
Genome-Wide Analysis of WRKY Gene Family and the Dynamic Responses of Key WRKY Genes Involved in Ostrinia furnacalis Attack in Zea mays


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification and Characterization of WRKY Genes in Maize
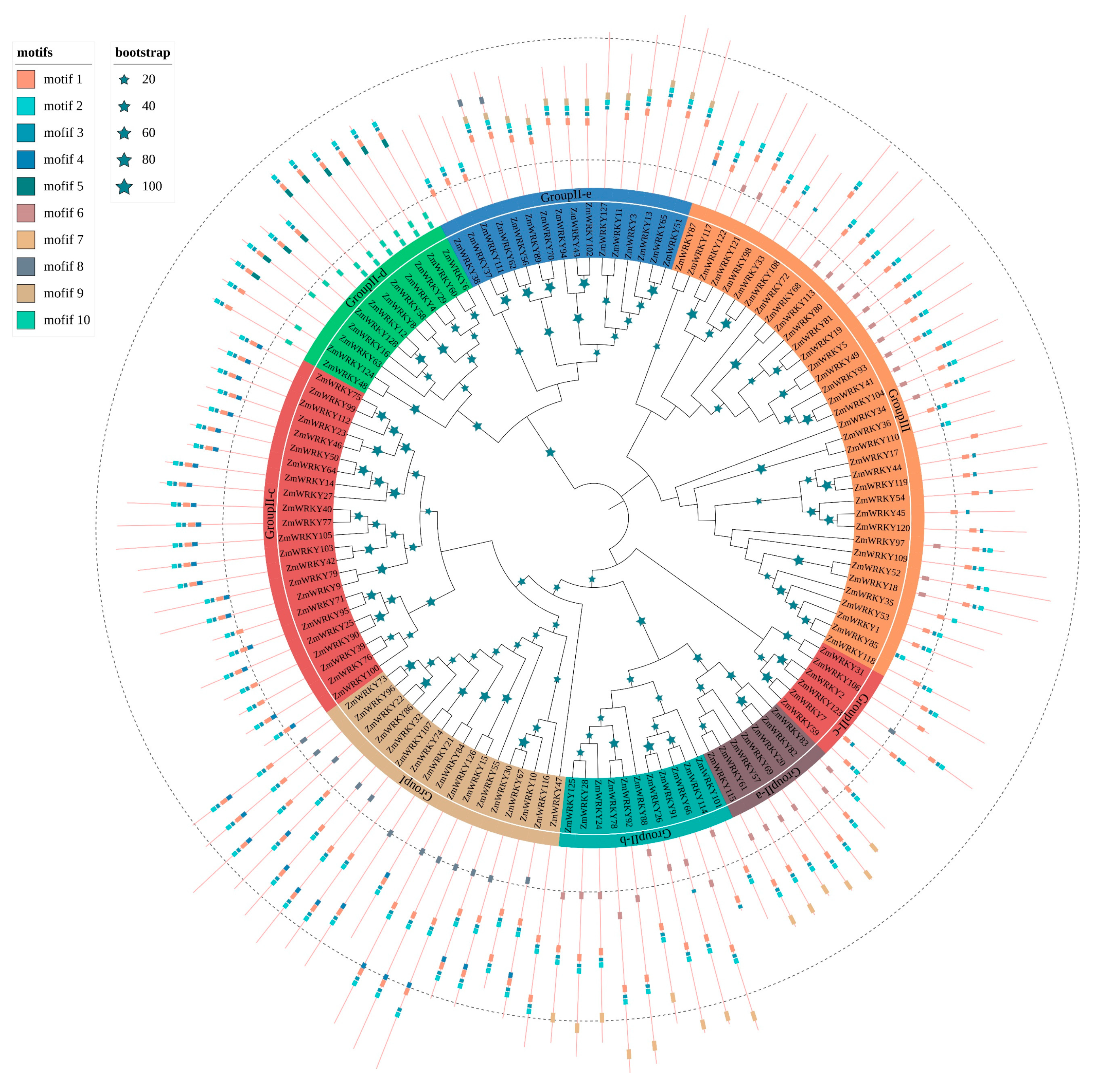
2.2. Classification, Phylogenetic Analysis, and Motif Composition of the ZmWRKY Genes
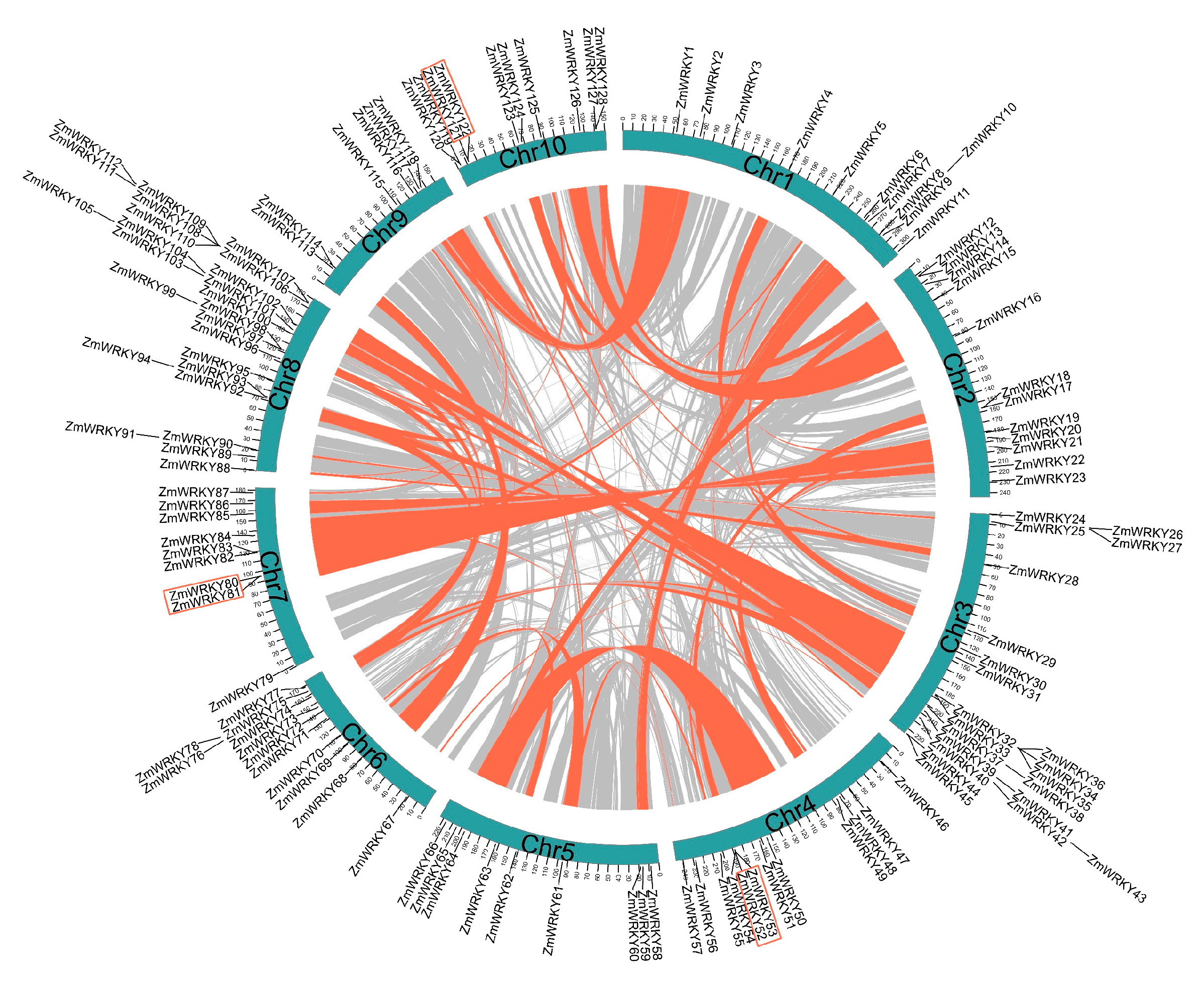
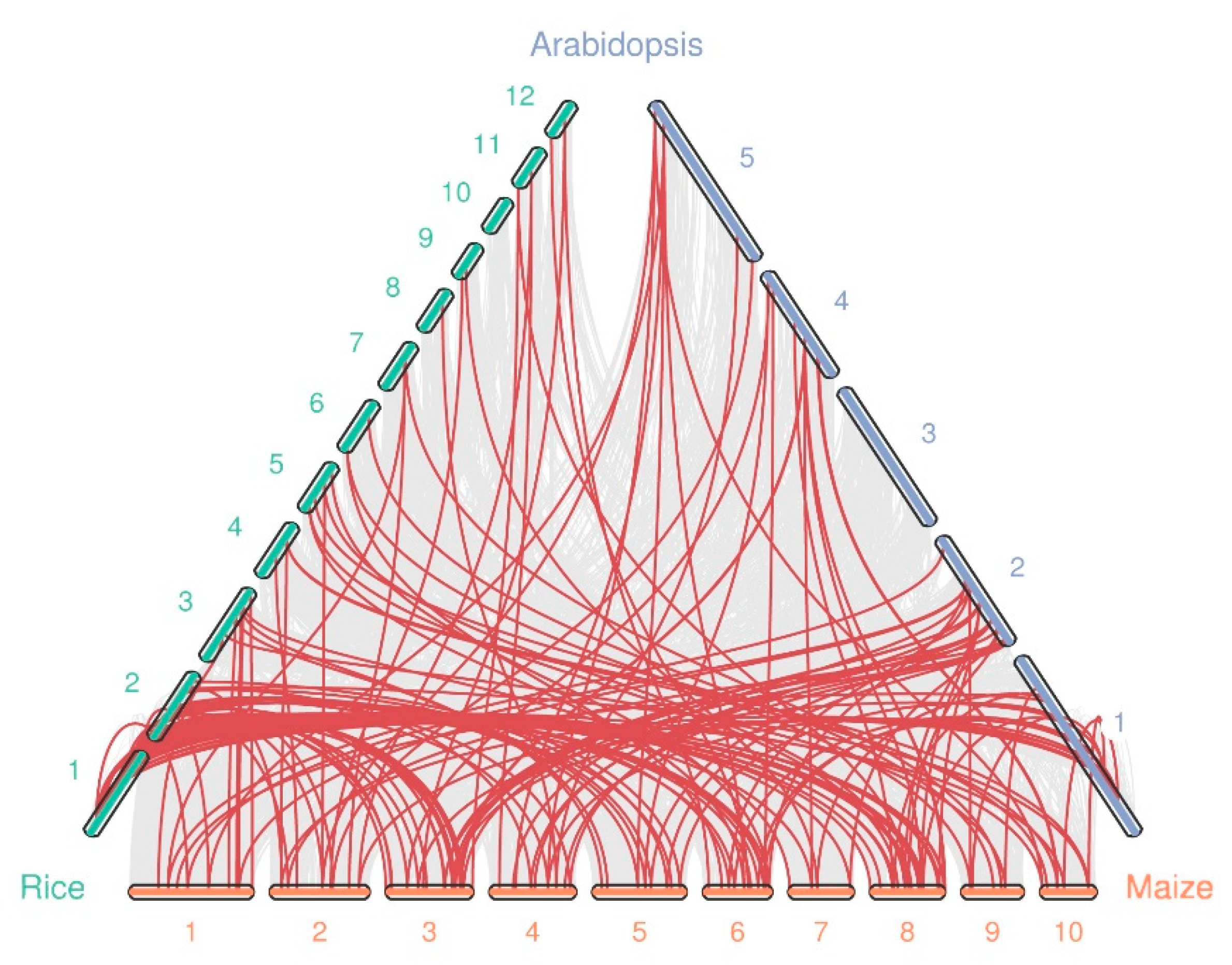
2.3. Chromosomal Location, Gene Duplication, and Collinearity Analysis of ZmWRKYs
2.4. Expression of ZmWRKY Genes in Response to O. furnacalis Feeding
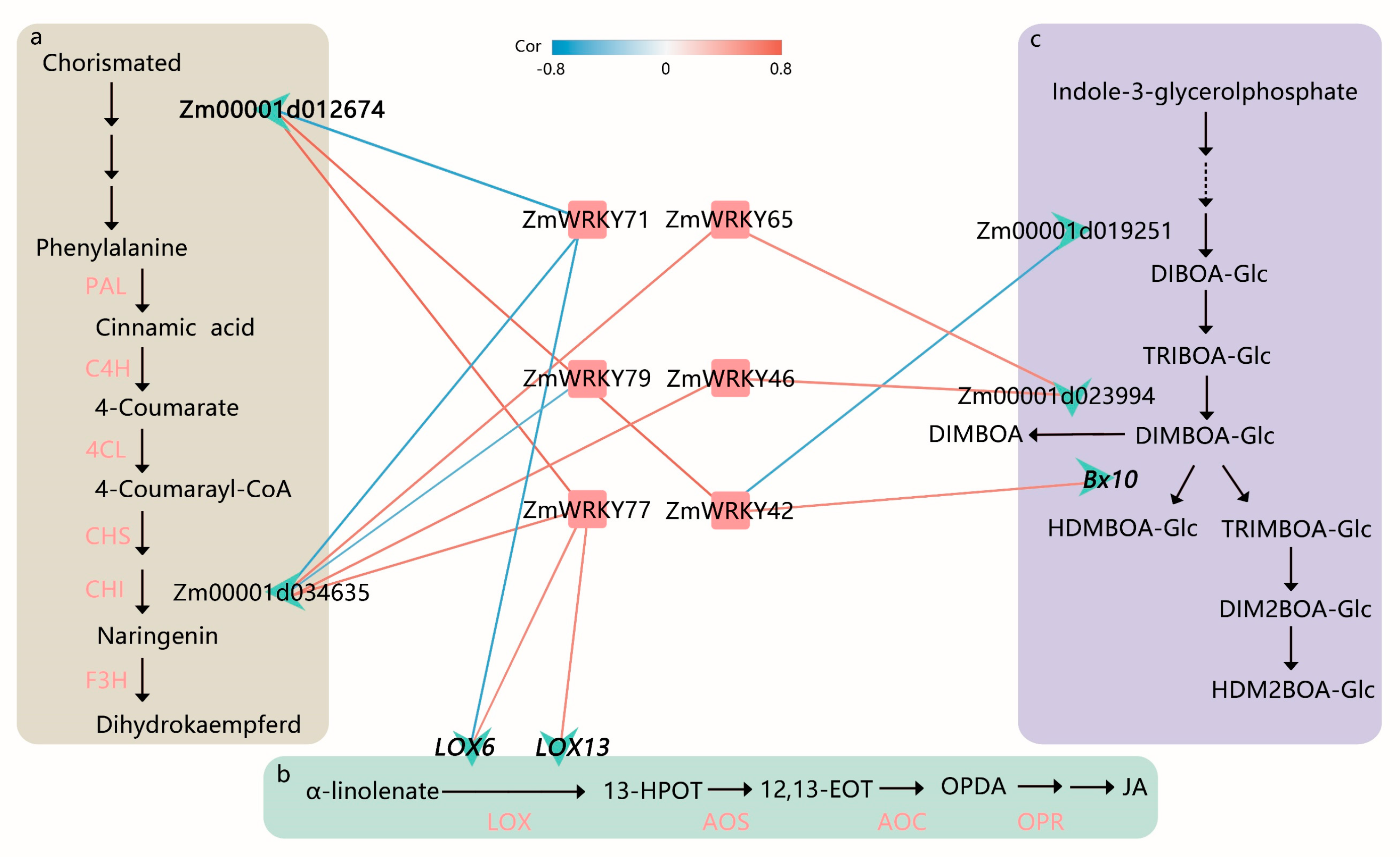
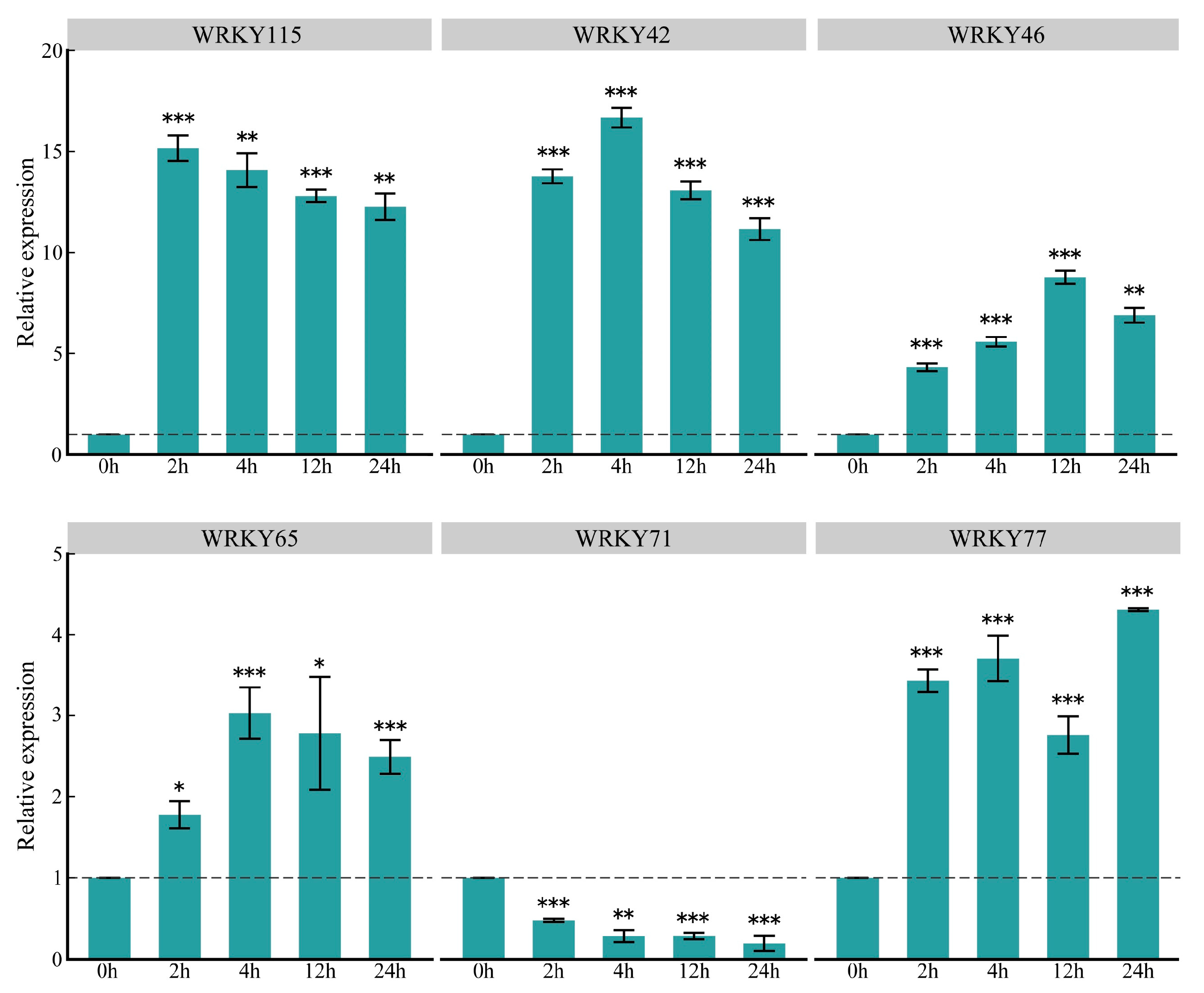
2.5. Prediction of Transcriptional Regulation and Differential Expression Analysis in Key ZmWRKY Genes
3. Discussion
4. Materials and Methods
4.1. Identification and Sequence Analysis of WRKY Genes in Maize
4.2. Sequence Alignment, Phylogenetic and Conserved Motifs Analysis
4.3. Chromosomal Location, Gene Duplication, and Synteny Analysis
4.4. RNA-Seq Analysis
4.5. Transcriptional Regulation Prediction and Functional Annotation
4.6. Plant Materials and qPCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, J.; Baldwin, I.T. New Insights into Plant Responses to the Attack from Insect Herbivores. Annu. Rev. Genet. 2010, 44, 1–24. [Google Scholar] [CrossRef]
- Spoel, S.H.; Johnson, J.S.; Dong, X. Regulation of tradeoffs between plant defenses against pathogens with different lifestyles. Proc. Natl. Acad. Sci. USA 2007, 104, 18842–18847. [Google Scholar] [CrossRef] [Green Version]
- Woldemariam, M.G.; Baldwin, I.T.; Galis, I. Transcriptional regulation of plant inducible defenses against herbivores: A mini-review. J. Plant Interact. 2011, 6, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, M.; Oelmüller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef] [Green Version]
- Romani, F.; Moreno, J.E. Molecular mechanisms involved in functional macroevolution of plant transcription factors. New Phytol. 2021, 230, 1345–1353. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY Transcription Factors: Molecular Regulation and Stress Responses in Plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Hu, Y.; Vannozzi, A.; Wu, K.; Cai, H.; Qin, Y.; Mullis, A.; Lin, Z.; Zhang, L. The WRKY Transcription Factor Family in Model Plants and Crops. Crit. Rev. Plant Sci. 2017, 36, 311–335. [Google Scholar] [CrossRef]
- Hu, L.; Ye, M.; Li, R.; Lou, Y. OsWRKY53, a versatile switch in regulating herbivore-induced defense responses in rice. Plant Signal. Behav. 2016, 11, e1169357. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hao, J.; Chen, X.; Hao, Z.; Wang, X.; Lou, Y.; Peng, Y.; Guo, Z. Overexpression of rice WRKY89 enhances ultraviolet B tolerance and disease resistance in rice plants. Plant Mol. Biol. 2007, 65, 799–815. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.K.; Atamian, H.S.; Kaloshian, I.; Eulgem, T. WRKY72-type transcription factors contribute to basal immunity in tomato and Arabidopsis as well as gene-for-gene resistance mediated by the tomato R gene Mi-1. Plant J. 2010, 63, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Van Eck, L.; Schultz, T.; Leach, J.E.; Scofield, S.R.; Peairs, F.B.; Botha, A.-M.; Lapitan, N.L. Virus-induced gene silencing of WRKY53 and an inducible phenylalanine ammonia-lyase in wheat reduces aphid resistance. Plant Biotechnol. J. 2010, 8, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Atamian, H.S.; Eulgem, T.; Kaloshian, I. SlWRKY70 is required for Mi-1-mediated resistance to aphids and nematodes in tomato. Planta 2012, 235, 299–309. [Google Scholar] [CrossRef]
- Ishiguro, S.; Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol. Genet. Genom. 1994, 244, 563–571. [Google Scholar] [CrossRef]
- Xie, T.; Chen, C.; Li, C.; Liu, J.; Liu, C.; He, Y. Genome-wide investigation of WRKY gene family in pineapple: Evolution and expression profiles during development and stress. BMC Genom. 2018, 19, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Li, C.; Wang, H.; Guo, Z. WRKY transcription factors: Evolution, binding, and action. Phytopathol. Res. 2019, 1, 13. [Google Scholar] [CrossRef]
- Li, J.; Xiong, Y.; Li, Y.; Ye, S.; Yin, Q.; Gao, S.; Yang, D.; Yang, M.; Palva, E.T.; Deng, X. Comprehensive Analysis and Functional Studies of WRKY Transcription Factors in Nelumbo nucifera. Int. J. Mol. Sci. 2019, 20, 5006. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Shi, H.; Xia, Z.; Tie, W.; Ding, Z.; Yan, Y.; Wang, W.; Hu, W.; Li, K. Genome-Wide Identification and Expression Analysis of the WRKY Gene Family in Cassava. Front. Plant Sci. 2016, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Lin, Z.; Li, X.; Zhao, Y.; Zhao, B.; Wu, G.; Ma, X.; Wang, H.; Xie, Y.; Li, Q.; et al. Genome-wide selection and genetic improvement during modern maize breeding. Nat. Genet. 2020, 52, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tan, D.; Zhang, L.; Zhang, X.; Han, Z. Phylogenetic analysis and drought-responsive expression profiles of the WRKY transcription factor family in maize. Agric. Gene 2017, 3, 99–108. [Google Scholar] [CrossRef]
- Hu, W.; Ren, Q.; Chen, Y.; Xu, G.; Qian, Y. Genome-wide identification and analysis of WRKY gene family in maize provide insights into regulatory network in response to abiotic stresses. BMC Plant Biol. 2021, 21, 427. [Google Scholar] [CrossRef]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S.; et al. Improved maize reference genome with single-molecule technologies. Nat. Cell Biol. 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Dai, W.; Zhang, C.; Wang, Y.; Wu, M.; Zhao, Y.; Ma, Q.; Xiang, Y.; Cheng, B. The maize WRKY transcription factor ZmWRKY17 negatively regulates salt stress tolerance in transgenic Arabidopsis plants. Planta 2017, 246, 1215–1231. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-T.; Ru, J.-N.; Liu, Y.-W.; Li, M.; Zhao, D.; Yang, J.-F.; Fu, J.-D.; Xu, Z.-S. Maize WRKY Transcription Factor ZmWRKY106 Confers Drought and Heat Tolerance in Transgenic Plants. Int. J. Mol. Sci. 2018, 19, 3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-Y.; He, K.-L.; Zhang, F.; Lu, X.; Babendreier, D. Mass rearing and release of Trichogramma for biological control of insect pests of corn in China. Biol. Control. 2014, 68, 136–144. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, w202–w208. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2019, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Hawkins, C.; Ginzburg, D.; Zhao, K.; Dwyer, W.; Xue, B.; Xu, A.; Rice, S.; Cole, B.; Paley, S.; Karp, P.; et al. Plant Metabolic Network 15: A resource of genome-wide metabolism databases for 126 plants and algae. J. Integr. Plant Biol. 2021, 63, 1888–1905. [Google Scholar] [CrossRef] [PubMed]
- Tzin, V.; Hojo, Y.; Strickler, S.R.; Bartsch, L.J.; Archer, C.M.; Ahern, K.R.; Zhou, S.; Christensen, S.A.; Galis, I.; Mueller, L.A.; et al. Rapid defense responses in maize leaves induced by Spodoptera exigua caterpillar feeding. J. Exp. Bot. 2017, 68, 4709–4723. [Google Scholar] [CrossRef]
- Ülker, B.; Somssich, I.E. WRKY transcription factors: From DNA binding towards biological function. Curr. Opin. Plant Biol. 2004, 7, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Deng, C.; Zhang, Y.; Cheng, Y.; Huo, Q.; Xue, L. The WRKY Transcription Factor Genes in Eggplant (Solanum melongena L.) and Turkey Berry (Solanum torvum Sw.). Int. J. Mol. Sci. 2015, 16, 7608–7626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, H.; Li, W.; Lin, Y.-L.; Gao, L.-Z. Genome-Wide Analysis of WRKY Genes and Their Response to Salt Stress in the Wild Progenitor of Asian Cultivated Rice, Oryza rufipogon. Front. Genet. 2020, 11, 359. [Google Scholar] [CrossRef]
- Dong, J.; Chen, C.; Chen, Z. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol. Biol. 2003, 51, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Zhang, X.; Pang, C.; Song, M.; Wei, H.; Fan, S.; Yu, S. Genome-wide analysis of the WRKY gene family in cotton. Mol. Genet. Genom. 2014, 289, 1103–1121. [Google Scholar] [CrossRef] [PubMed]
- Ning, P.; Liu, C.; Kang, J.; Lv, J. Genome-wide analysis of WRKY transcription factors in wheat (Triticum aestivum L.) and differential expression under water deficit condition. PeerJ 2017, 5, e3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wang, N.; Hu, R.; Xiang, F. Genome-wide identification of soybean WRKY transcription factors in response to salt stress. SpringerPlus 2016, 5, 920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Li, X.; Cheng, L.; Liu, Y.; Wang, H.; Ke, D.; Yuan, H.; Zhang, L.; Wang, L. Genome-Wide Analysis of Soybean JmjC Domain-Containing Proteins Suggests Evolutionary Conservation Following Whole-Genome Duplication. Front. Plant Sci. 2016, 7, 1800. [Google Scholar] [CrossRef] [Green Version]
- Blanc, G.; Wofe, K.H. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 2004, 16, 1679–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangelsen, E.; Kilian, J.; Berendzen, K.W.; Kolukisaoglu, Ü.H.; Harter, K.; Jansson, C.; Wanke, D. Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots. BMC Genom. 2008, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mur, L.A.J.; Prats, E.; Pierre, S.; Hall, M.A.; Hebelstrup, K.H. Integrating nitric oxide into salicylic acid and jasmonic acid/ethylene plant defense pathways. Front. Plant Sci. 2013, 4, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Li, M.; Xiong, Y.; Wu, J.; Da Silva, J.A.T.; Ma, G.; Da Silva, J.T. Genome-Wide Characterization, Expression Profile Analysis of WRKY Family Genes in Santalum album and Functional Identification of Their Role in Abiotic Stress. Int. J. Mol. Sci. 2019, 20, 5676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.-B.; Liu, Y.-Q.; Chen, D.-Y.; Chen, F.-Y.; Fang, X.; Hong, G.-J.; Wang, L.-J.; Wang, J.-W.; Chen, X.-Y. Jasmonate response decay and defense metabolite accumulation contributes to age-regulated dynamics of plant insect resistance. Nat. Commun. 2017, 8, 13925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- War, A.R.; Hussain, B.; Sharma, H.C. Induced resistance in groundnut by jasmonic acid and salicylic acid through alteration of trichome density and oviposition by Helicoverpa armigera (Lepidoptera: Noctuidae). AoB Plants 2013, 5, 053. [Google Scholar] [CrossRef] [Green Version]
- Skibbe, M.; Qu, N.; Galis, I.; Baldwin, I. Induced plant defenses in the natural environment: Nicotiana attenuate WRKY3 and WRKY6 coordinate responses to herbivory. Plant Cell 2008, 20, 1984–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Zhang, J.; Li, J.; Zhou, G.; Wang, Q.; Bian, W.; Erb, M.; Lou, Y. Prioritizing plant defence over growth through WRKY regulation facilitates infestation by non-target herbivores. eLife 2015, 4, e04805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Memelink, J. Jasmonate-responsive transcription factors regulating plant secondary metabolism. Biotechnol. Adv. 2016, 34, 441–449. [Google Scholar] [CrossRef]
- Guo, J.; Qi, J.; He, K.; Wu, J.; Bai, S.; Zhang, T.; Zhao, J.; Wang, Z. The Asian corn borer Ostrinia furnacalis feeding increases the direct and indirect defence of mid-whorl stage commercial maize in the field. Plant Biotechnol. J. 2019, 17, 88–102. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, A.; Ishihara, A.; Hasegawa, M.; Kodama, O.; Iwamura, H. Induced accumulation of 2-hydroxy-4,7-dimethoxy-1,4-benzoxazin-3-one glucoside (HDMBOA-Glc) in maize leaves. Phytochemistry 2001, 56, 669–675. [Google Scholar] [CrossRef]
- Glauser, G.; Marti, G.; Villard, N.; Doyen, G.A.; Wolfender, J.-L.; Turlings, T.C.; Erb, M. Induction and detoxification of maize 1,4-benzoxazin-3-ones by insect herbivores. Plant J. 2011, 68, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.; Schullehner, K.; Dick, R.; Fiesselmann, A.; Gierl, A. Benzoxazinoid biosynthesis, a model for evolution of secondary metabolic pathways in plants. Phytochemistry 2009, 70, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Malook, S.U.; Qi, J.; Hettenhausen, C.; Xu, Y.; Zhang, C.; Zhang, J.; Lu, C.; Li, J.; Wang, L.; Wu, J. The oriental armyworm (Mythimna separata) feeding induces systemic defence responses within and between maize leaves. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180307. [Google Scholar] [CrossRef] [Green Version]
- Mithöfer, A.; Boland, W. Plant Defense Against Herbivores: Chemical Aspects. Annu. Rev. Plant Biol. 2012, 63, 431–450. [Google Scholar] [CrossRef] [Green Version]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, A.; Li, P.; Jiang, J.; Chen, S.; Li, H.; Zeng, J.; Shao, Y.; Zhu, L.; Zhang, Z.; Chen, F. Phylogenetic and Transcription Analysis of Chrysanthemum WRKY Transcription Factors. Int. J. Mol. Sci. 2014, 15, 14442–14455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.-M.; Venugopal, S.; Navarre, D.; Kachroo, A. Low Oleic Acid-Derived Repression of Jasmonic Acid-Inducible Defense Responses Requires the WRKY50 and WRKY51 Proteins. Plant Physiol. 2011, 155, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, W.; De Smet, I.; Lewis, D.R.; Löfke, C.; Jansen, L.; Goeminne, G.; Bossche, R.V.; Karimi, M.; De Rybel, B.; Vanholme, B.; et al. Transcription factor WRKY23 assists auxin distribution patterns during Arabidopsis root development through local control on flavonol biosynthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 1554–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Chen, C.; Fan, B.; Chen, Z. Physical and Functional Interactions between Pathogen-Induced Arabidopsis WRKY18, WRKY40, and WRKY60 Transcription Factors. Plant Cell 2006, 18, 1310–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appel, H.M.; Fescemyer, H.; Ehlting, J.; Weston, D.; Rehrig, E.; Joshi, T.; Xu, D.; Bohlmann, J.; Schultz, J. Transcriptional responses of Arabidopsis thaliana to chewing and sucking insect herbivores. Front. Plant Sci. 2014, 5, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, F.; Bodenhausen, N.; Lassueur, S.; Masclaux, F.G.; Reymond, P. Differential Contribution of Transcription Factors to Arabidopsis thaliana Defense Against Spodoptera littoralis. Front. Plant Sci. 2013, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Anders, S.; Huber, W. Differential analysis of count data-the DESeq2 package. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for Visualization and Analysis of Biological Networks. In Data Mining in Proteomics; Springer: Berlin/Heidelberg, Germany, 2011; Volume 696, pp. 291–303. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Y.; Guo, J.; Zhang, T.; Bai, S.; He, K.; Wang, Z. Genome-Wide Analysis of WRKY Gene Family and the Dynamic Responses of Key WRKY Genes Involved in Ostrinia furnacalis Attack in Zea mays. Int. J. Mol. Sci. 2021, 22, 13045. https://doi.org/10.3390/ijms222313045
Tang Y, Guo J, Zhang T, Bai S, He K, Wang Z. Genome-Wide Analysis of WRKY Gene Family and the Dynamic Responses of Key WRKY Genes Involved in Ostrinia furnacalis Attack in Zea mays. International Journal of Molecular Sciences. 2021; 22(23):13045. https://doi.org/10.3390/ijms222313045
Chicago/Turabian StyleTang, Yin, Jingfei Guo, Tiantao Zhang, Shuxiong Bai, Kanglai He, and Zhenying Wang. 2021. "Genome-Wide Analysis of WRKY Gene Family and the Dynamic Responses of Key WRKY Genes Involved in Ostrinia furnacalis Attack in Zea mays" International Journal of Molecular Sciences 22, no. 23: 13045. https://doi.org/10.3390/ijms222313045