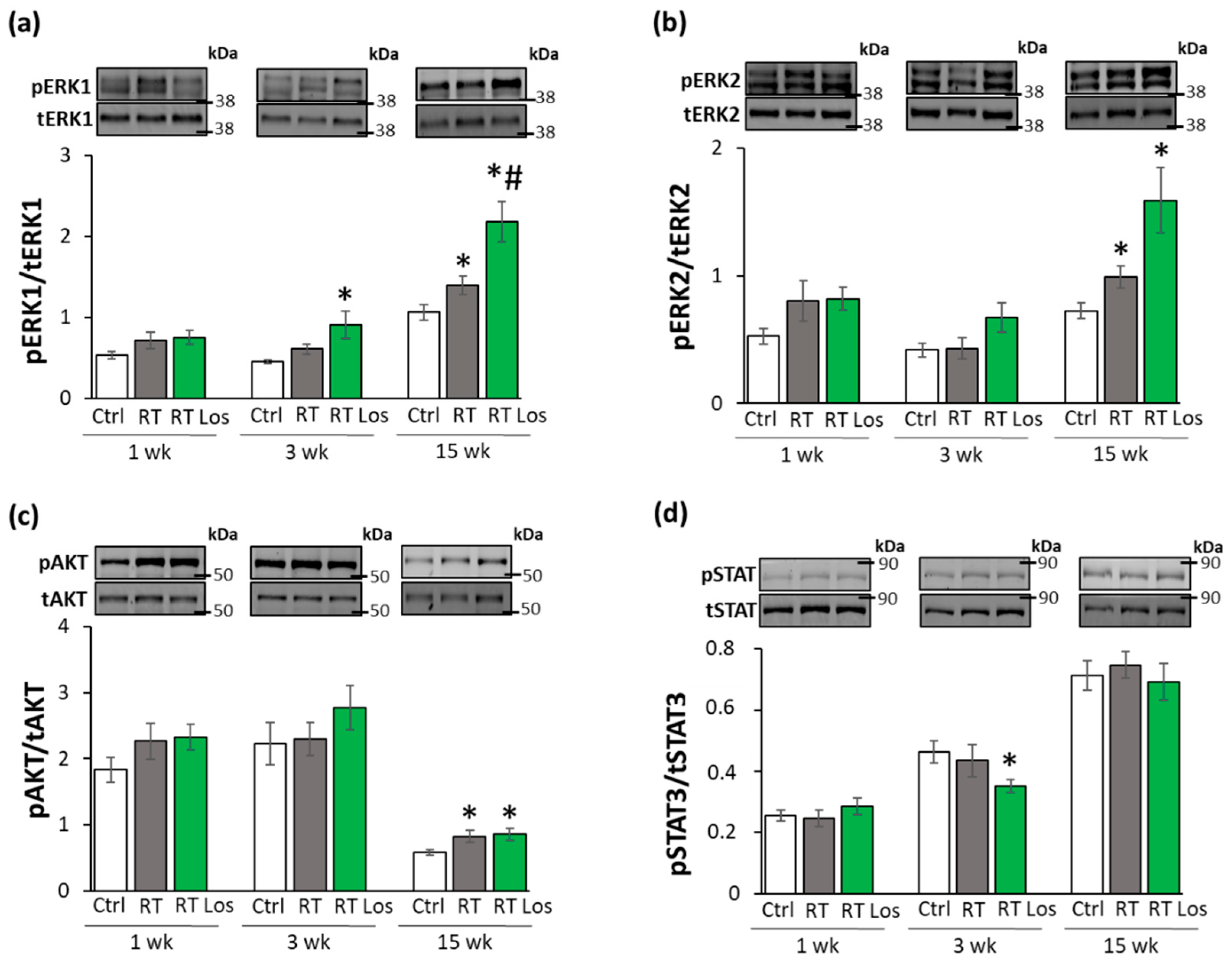
2.8. ERK1,2- and AKT-Mediated Pathways Might Be Involved in Compensatory Hypertrophy after RT at Week 15
3. Discussion
4. Materials and Methods
4.1. Ethics Approval
4.2. Animals
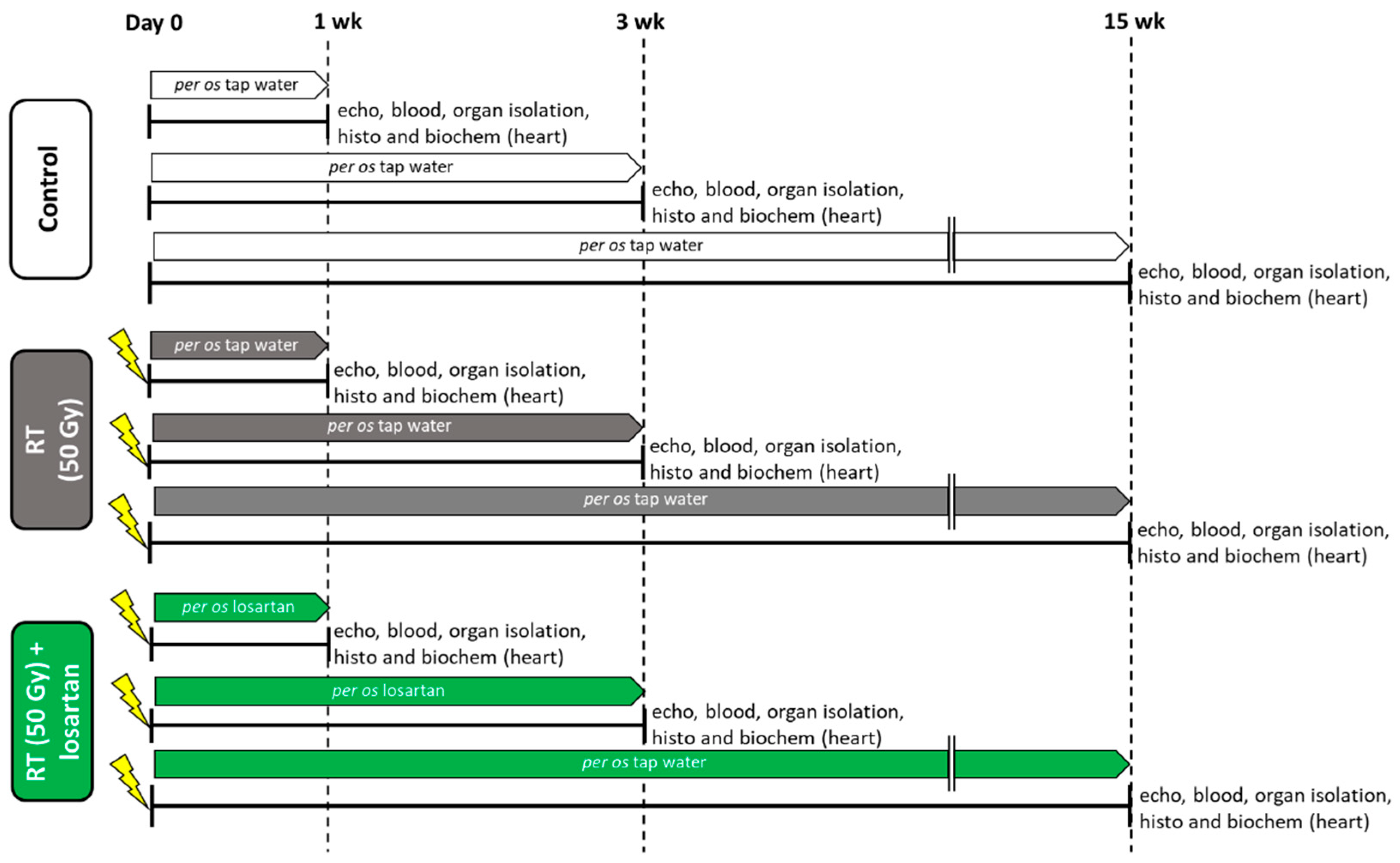
4.3. Experimental Setup
4.4. Heart Irradiation
4.5. Transthoracic Echocardiography
4.6. Blood Parameters
4.7. Tissue Harvesting
4.8. Hematoxylin-Eosin and Picrosirius Red and Fast Green Stainings
4.9. mRNA Expression Profiling by qRT-PCR
4.10. Western Blot
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Cardiovascular Diseases (CVDs). 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 20 November 2021).
- WHO. WHO Cancer. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 20 November 2021).
- Miller Mph, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [Green Version]
- Baskar, R.; Itahana, K. Radiation therapy and cancer control in developing countries: Can we save more lives? Int. J. Med. Sci. 2017, 14, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Menezes, K.M.; Wang, H.; Hada, M.; Saganti, P.B. Radiation Matters of the Heart: A Mini Review. Front. Cardiovasc. Med. 2018, 5, 83. [Google Scholar] [CrossRef]
- Taylor, C.; Correa, C.; Duane, F.K.; Aznar, M.C.; Anderson, S.; Bergh, J.; Dodwell, D.; Ewertz, M.; Gray, R.; Jagsi, R.; et al. Estimating the Risks of Breast Cancer Radiotherapy: Evidence from Modern Radiation Doses to the Lungs and Heart and From Previous Randomized Trials. J. Clin. Oncol. 2017, 35, 1641–1649. [Google Scholar] [CrossRef]
- van Nimwegen, F.A.; Schaapveld, M.; Cutter, D.J.; Janus, C.P.; Krol, A.D.; Hauptmann, M.; Kooijman, K.; Roesink, J.; van der Maazen, R.; Darby, S.C.; et al. Radiation Dose-Response Relationship for Risk of Coronary Heart Disease in Survivors of Hodgkin Lymphoma. J. Clin. Oncol. 2016, 34, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Sárközy, M.; Varga, Z.; Gáspár, R.; Szűcs, G.; Kovács, M.G.; Kovács, Z.Z.A.; Dux, L.; Kahán, Z.; Csont, T. Pathomechanisms and therapeutic opportunities in radiation-induced heart disease: From bench to bedside. Clin. Res. Cardiol. 2021, 110, 507–531. [Google Scholar] [CrossRef] [PubMed]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 22016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef] [PubMed]
- Rassaf, T.; Committee for Clinical Cardiovascular Medicine of the German Cardiac Society; Totzeck, M.; Backs, J.; Bokemeyer, C.; Hallek, M.; Hilfiker-Kleiner, D.; Hochhaus, A.; Lüftner, D.; Müller, O.J.; et al. Onco-Cardiology: Consensus Paper of the German Cardiac Society, the German Society for Pediatric Cardiology and Congenital Heart Defects and the German Society for Hematology and Medical Oncology. Clin. Res. Cardiol. 2020, 109, 1197–1222. [Google Scholar] [CrossRef]
- Andratschke, N.; Maurer, J.; Molls, M.; Trott, K.-R. Late radiation-induced heart disease after radiotherapy. Clinical importance, radiobiological mechanisms and strategies of prevention. Radiother. Oncol. 2011, 100, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Taunk, N.; Haffty, B.G.; Kostis, J.B.; Egoyal, S. Radiation-Induced Heart Disease: Pathologic Abnormalities and Putative Mechanisms. Front. Oncol. 2015, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapio, S. Pathology and biology of radiation-induced cardiac disease. J. Radiat. Res. 2016, 57, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Zeng, Y. Does angiotensin II–aldosterone have a role in radiation-induced heart disease? Med. Hypotheses 2009, 72, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Wu, R. Expression of Angiotensin II and Aldosterone in Radiation-induced Lung Injury. Cancer Biol. Med. 2012, 9, 254–260. [Google Scholar] [CrossRef]
- Pinter, M.; Kwanten, W.J.; Jain, R.K. Renin–Angiotensin System Inhibitors to Mitigate Cancer Treatment–Related Adverse Events. Clin. Cancer Res. 2018, 24, 3803–3812. [Google Scholar] [CrossRef] [Green Version]
- Gulati, G.; Heck, S.L.; Ree, A.H.; Hoffmann, P.; Schulz-Menger, J.; Fagerland, M.W.; Gravdehaug, B.; Von Knobelsdorff-Brenkenhoff, F.; Bratland, Å.; Storås, T.H.; et al. Prevention of cardiac dysfunction during adjuvant breast cancer therapy (PRADA): A 2 × 2 factorial, randomized, placebo-controlled, double-blind clinical trial of candesartan and metoprolol. Eur. Heart J. 2016, 37, 1671–1680. [Google Scholar] [CrossRef] [Green Version]
- Sio, T.T.; Liang, J.J.; Chang, K.; Jayakrishnan, R.; Novotny, P.J.; Prasad, A.; Miller, R.C. Dosimetric Correlate of Cardiac-Specific Survival Among Patients Undergoing Coronary Artery Stenting After Thoracic Radiotherapy for Cancer. Am. J. Clin. Oncol. 2017, 40, 133–139. [Google Scholar] [CrossRef]
- Ahamed, J.; Laurence, J. Role of Platelet-Derived Transforming Growth Factor-β1 and Reactive Oxygen Species in Radiation-Induced Organ Fibrosis. Antioxid. Redox Signal. 2017, 27, 977–988. [Google Scholar] [CrossRef]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Boerma, M. Potential Targets for Intervention in Radiation-Induced Heart Disease. Curr. Drug Targets 2010, 11, 1405–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froogh, G.; Pinto, J.T.; Le, Y.; Kandhi, S.; Aleligne, Y.; Huang, A.; Sun, D. Chymase-dependent production of angiotensin II: An old enzyme in old hearts. Am. J. Physiol. Circ. Physiol. 2017, 312, H223–H231. [Google Scholar] [CrossRef]
- Liu, P.; Sun, M.; Sader, S. Matrix metalloproteinases in cardiovascular disease. Can. J. Cardiol. 2006, 22, 25B–30B. [Google Scholar] [CrossRef] [Green Version]
- Saadat, S.; Noureddini, M.; Mahjoubin-Tehran, M.; Nazemi, S.; Shojaie, L.; Aschner, M.; Maleki, B.; Abbasi-Kolli, M.; Moghadam, H.R.; Alani, B.; et al. Pivotal Role of TGF-β/Smad Signaling in Cardiac Fibrosis: Non-coding RNAs as Effectual Players. Front. Cardiovasc. Med. 2021, 7, 256. [Google Scholar] [CrossRef] [PubMed]
- Finnson, K.; Almadani, Y.; Philip, A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: Mechanisms and targets. Semin. Cell Dev. Biol. 2020, 101, 115–122. [Google Scholar] [CrossRef]
- Ye, S.; Luo, W.; Khan, Z.A.; Wu, G.; Xuan, L.; Shan, P.; Lin, K.; Chen, T.; Wang, J.; Hu, X.; et al. Celastrol Attenuates Angiotensin II–Induced Cardiac Remodeling by Targeting STAT3. Circ. Res. 2020, 126, 1007–1023. [Google Scholar] [CrossRef]
- O’Donnell, L.; O’Neill, T.; Toner, M.; O’Briain, S.; Graham, I. Myocardial hypertrophy, fibrosis and infarction following exposure of the heart to radiation for Hodgkin’s disease. Postgrad. Med. J. 1986, 62, 1055–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monceau, V.; Llach, A.; Azria, D.; Bridier, A.; Petit, B.; Mazevet, M.; Strup-Perrot, C.; To, T.-H.-V.; Calmels, L.; Germaini, M.-M.; et al. Epac contributes to cardiac hypertrophy and amyloidosis induced by radiotherapy but not fibrosis. Radiother. Oncol. 2014, 111, 63–71. [Google Scholar] [CrossRef]
- Saiki, H.; Moulay, G.; Guenzel, A.J.; Liu, W.; Decklever, T.D.; Classic, K.L.; Pham, L.; Chen, H.H.; Burnett, J.C.; Russell, S.J.; et al. Experimental cardiac radiation exposure induces ventricular diastolic dysfunction with preserved ejection fraction. Am. J. Physiol. Circ. Physiol. 2017, 313, H392–H407. [Google Scholar] [CrossRef] [Green Version]
- Cuomo, J.R.; Sharma, G.K.; Conger, P.D.; Weintraub, N.L. Novel concepts in radiation-induced cardiovascular disease. World J. Cardiol. 2016, 8, 504–519. [Google Scholar] [CrossRef]
- Wang, H.; Wei, J.; Zheng, Q.; Meng, L.; Xin, Y.; Yin, X.; Jiang, X. Radiation-induced heart disease: A review of classification, mechanism and prevention. Int. J. Biol. Sci. 2019, 15, 2128–2138. [Google Scholar] [CrossRef] [Green Version]
- Hardy, D.; Liu, C.-C.; Cormier, J.N.; Xia, R.; Du, X.L. Cardiac toxicity in association with chemotherapy and radiation therapy in a large cohort of older patients with non-small-cell lung cancer. Ann. Oncol. 2010, 21, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- van den Bogaard, V.A.B.; Ta, B.D.P.; van der Schaaf, A.; Bouma, A.B.; Middag, A.M.H.; Bantema-Joppe, E.J.; Van Dijk, L.V.; Van Dijk-Peters, F.B.J.; Marteijn, L.A.W.; de Bock, G.H.; et al. Validation and Modification of a Prediction Model for Acute Cardiac Events in Patients With Breast Cancer Treated With Radiotherapy Based on Three-Dimensional Dose Distributions to Cardiac Substructures. J. Clin. Oncol. 2017, 35, 1171–1178. [Google Scholar] [CrossRef]
- Boerma, M.; Roberto, K.A.; Hauer-Jensen, M. Prevention and Treatment of Functional and Structural Radiation Injury in the Rat Heart by Pentoxifylline and Alpha-Tocopherol. Int. J. Radiat. Oncol. 2008, 72, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiscsatári, L.; Sárközy, M.; Kővári, B.P.; Varga, Z.; Gömöri, K.; Morvay, N.; Leprán, I.; Hegyesi, H.; Fábián, G.; Cserni, B.; et al. High-dose Radiation Induced Heart Damage in a Rat Model. In Vivo 2016, 30, 623–631. [Google Scholar]
- Sárközy, M.; Gáspár, R.; Zvara, Á.; Kiscsatári, L.; Varga, Z.; Kővári, B.; Kovács, M.G.; Szűcs, G.; Fábián, G.; Diószegi, P.; et al. Selective Heart Irradiation Induces Cardiac Overexpression of the Pro-hypertrophic miR-212. Front. Oncol. 2019, 9, 598. [Google Scholar] [CrossRef] [Green Version]
- Boerma, M.; Wang, J.; Kulkarni, A.; Roberto, K.A.; Qiu, X.; Kennedy, R.H.; Hauer-Jensen, M. Influence of Endothelin 1 Receptor Inhibition on Functional, Structural and Molecular Changes in the Rat Heart after Irradiation. Radiat. Res. 2008, 170, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Meléndez, G.C.; McLarty, J.L.; Levick, S.; Du, Y.; Janicki, J.S.; Brower, G. Interleukin 6 Mediates Myocardial Fibrosis, Concentric Hypertrophy, and Diastolic Dysfunction in Rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Schultz-Hector, S. Radiation-induced Heart Disease: Review of Experimental Data on Dose Reponse and Pathogenesis. Int. J. Radiat. Biol. 1992, 61, 149–160. [Google Scholar] [CrossRef]
- Farhood, B.; Khodamoradi, E.; Hoseini-Ghahfarokhi, M.; Motevaseli, E.; Mahyari, H.M.; Musa, A.E.; Najafi, M. TGF-β in radiotherapy: Mechanisms of tumor resistance and normal tissues injury. Pharmacol. Res. 2020, 155, 104745. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef]
- Miyazaki, M.; Takai, S. Tissue Angiotensin II Generating System by Angiotensin-Converting Enzyme and Chymase. J. Pharmacol. Sci. 2006, 100, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Company, C.; Piqueras, L.; Abu Nabah, Y.N.; Escudero, P.; Blanes, J.I.; Jose, P.J.; Morcillo, E.J.; Sanz, M.-J. Contributions of ACE and mast cell chymase to endogenous angiotensin II generation and leucocyte recruitment in vivo. Cardiovasc. Res. 2011, 92, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Boerma, M.; Zurcher, C.; Esveldt, I.; Schutte-Bart, C.; Wondergem, J. Histopathology of ventricles, coronary arteries and mast cell accumulation in transverse and longitudinal sections of the rat heart after irradiation. Oncol. Rep. 2004, 12, 213–219. [Google Scholar] [CrossRef]
- Yarom, R.; Harper, I.S.; Wynchank, S.; Van Schalkwyk, D.; Madhoo, J.; Williams, K.; Salie, R.; Genade, S.; Lochner, A. Effect of Captopril on Changes in Rats’ Hearts Induced by Long-Term Irradiation. Radiat. Res. 1993, 133, 187–197. [Google Scholar] [CrossRef]
- Schultz, J.E.J.; Witt, S.A.; Glascock, B.J.; Nieman, M.L.; Reiser, P.J.; Nix, S.L.; Kimball, T.R.; Doetschman, T. TGF-β1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Investig. 2002, 109, 787–796. [Google Scholar] [CrossRef]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Böhm, M.; Nickenig, G. Interleukin-6 Induces Oxidative Stress and Endothelial Dysfunction by Overexpression of the Angiotensin II Type 1 Receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Miguel-Carrasco, J.L.; Beaumont, J.; José, G.S.; Moreno, M.; López, B.; Gonzalez, A.; Zalba, G.; Díez, J.; Fortuño, A.; Ravassa, S. Mechanisms underlying the cardiac antifibrotic effects of losartan metabolites. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Peng, Z.; Zu, C.; Ma, J.; Lu, S.; Zhong, J.; Zhang, S. Losartan Attenuates Myocardial Endothelial-To-Mesenchymal Transition in Spontaneous Hypertensive Rats via Inhibiting TGF-β/Smad Signaling. PLoS ONE 2016, 11, e0155730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Elimban, V.; Xu, Y.J.; Zhang, M.; Nijjar, M.S.; Dhalla, N.S. Alterations of Cardiac ERK1/2 Expression and Activity Due to Volume Overload Were Attenuated by the Blockade of RAS. J. Cardiovasc. Pharmacol. Ther. 2010, 15, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhang, J.; Wang, H.; Gao, P.; Singh, M.; Shen, K.; Fang, N. Angiotensin II downregulates catalase expression and activity in vascular adventitial fibroblasts through an AT1R/ERK1/2-dependent pathway. Mol. Cell. Biochem. 2011, 358, 21–29. [Google Scholar] [CrossRef]
- Xiong, W.; Meisinger, T.; Knispel, R.; Worth, J.M.; Baxter, B.T. MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ. Res. 2012, 110, e92–e101. [Google Scholar] [CrossRef] [Green Version]
- Asano, K.; Dutcher, D.L.; Port, J.D.; Minobe, W.A.; Tremmel, K.D.; Roden, R.L.; Bohlmeyer, T.J.; Bush, E.W.; Jenkin, M.J.; Abraham, W.T.; et al. Selective Downregulation of the Angiotensin II AT 1 -Receptor Subtype in Failing Human Ventricular Myocardium. Circulation 1997, 95, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Hermans, M.A.W.; Van Lennep, J.E.R.; Van Daele, P.L.A.; Bot, I. Mast Cells in Cardiovascular Disease: From Bench to Bedside. Int. J. Mol. Sci. 2019, 20, 3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Gao, W.; Xiao, K.; Liu, Q.; Jia, R. Interaction between interleukin-6 and angiotensin II receptor 1 in the hypothalamic paraventricular nucleus contributes to progression of heart failure. Mol. Med. Rep. 2017, 15, 4259–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Louie, A.; Higginson, D.; Palma, D.; Colaco, R.; Sahgal, A. Stereotactic Radiosurgery and Stereotactic Body Radiotherapy in the Management of Oligometastatic Disease. Clin. Oncol. 2020, 32, 713–727. [Google Scholar] [CrossRef]
- Sárközy, M.; Márványkövi, F.M.; Szűcs, G.; Kovács, Z.Z.A.; Szabó, M.R.; Gáspár, R.; Siska, A.; Kővári, B.; Cserni, G.; Földesi, I.; et al. Ischemic preconditioning protects the heart against ischemia-reperfusion injury in chronic kidney disease in both males and females. Biol. Sex Differ. 2021, 12, 1–20. [Google Scholar] [CrossRef]
- Kovács, Z.Z.A.; Szűcs, G.; Freiwan, M.; Kovács, M.G.; Márványkövi, F.M.; Dinh, H.; Siska, A.; Farkas, K.; Kovács, F.; Kriston, A.; et al. Comparison of the antiremodeling effects of losartan and mirabegron in a rat model of uremic cardiomyopathy. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Sárközy, M.; Gáspár, R.; Zvara, Á.; Siska, A.; Kővári, B.P.; Szűcs, G.; Márványkövi, F.; Kovács, M.G.; Diószegi, P.; Bodai, L.; et al. Chronic kidney disease induces left ventricular overexpression of the pro-hypertrophic microRNA-212. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (Unit) | Week 1 | Week 3 | Week 15 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | RT | RT Los | Control | RT | RT Los | Control | RT | RT Los | |
| Body weight at the beginning (g) | 233 ± 8 | 233 ± 5 | 232 ± 4 * | 282 ± 5 | 281 ± 7 | 282 ± 6 | 261 ± 3 | 263 ± 4 | 261 ± 5 |
| Body weight at the endpoint (g) | 269 ± 7 | 258 ± 6 | 251 ± 5 * | 382 ± 9.57 | 338 ± 5.22 * | 344 ± 8.8 * | 525 ± 14 | 377 ± 29 * | 414 ± 17 * |
| Tibia length (cm) | 3.39 ± 0.06 | 3.36 ± 0.03 | 3.33 ± 0.04 | 3.83 ± 0.07 | 3.73 ± 0.05 | 3.86 ± 0.09 | 4.5 ± 0.04 | 4.25 ± 0.08 * | 4.28 ± 0.04 * |
| Heart weight (g) | 0.91 ± 0.02 | 0.87 ± 0.03 | 0.87 ± 0.02 | 1.16 ± 0.03 | 1.1 ± 0.04 | 1.06 ± 0.05 | 2.24 ± 0.11 | 1.75 ± 0.04 * | 1.63 ± 0.11 * |
| Left ventricle weight (g) | 0.62 ± 0.01 | 0.57 ± 0.02 * | 0.55 ± 0.02 * | 0.77 ± 0.02 | 0.69 ± 0.02 * | 0.66 ± 0.02 * | 1.3 ± 0.05 | 0.99 ± 0.03 * | 0.88 ± 0.06 * |
| Right ventricle weight (g) | 0.17 ± 0.01 | 0.17 ± 0.01 | 0.18 ± 0.01 | 0.24 ± 0.01 | 0.25 ± 0.01 | 0.23 ± 0.02 | 0.35 ± 0.01 | 0.33 ± 0.01 | 0.32 ± 0.02 |
| Lung weight (g) | 1.3 ± 0.02 | 1.29 ± 0.05 | 1.3 ±0.03 | 1.7 ± 0.12 | 2.98 ± 0.26 * | 2.47 ± 0.29 * | 1.64 ± 0.04 | 1.88 ± 0.32 | 1.50 ± 0.1 |
| Parameter (Unit) | Week 1 | Week 3 | Week 15 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | RT | RT Los | Control | RT | RT Los | Control | RT | RT Los | |
| White blood cell counts (109/L) | 5.79 ± 0.43 | 6.18 ± 0.7 | 7.15 ± 0.36 * | 4.44 ± 0.56 | 6.77 ± 0.8 * | 7.16 ± 0.7 * | 6.09 ± 0.58 | 6 ± 0.39 | 6.51 ± 0.52 |
| Platelet counts (109/L) | 569 ± 8 | 587 ± 20 | 643 ± 33 | 559 ± 27 | 646 ± 35 | 673 ± 31 * | 636 ± 17 | 586 ± 67 | 736 ± 53 |
| Red blood cell counts (1012/L) | 6.69 ± 0.11 | 7.37 ± 0.29 * | 7.74 ± 0.2 * | 7.54 ± 0.15 | 7.54 ± 0.18 | 7.95 ± 0.18 | 8.30 ± 0.15 | 9.16 ± 0.44 | 9.50 ± 0.3 * |
| Hemoglobin (g/L) | 130 ± 3 | 143 ± 5 | 146 ± 3 * | 143 ± 3 | 142 ± 3 | 152 ± 3 | 146 ± 2 | 165 ± 8 * | 175 ± 7 * |
| Hematocrit (L/L) | 0.41 ± 0.01 | 0.44 ± 0.02 | 0.45 ± 0.01 | 0.44 ± 0.01 | 0.45 ± 0.01 | 0.47 ± 0.01 | 0.43 ± 0.01 | 0.49 ± 0.02 * | 0.50 ± 0.02 * |
| Parameter (Unit) | Week 1 | Week 3 | Week 15 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | RT | RT Los | Control | RT | RT Los | Control | RT | RT Los | |
| E velocity (m/s) | 1.00 ± 0.04 | 0.94 ± 0.05 | 0.90 ± 0.04 | 1.00 ± 0.04 | 1.05 ± 0.03 | 0.93 ± 0.04 # | 1.01 ± 0.02 | 0.81 ± 0.10 * | 0.75 ± 0.05 * |
| e’ (m/s) | 0.060 ± 0.002 | 0.053 ± 0.001 * | 0.063 ± 0.005 | 0.066 ± 0.006 | 0.038 ± 0.002 * | 0.056 ± 0.005 # | 0.077 ± 0.007 | 0.035 ± 0.002 * | 0.042 ± 0.002 * |
| SWTs (mm) | 2.70 ± 0.10 | 2.84 ± 0.16 | 2.91 ± 0.10 | 3.14 ± 0.15 | 3.47 ± 0.12 | 3.18 ± 0.14 | 3.58 ± 0.12 | 4.64 ± 0.17 * | 4.29 ± 0.32 |
| SWTd (mm) | 1.61 ± 0.10 | 1.60 ± 0.09 | 1.90 ± 0.06 | 1.79 ± 0.12 | 1.73 ± 0.06 | 1.77 ± 0.14 | 1.83 ± 0.04 | 3.23 ± 0.33* | 3.12 ± 0.28 |
| AWTs (mm) | 2.64 ± 0.17 | 2.61 ± 0.09 | 2.92 ± 0.20 | 2.85 ± 0.17 | 3.19 ± 0.18 | 2.89 ± 0.27 | 3.3 ± 0.23 | 4.56 ± 0.28 * | 4.51 ± 0.17 * |
| AWTd (mm) | 1.63 ± 0.17 | 1.51 ± 0.07 | 1.75 ± 0.09 | 1.61 ± 0.07 | 1.67 ± 0.11 | 1.67 ± 0.09 | 1.99 ± 0.13 | 3.14 ± 0.31 * | 3.04 ± 0.17 * |
| IWTs (mm) | 2.24 ± 0.13 | 2.47 ± 0.08 | 2.70 ± 0.10 * | 2.67 ± 0.07 | 3.32 ± 0.17 * | 2.91 ± 0.23 | 3.44 ± 0.25 | 4.56 ± 0.09 * | 3.69 ± 0.31 |
| IWTd (mm) | 1.48 ± 0.03 | 1.51 ± 0.09 | 1.67 ± 0.09 | 1.73 ± 0.07 | 1.98 ± 0.07 * | 1.90 ± 0.15 | 2.01 ± 0.12 | 3.67 ± 0.12 * | 2.72 ± 0.26 *# |
| LVEDD (mm) | 7.73 ± 0.22 | 7.34 ± 0.27 | 6.98 ± 0.16 * | 8.33 ± 0.2 | 7.4 ± 0.14 * | 7.11 ± 0.29 * | 8.44 ± 0.33 | 4.85 ± 0.17 * | 5.67 ± 0.29 *# |
| LVESD (mm) | 4.75 ± 0.21 | 4.05 ± 0.28 | 3.68 ± 0.21 * | 4.83 ± 0.17 | 3.40 ± 0.20 * | 3.25 ± 0.25 * | 4.26 ± 0.55 | 0.98 ± 0.15 * | 1.63 ± 0.16 * |
| FS (%) | 39 ± 2 | 45 ± 2 * | 48 ± 2 * | 42 ± 2 | 54 ± 2 * | 54 ± 2 * | 50 ± 5 | 80 ± 3 * | 71 ± 3 * |
| EF (%) | 56 ± 2 | 57 ± 3 | 52 ± 1 | 53 ± 2 | 53 ± 3 | 53 ± 1 | 53 ± 2 | 57 ± 2 | 55 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, M.G.; Kovács, Z.Z.A.; Varga, Z.; Szűcs, G.; Freiwan, M.; Farkas, K.; Kővári, B.; Cserni, G.; Kriston, A.; Kovács, F.; et al. Investigation of the Antihypertrophic and Antifibrotic Effects of Losartan in a Rat Model of Radiation-Induced Heart Disease. Int. J. Mol. Sci. 2021, 22, 12963. https://doi.org/10.3390/ijms222312963
Kovács MG, Kovács ZZA, Varga Z, Szűcs G, Freiwan M, Farkas K, Kővári B, Cserni G, Kriston A, Kovács F, et al. Investigation of the Antihypertrophic and Antifibrotic Effects of Losartan in a Rat Model of Radiation-Induced Heart Disease. International Journal of Molecular Sciences. 2021; 22(23):12963. https://doi.org/10.3390/ijms222312963
Chicago/Turabian StyleKovács, Mónika Gabriella, Zsuzsanna Z. A. Kovács, Zoltán Varga, Gergő Szűcs, Marah Freiwan, Katalin Farkas, Bence Kővári, Gábor Cserni, András Kriston, Ferenc Kovács, and et al. 2021. "Investigation of the Antihypertrophic and Antifibrotic Effects of Losartan in a Rat Model of Radiation-Induced Heart Disease" International Journal of Molecular Sciences 22, no. 23: 12963. https://doi.org/10.3390/ijms222312963
APA StyleKovács, M. G., Kovács, Z. Z. A., Varga, Z., Szűcs, G., Freiwan, M., Farkas, K., Kővári, B., Cserni, G., Kriston, A., Kovács, F., Horváth, P., Földesi, I., Csont, T., Kahán, Z., & Sárközy, M. (2021). Investigation of the Antihypertrophic and Antifibrotic Effects of Losartan in a Rat Model of Radiation-Induced Heart Disease. International Journal of Molecular Sciences, 22(23), 12963. https://doi.org/10.3390/ijms222312963