
Design of Biopolymer-Based Interstitial Therapies for the Treatment of Glioblastoma


Abstract
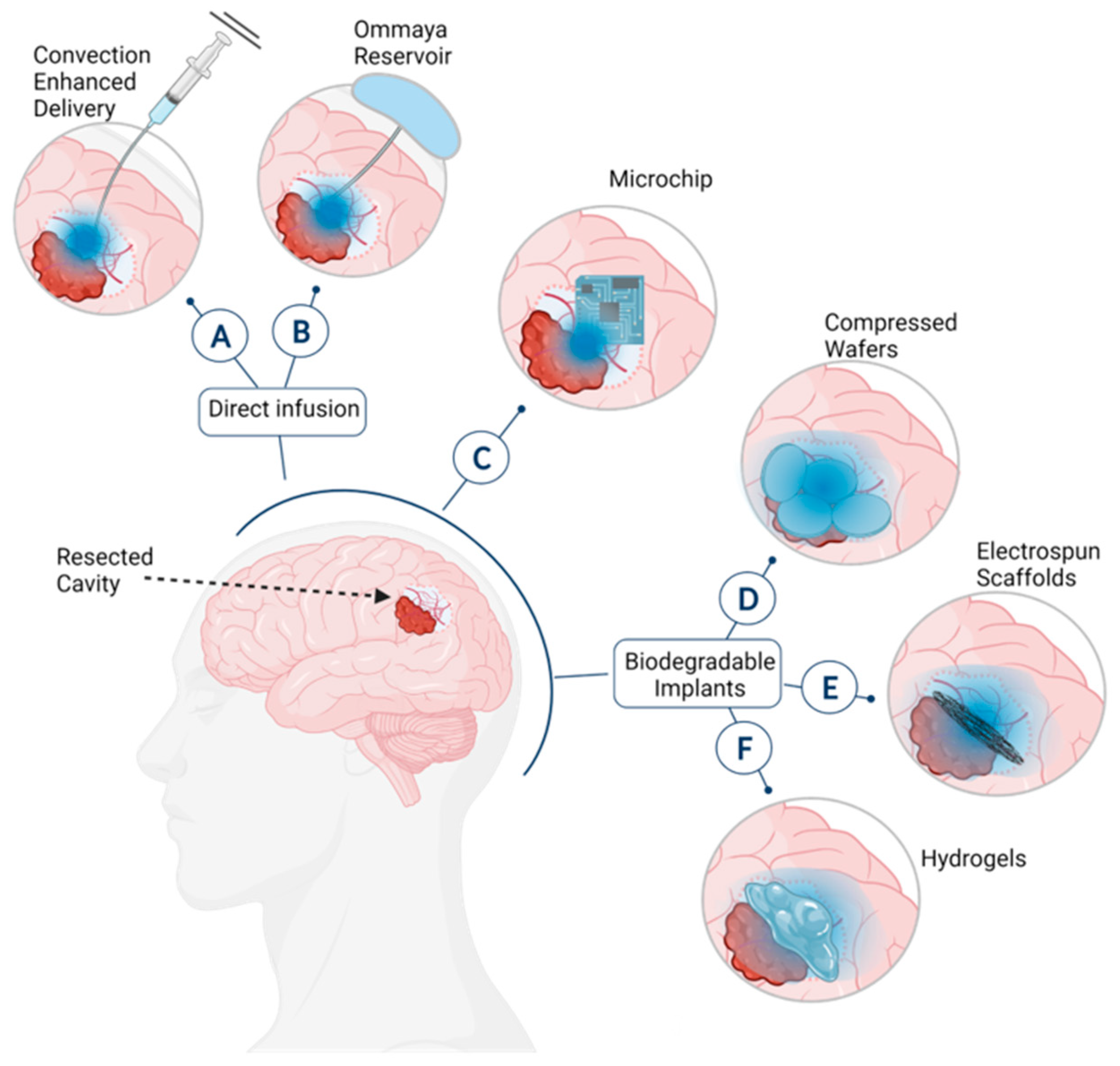
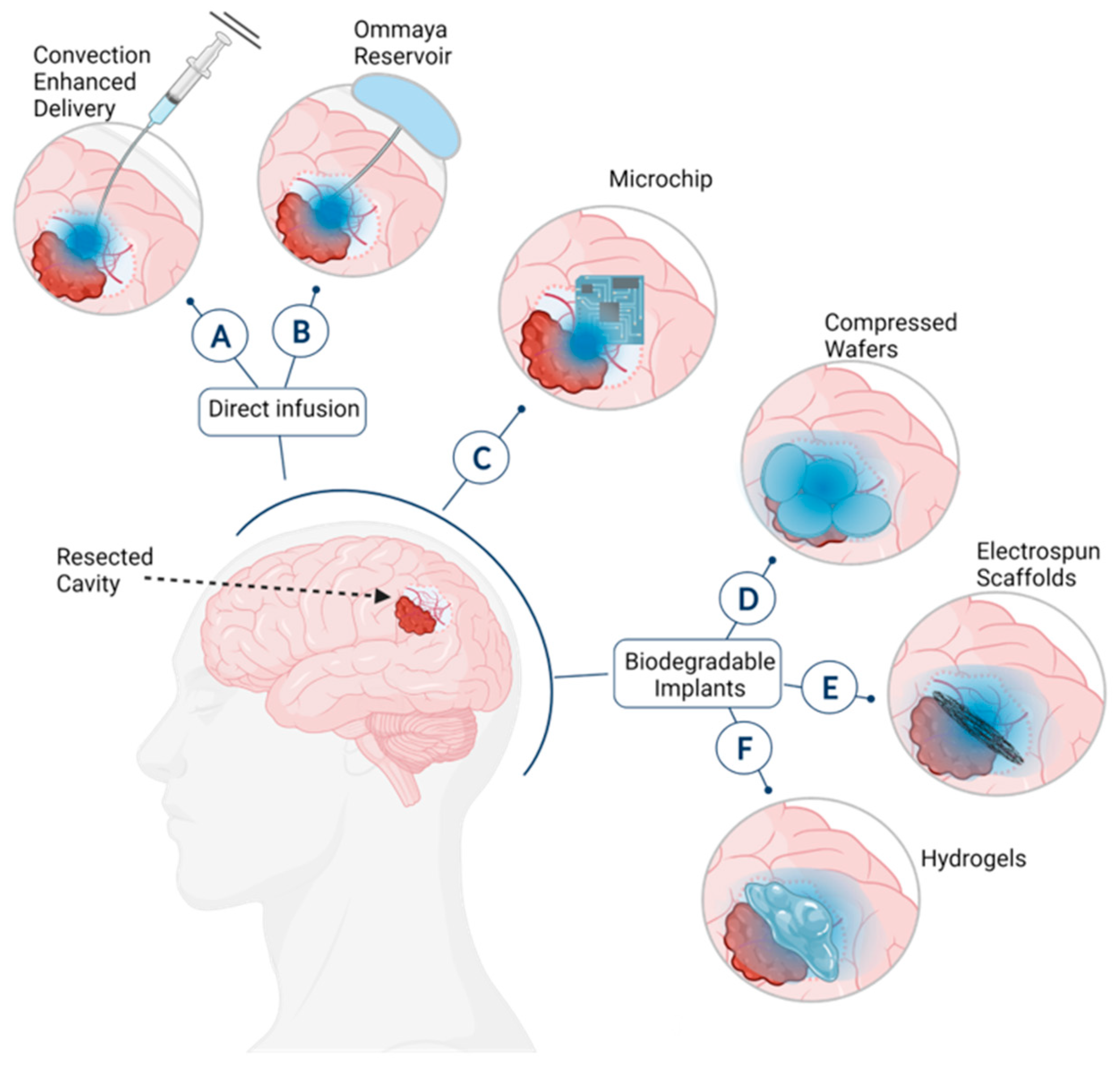
:1. Introduction
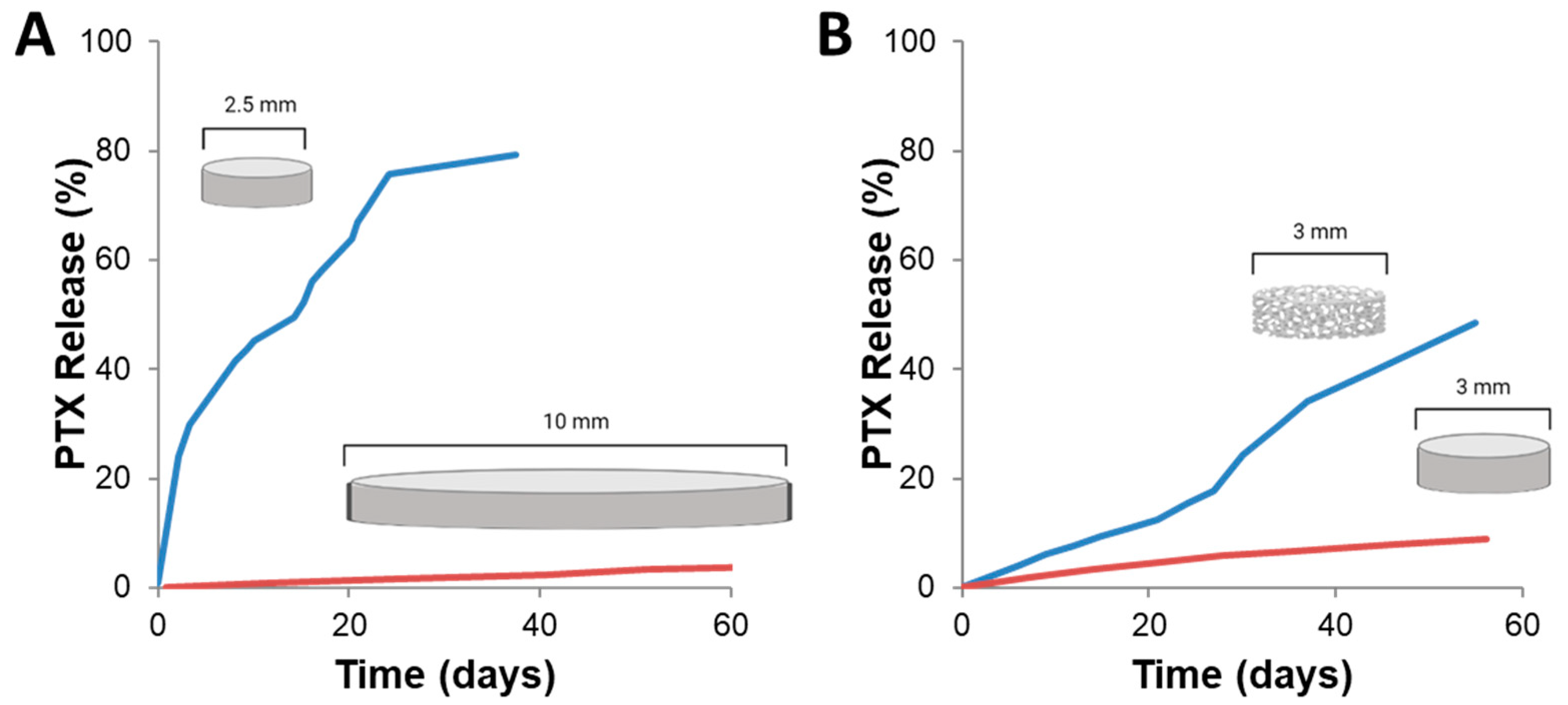
2. Development and Overview of Gliadel
2.1. Gliadel® Development
2.2. Current Clinical Use, Benefits, and Complications
3. Polymer
3.1. Polyanhydrides
3.2. Polyesters
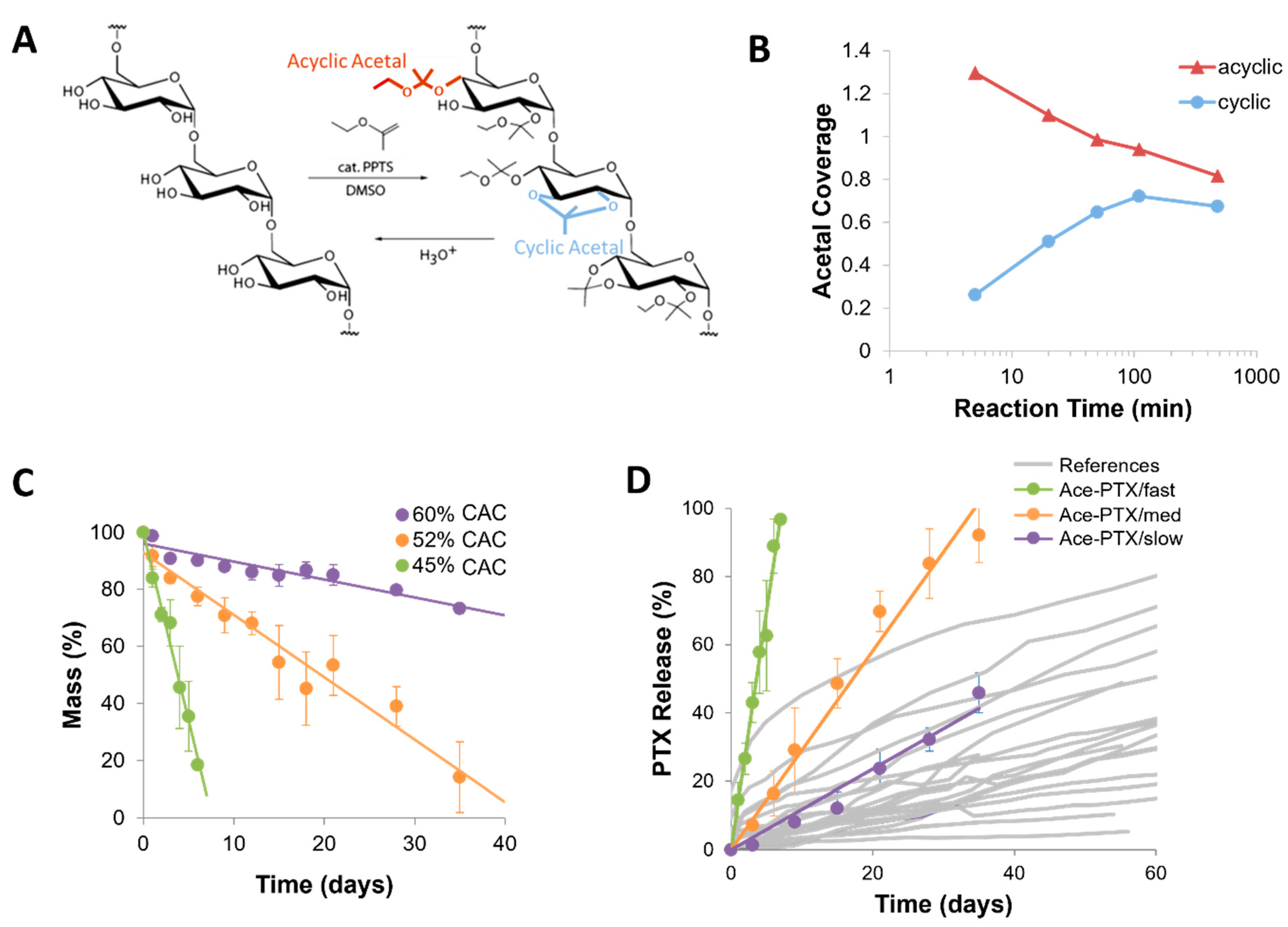
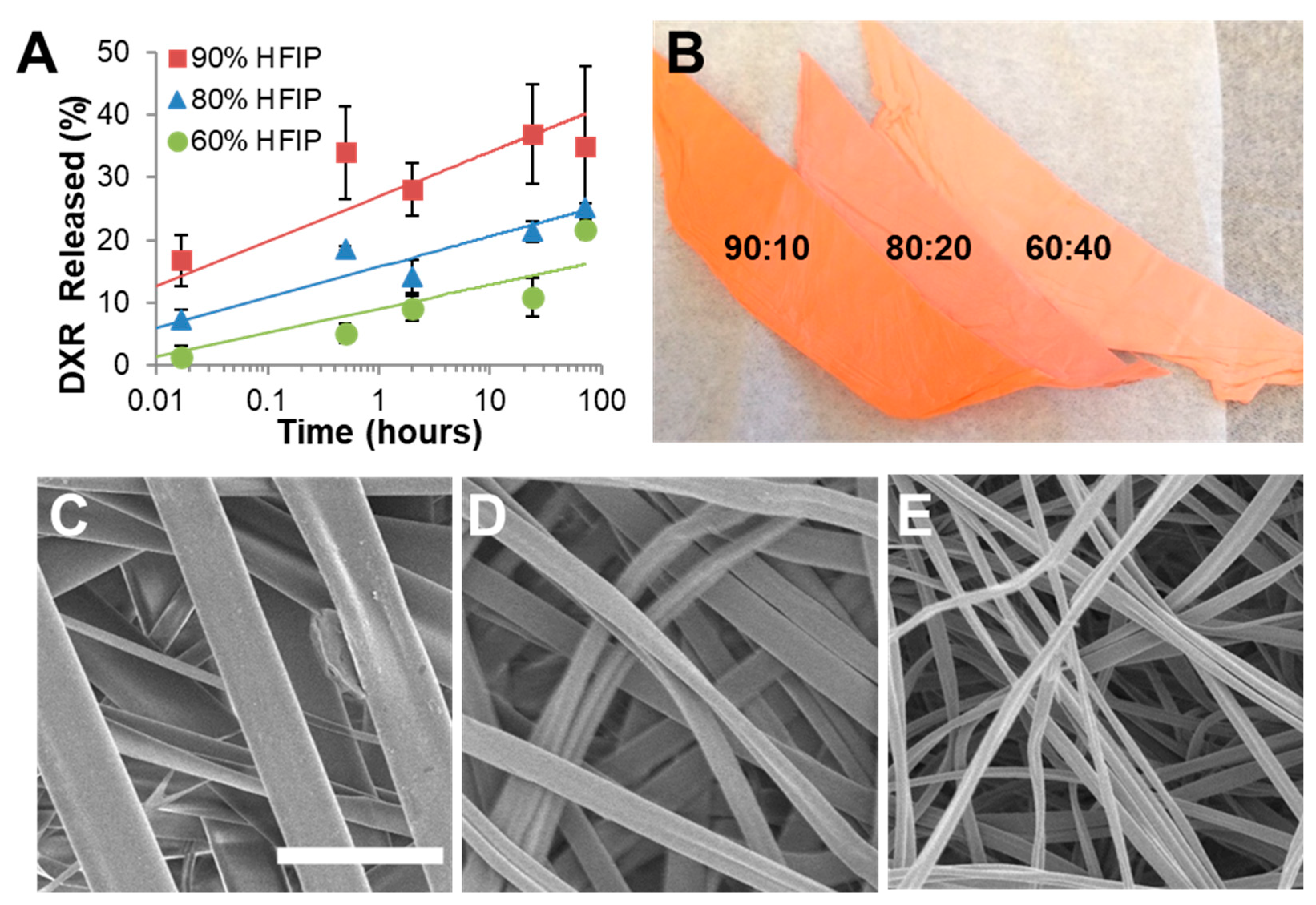
3.3. Acetalated Dextran
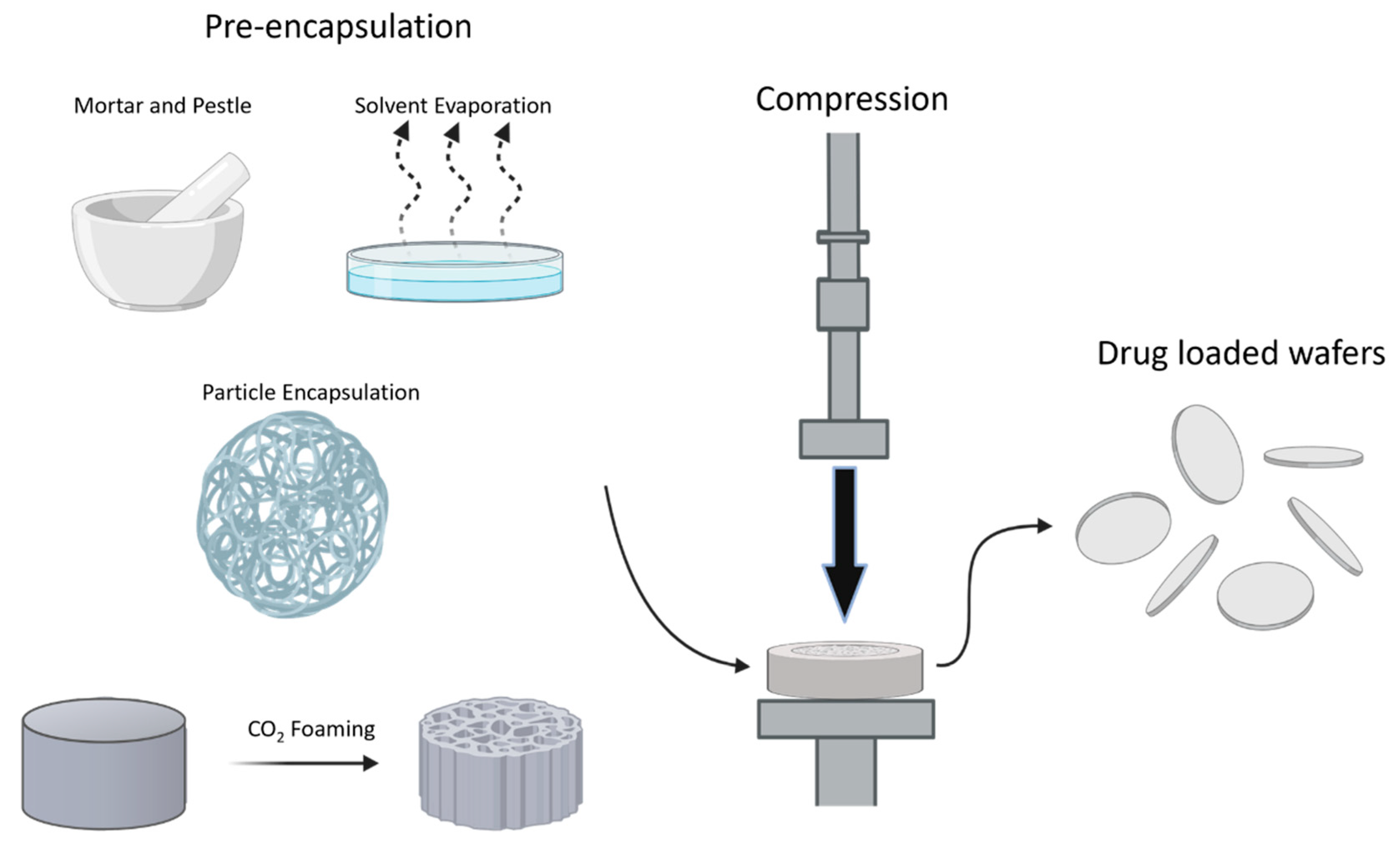
4. Formulation
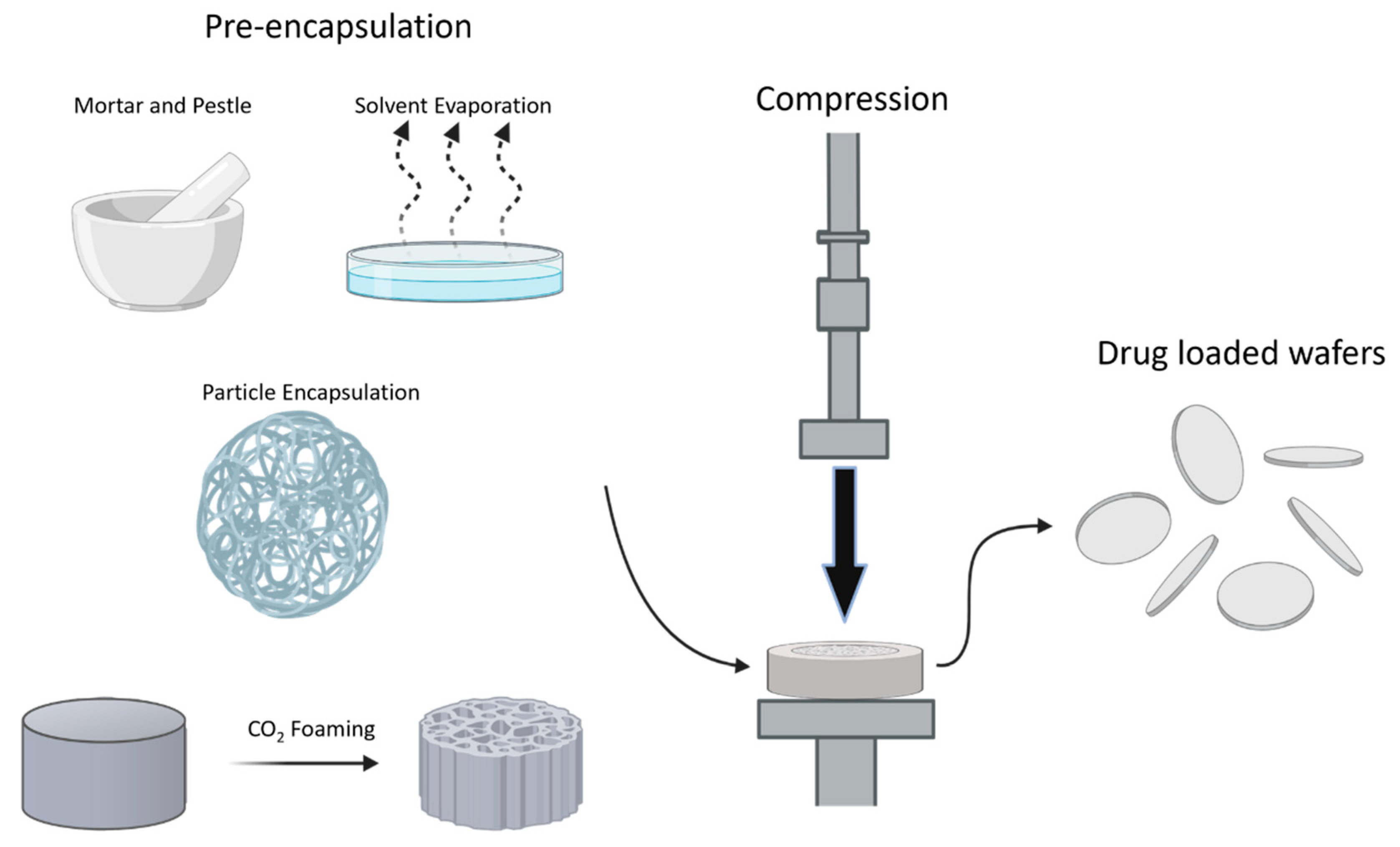
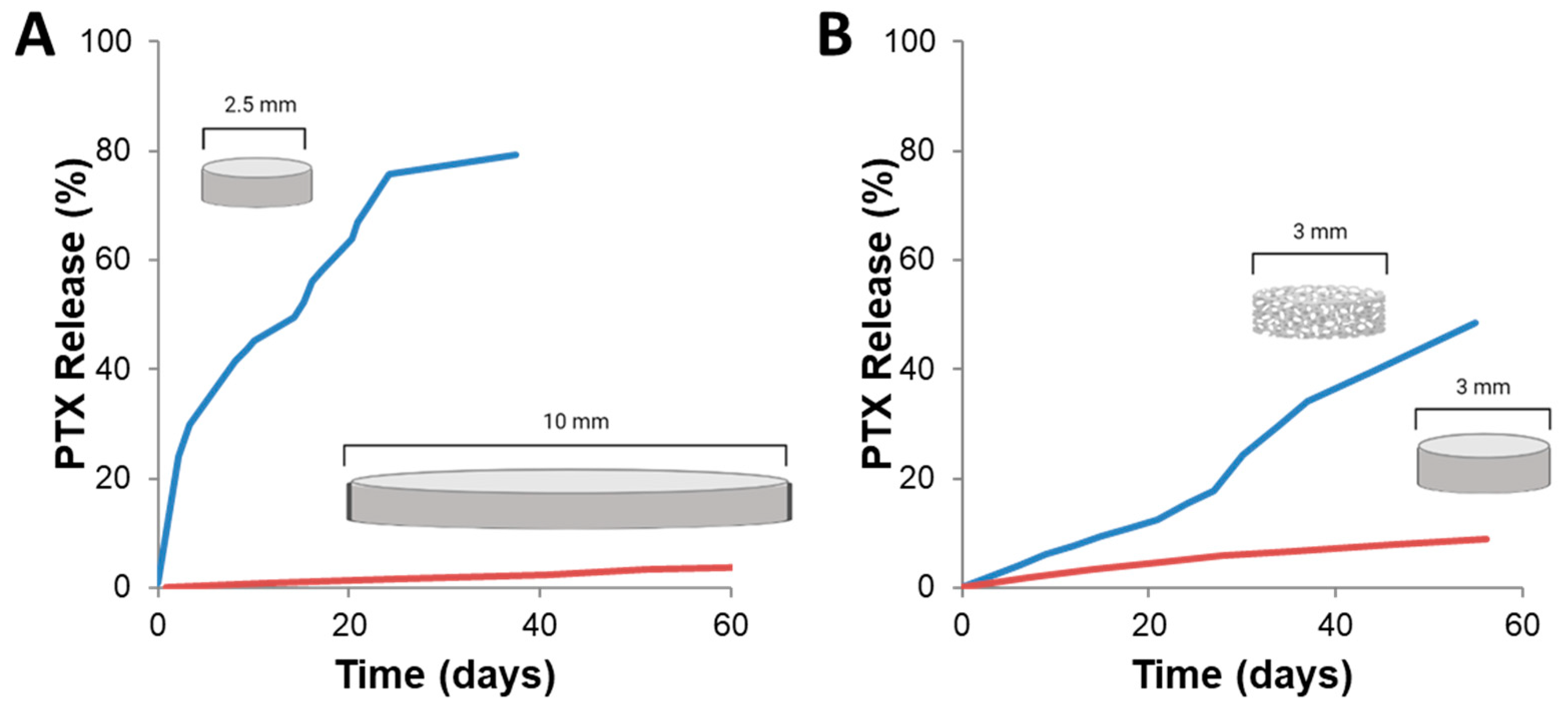
4.1. Compression Molding
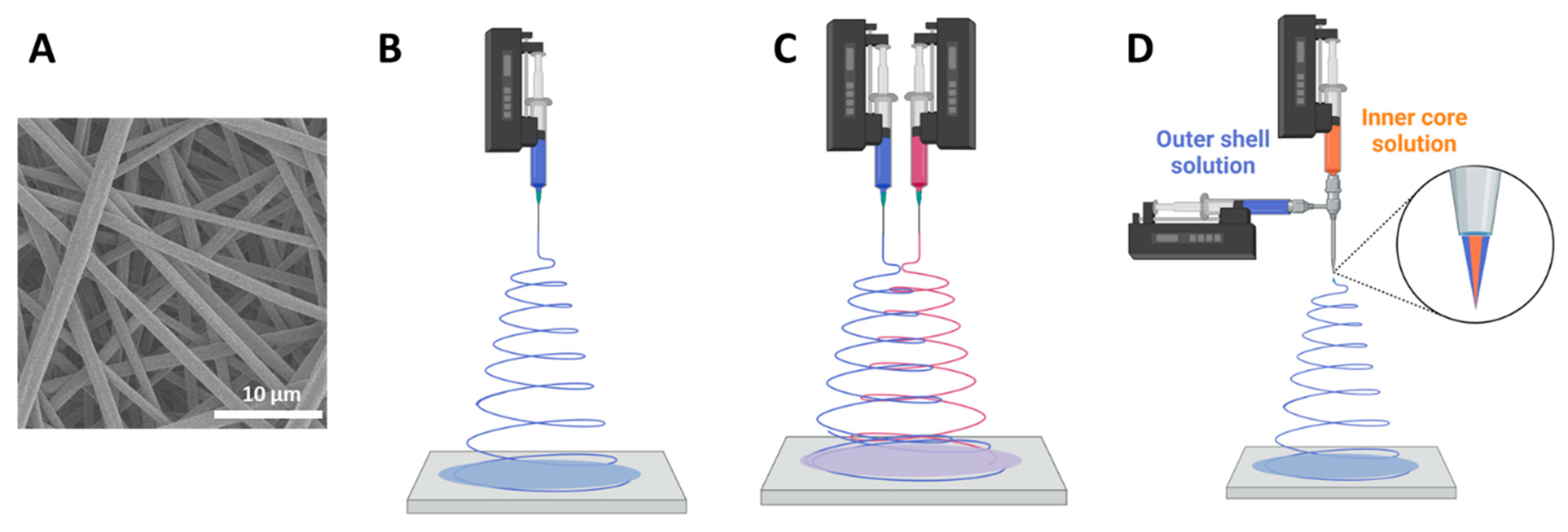
4.2. Electrospinning

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Drug | Needle | Morphology |
|---|---|---|---|
| Ace-DEX | DXR [49], Everolimus [54], PTX [27,54] | Uniaxial | Microfibers |
| PCL | Daunorubicin HCl [100], Methiopropamine [101], TMZ [98] | Uniaxial [100] Coaxial [101] Multiaxial [98] | Microfibers |
| PCL & alginate | TMZ & NGF [99] | Uniaxial | Multilayer fibers glued with gel |
| PCL & gelatin | Camptothecin [102] | Uniaxial | Nanofibers |
| PCL & PVP | MPA [101] | Coaxial | Microfibers |
| PCL-Diol-b-PU | TMZ [90,91] | Uniaxial | Microfibers |
| PCL-Diol-b-PU & chitosan | TMZ [90] | Uniaxial | TMZ loaded chitosan NP in fibers |
| PCL-PEG-PCL | Curcumin [92] | Uniaxial | Microfibers |
| PLA | DXR [49,103], PTX [27], TMZ [98] | Uniaxial [27,49,103] Multiaxial [98] | Microfibers |
| PLA-PEG | BCNU [104], DXR [103,105], PTX [105] | Uniaxial | Microfibers |
| PLA-PEO | Rapamycin [106] | Uniaxial | Nanofibers |
| PLGA | BCNU [107], Cisplatin [107], Combrestastatin [107], Irinotecan [107], PTX [28,29,95,108], TMZ [98] | Uniaxial | Nanofibers [98,107], Microfibers [28,29,95,108] |
| PPC & alginate | PTX [109], TMZ [109] | Uniaxial | Microparticles in microfibers |
| PVA | Dacarbazine [110] | Uniaxial | Nanofibers |
| PVP | Methiopropamine [101] | Uniaxial | Microfibers |
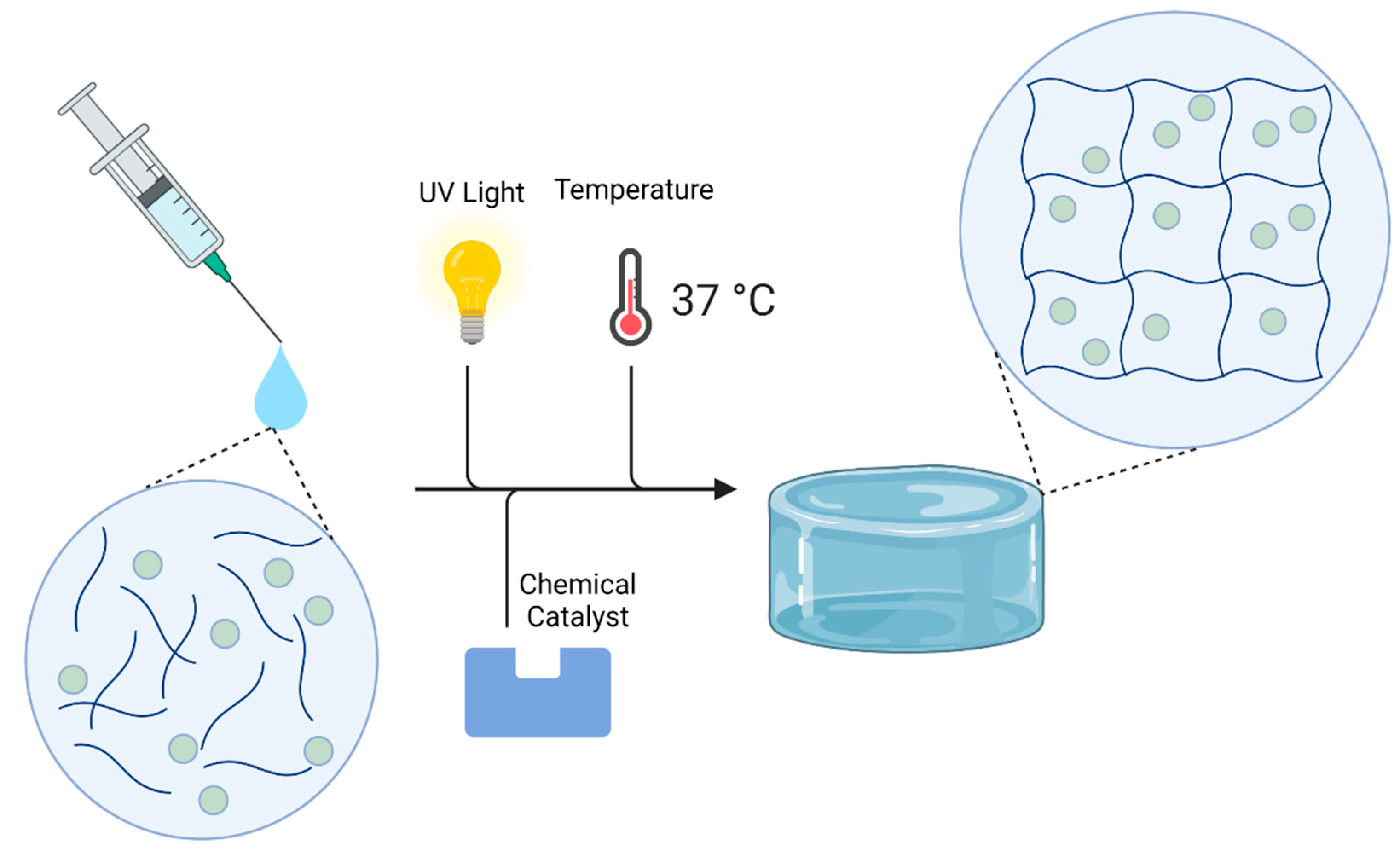
4.3. Hydrogel Synthesis
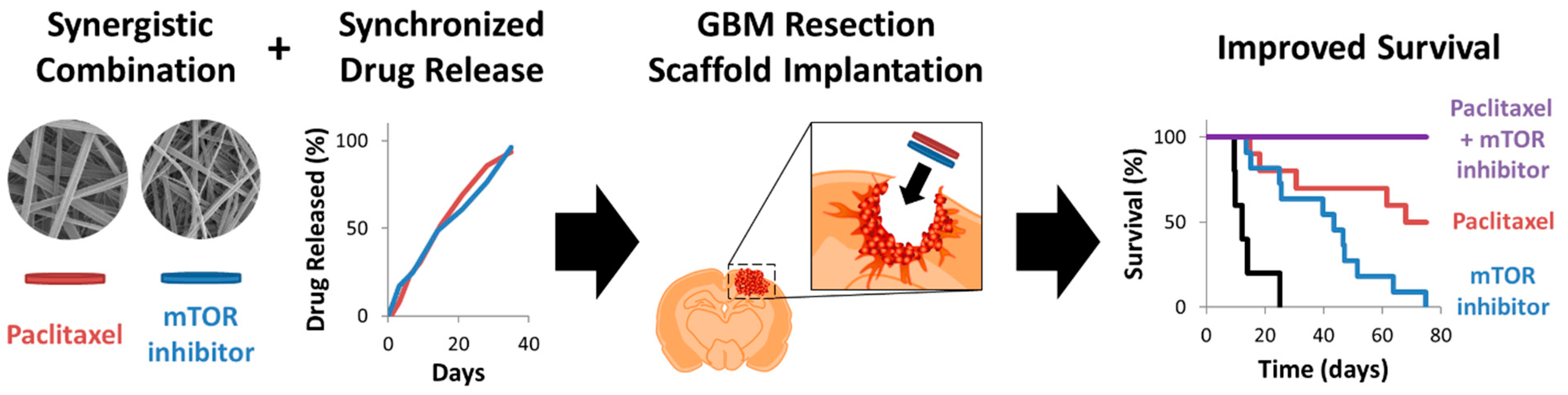
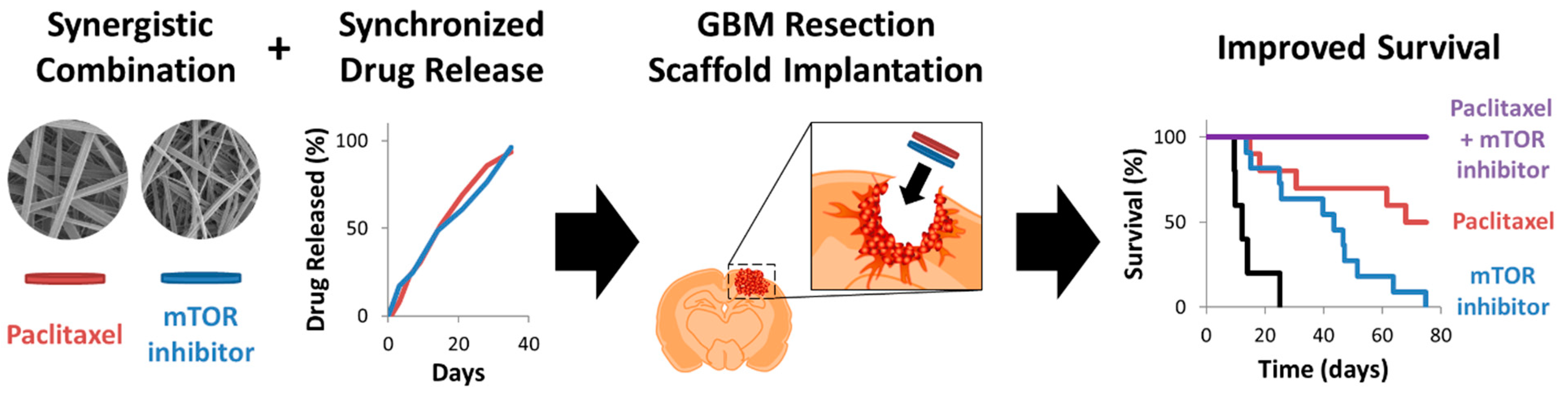
4.4. Combination Therapy
| Drugs | Device Design | In Vitro Release Kinetics | Model (outcome) | Ref |
|---|---|---|---|---|
| BCNU, TMZ | Co-loaded in compressed polymer wafer | BCNU: ---- | Orthotopic, F344 rat no resection (survival) | [74] |
| TMZ: linear release 100% at 35 days | ||||
| PTX, TMZ | PTX-loaded alginate microparticles electrospun into TMZ-loaded polymer fiber scaffold | PTX: linear release 100% at 7 days | ---- | [109] |
| TMZ: linear release 100% at 5 days | ||||
| PTX, TMZ | PTX-loaded polymer microparticles incorporated in photopolymerizable hydrogel containing TMZ | ---- | Orthotopic, nude mouse, tumor resection (survival) | [114] |
| Plasmid DNA for RNAi of MMP2, PTX | Plasmid DNA-loaded polymer nanoparticles electrospun with PTX into polymer fiber scaffold | Plasmid DNA: ~15% release over 42 days | Orthotopic, nude mouse, no resection (tumor growth) | [108] |
| PTX: ~10% release over 42 days | ||||
| EPR, PTX | PTX-loaded BSA nanoparticles incorporated in thermosensitive hydrogel containing EPR | EPR: ~80% at 12 days | Orthotopic, nude mouse, no resection (survival) | [118] |
| PTX: ~50% at 12 days | ||||
| BCNU, CIS, CA-4 irinotecan | BCNU, CIS, and irinotecan electrospun into polymer fiber layer, followed by layer of CA-4 within polymer fibers | ---- | Orthotopic, Wistar rat, no resection (survival) | [107] |
| siRNA, MIT, CXCL10 | siRNA loaded in MOF suspended in hydrogel containing MIT and CXCL10 | siRNA: linear release 100% at 15 days | Orthotopic, C57BL6 mouse, no resection (survival) | [137] |
| MIT: linear release 100% at 18 days | ||||
| CXCL10: linear release 100% at 12 days | ||||
| BCNU, TMZ | Co-loaded in compressed polymer wafer | ---- | Orthotopic, F344 rat, no resection (survival) | [64] |
| PTX, EVR | Separately electrospun polymer fiber scaffolds | PTX: linear release 100% at 35 days | Orthotopic, nude mouse, tumor resection (survival) | [54] |
| EVR: linear release 100% at 35 days |
5. Controlling Release Kinetics
6. Drug Properties on Release Kinetics
6.1. Polymer Properties on Release Kinetics
6.2. Effect of Formulation Process on Release Kinetics
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2011–2015. Neuro-Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hayat, M.A. Tumors of the Central Nervous System, Volume 2: Gliomas: Glioblastoma; Springer Science+Business Media: Berlin/Heidelberg, Germany, 2011. [Google Scholar] [CrossRef]
- Vleeschouwer, S.D. Glioblastoma: To Target the Tumor cell or the Microenvironment? In Glioblastoma: Internet; Codon Publications: Brisbane, Australia, 2017; Chapter 16. [Google Scholar]
- Urbańska, K.; Sokołowska, J.; Szmidt, M.; Sysa, P. Glioblastoma multiforme—An overview. Contemp. Oncol. 2014, 18, 307–312. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Gerlinger, M.; Futreal, P.A.; Pusztai, L.; Swanton, C. Intratumor heterogeneity: Seeing the wood for the trees. Sci. Transl. Med. 2012, 4, 127ps110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bonis, P.; Anile, C.; Pompucci, A.; Fiorentino, A.; Balducci, M.; Chiesa, S.; Lauriola, L.; Maira, G.; Mangiola, A. The influence of surgery on recurrence pattern of glioblastoma. Clin. Neurol. Neurosurg. 2013, 115, 37–43. [Google Scholar] [CrossRef]
- Daneman, R. The blood-brain barrier in health and disease. Ann. Neurol. 2012, 72, 648–672. [Google Scholar] [CrossRef]
- Gloor, S.M.; Wachtel, M.; Bolliger, M.F.; Ishihara, H.; Landmann, R.; Frei, K. Molecular and cellular permeability control at the blood-brain barrier. Brain Res. Brain Res. Rev. 2001, 36, 258–264. [Google Scholar] [CrossRef]
- Kumari, S.; Ahsan, S.M.; Kumar, J.M.; Kondapi, A.K.; Rao, N.M. Overcoming blood brain barrier with a dual purpose Temozolomide loaded Lactoferrin nanoparticles for combating glioma (SERP-17-12433). Sci. Rep. 2017, 7, 6602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, S.A.; Batara, J.F. Current management of glioblastoma multiforme. Semin. Oncol. 2004, 31, 635–644. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.D.; Wirth, M.; Statkevich, P.; Reidenberg, P.; Alton, K.; Sartorius, S.E.; Dugan, M.; Cutler, D.; Batra, V.; Grochow, L.B.; et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. 1999, 5, 309–317. [Google Scholar]
- Grossman, S.A.; O’Neill, A.; Grunnet, M.; Mehta, M.; Pearlman, J.L.; Wagner, H.; Gilbert, M.; Newton, H.B.; Hellman, R. Phase III study comparing three cycles of infusional carmustine and cisplatin followed by radiation therapy with radiation therapy and concurrent carmustine in patients with newly diagnosed supratentorial glioblastoma multiforme: Eastern cooperative oncology group trial 2394. J. Clin. Oncol. 2003, 21, 1485–1491. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, R.; Sun, T.; Rashid, M.H.; Israel, L.L.; Ramesh, A.; Davani, S.; Black, K.L.; Ljubimov, A.V.; Holler, E.; Ljubimova, J.Y. Multifunctional nanopolymers for blood–brain barrier delivery and inhibition of glioblastoma growth through EGFR/EGFRvIII, c-Myc, and PD-1. Nanomaterials 2021, 11, 2892. [Google Scholar] [CrossRef]
- Jahangiri, A.; Chin, A.T.; Flanigan, P.M.; Chen, R.; Bankiewicz, K.; Aghi, M.K. Convection-Enhanced delivery in glioblastoma: A review of preclinical and clinical studies. J. Neurosurg. 2017, 126, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.-C.; Liu, S.-J.; Lo, W.-L.; Chen, S.-M.; Tang, Y.-L.; Tseng, Y.-Y. Enhanced anti-tumor activity in mice with temozolomide-resistant human glioblastoma cell line-derived xenograft using SN-38-Incorporated polymeric microparticle. Int. J. Mol. Sci. 2021, 22, 5557. [Google Scholar] [CrossRef]
- Patchell, R.A.; Regine, W.F.; Ashton, P.; Tibbs, P.A.; Wilson, D.; Shappley, D.; Young, B. A phase I trial of continuously infused intratumoral bleomycin for the treatment of recurrent glioblastoma multiforme. J. Neuro-Oncol. 2002, 60, 37–42. [Google Scholar] [CrossRef]
- Chamberlain, M.C.; Kormanik, P.A.; Barba, D. Complications associated with intraventricular chemotherapy in patients with leptomeningeal metastases. J. Neurosurg. 1997, 87, 694–699. [Google Scholar] [CrossRef] [Green Version]
- Obbens, E.A.M.T.; Leavens, M.E.; Bed, J.W.; Lee, Y.-Y. Ommaya reservoirs in 387 cancer patients: A 15-year experience. Neurology 1985, 35, 1274. [Google Scholar] [CrossRef]
- Bosse, R.; Doonan, B.; Ali, A.; Barish, J.; Narayan, P.; Welniak, S.; Jester, G.; Delaune, J.; Heldermon, C.D. A retrospective review of complication rates of Ommaya reservoir placement for intrathecal medication administration. J. Clin. Oncol. 2018, 36, e18532. [Google Scholar] [CrossRef]
- John, T.; Santini, A.C.R.; Scheidt, R.A.; Cima, M.J.; Langer, R.S. Microchip technology in drug delivery. Ann. Med. 2000, 32, 377–379. [Google Scholar]
- Brem, H.; Mahaley, M.S., Jr.; Vick, N.A.; Black, K.L.; Schold, S.C., Jr.; Burger, P.C.; Friedman, A.H.; Ciric, I.S.; Eller, T.W.; Cozzens, J.W.; et al. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J. Neurosurg. 1991, 74, 441–446. [Google Scholar] [CrossRef]
- Graham-Gurysh, E.G.; Moore, K.; Schorzman, A.N.; Lee, T.; Zamboni, W.C.; Hingtgen, S.; Bachelder, E.M.; Ainslie, K.M. Tumor responsive and tunable polymeric platform for optimized delivery of paclitaxel to treat glioblastoma. ACS Appl. Mater. Inter. 2020, 12, 19345–19356. [Google Scholar] [CrossRef]
- Ranganath, S.H.; Fu, Y.; Arifin, D.Y.; Kee, I.; Zheng, L.; Lee, H.S.; Chow, P.K.; Wang, C.H. The use of submicron/nanoscale PLGA implants to deliver paclitaxel with enhanced pharmacokinetics and therapeutic efficacy in intracranial glioblastoma in mice. Biomaterials 2010, 31, 5199–5207. [Google Scholar] [CrossRef]
- Ranganath, S.H.; Wang, C.H. Biodegradable microfiber implants delivering paclitaxel for post-surgical chemotherapy against malignant glioma. Biomaterials 2008, 29, 2996–3003. [Google Scholar] [CrossRef]
- Ong, B.Y.; Ranganath, S.H.; Lee, L.Y.; Lu, F.; Lee, H.S.; Sahinidis, N.V.; Wang, C.H. Paclitaxel delivery from PLGA foams for controlled release in post-surgical chemotherapy against glioblastoma multiforme. Biomaterials 2009, 30, 3189–3196. [Google Scholar] [CrossRef]
- Walter, K.A.; Cahan, M.A.; Gur, A.; Tyler, B.; Hilton, J.; Colvin, O.M.; Burger, P.C.; Domb, A.; Brem, H. Interstitial taxol delivered from a biodegradable polymer implant against experimental malignant glioma. Cancer Res. 1994, 54, 2207–2212. [Google Scholar] [PubMed]
- Brem, H.; Gabikian, P. Biodegradable polymer implants to treat brain tumors. J. Control Release 2001, 74, 63–67. [Google Scholar] [CrossRef]
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-Oncol. 2003, 5, 79–88. [Google Scholar] [CrossRef]
- Bregy, A.; Shah, A.H.; Diaz, M.V.; Pierce, H.E.; Ames, P.L.; Diaz, D.; Komotar, R.J. The role of Gliadel wafers in the treatment of high-grade gliomas. Expert Rev. Anticancer Ther. 2013, 13, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.; Kanu, O.O.; Mehta, A.I.; Di, C.; Lin, N.; Mattox, A.K.; Bigner, D.D. Glioblastoma multiforme: A review of where we have been and where we are going. Expert Opin. Investig. Drugs 2009, 18, 1061–1083. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.W.; D’Amore, P.D.; Marletta, M.; Langer, R. Bioerodible polyanhydrides as drug-carrier matrices. II. Biocompatibility and chemical reactivity. J. Biomed. Mater. Res. 1986, 20, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.W.; Brott, B.C.; Langer, R. Bioerodible polyanhydrides as drug-carrier matrices. I: Characterization, degradation, and release characteristics. J. Biomed. Mater. Res. 1985, 19, 941–955. [Google Scholar] [CrossRef]
- Brem, H.; Kader, A.; Epstein, J.I.; Tamargo, R.J.; Domb, A.; Langer, R.; Leong, K.W. Biocompatibility of a biodegradable, controlled-release polymer in the rabbit brain. Sel. Cancer Ther. 1989, 5, 55–65. [Google Scholar] [CrossRef]
- Tamargo, R.J.; Epstein, J.I.; Reinhard, C.S.; Chasin, M.; Brem, H. Brain biocompatibility of a biodegradable, controlled-release polymer in rats. J. Biomed. Mater. Res. 1989, 23, 253–266. [Google Scholar] [CrossRef]
- Grossman, S.A.; Reinhard, C.; Colvin, O.M.; Chasin, M.; Brundrett, R.; Tamargo, R.J.; Brem, H. The intracerebral distribution of BCNU delivered by surgically implanted biodegradable polymers. J. Neurosurg. 1992, 76, 640–647. [Google Scholar] [CrossRef]
- Tamargo, R.J.; Myseros, J.S.; Epstein, J.I.; Yang, M.B.; Chasin, M.; Brem, H. Interstitial chemotherapy of the 9L gliosarcoma: Controlled release polymers for drug delivery in the brain. Cancer Res. 1993, 53, 329–333. [Google Scholar] [PubMed]
- Brem, H.; Tamargo, R.J.; Olivi, A.; Pinn, M.; Weingart, J.D.; Wharam, M.; Epstein, J.I. Biodegradable polymers for controlled delivery of chemotherapy with and without radiation therapy in the monkey brain. J. Neurosurg. 1994, 80, 283–290. [Google Scholar] [CrossRef]
- Buahin, K.G.; Brem, H. Interstitial chemotherapy of experimental brain tumors: Comparison of intratumoral injection versus polymeric controlled release. J. Neurooncol. 1995, 26, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Brem, H.; Piantadosi, S.; Burger, P.C.; Walker, M.; Selker, R.; Vick, N.A.; Black, K.; Sisti, M.; Brem, S.; Mohr, G.; et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet 1995, 345, 1008–1012. [Google Scholar] [CrossRef]
- Food and Drug Administration. GLIADEL WAFER (Carmustine Implant) for Intracranial Use; Food and Drug Administration: Montgomery, AL, USA, 2013. [Google Scholar]
- Chowdhary, S.A.; Ryken, T.; Newton, H.B. Survival outcomes and safety of carmustine wafers in the treatment of high-grade gliomas: A meta-analysis. J. Neurooncol. 2015, 122, 367–382. [Google Scholar] [CrossRef] [Green Version]
- Gutenberg, A.; Lumenta, C.B.; Braunsdorf, W.E.; Sabel, M.; Mehdorn, H.M.; Westphal, M.; Giese, A. The combination of carmustine wafers and temozolomide for the treatment of malignant gliomas. A comprehensive review of the rationale and clinical experience. J. Neurooncol. 2013, 113, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Ashby, L.S.; Smith, K.A.; Stea, B. Gliadel wafer implantation combined with standard radiotherapy and concurrent followed by adjuvant temozolomide for treatment of newly diagnosed high-grade glioma: A systematic literature review. World J. Surg. Oncol. 2016, 14, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham-Gurysh, E.; Moore, K.M.; Satterlee, A.B.; Sheets, K.T.; Lin, F.-C.; Bachelder, E.M.; Miller, C.R.; Hingtgen, S.D.; Ainslie, K.M. Sustained delivery of doxorubicin via acetalated dextran scaffold prevents glioblastoma recurrence after surgical resection. Mol. Pharm. 2018, 15, 1309–1318. [Google Scholar] [CrossRef]
- Arifin, D.Y.; Lee, K.Y.; Wang, C.H. Chemotherapeutic drug transport to brain tumor. J. Control Release 2009, 137, 203–210. [Google Scholar] [CrossRef]
- Arifin, D.Y.; Lee, K.Y.; Wang, C.H.; Smith, K.A. Role of convective flow in carmustine delivery to a brain tumor. Pharm. Res. 2009, 26, 2289–2302. [Google Scholar] [CrossRef] [PubMed]
- Bobola, M.S.; Silber, J.R.; Ellenbogen, R.G.; Geyer, J.R.; Blank, A.; Goff, R.D. O6-Methylguanine-DNA methyltransferase, O6-benzylguanine, and resistance to clinical alkylators in pediatric primary brain tumor cell lines. Clin. Cancer Res. 2005, 11, 2747–2755. [Google Scholar] [CrossRef] [Green Version]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef]
- Cahan, M.A.; Walter, K.A.; Colvin, O.M.; Brem, H. Cytotoxicity of taxol in vitro against human and rat malignant brain tumors. Cancer Chemother. Pharmacol. 1994, 33, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Graham-Gurysh, E.G.; Murthy, A.B.; Moore, K.M.; Hingtgen, S.D.; Bachelder, E.M.; Ainslie, K.M. Synergistic drug combinations for a precision medicine approach to interstitial glioblastoma therapy. J. Control Release 2020, 323, 282–292. [Google Scholar] [CrossRef]
- Fung, L.K.; Ewend, M.G.; Sills, A.; Sipos, E.P.; Thompson, R.; Watts, M.; Colvin, O.M.; Brem, H.; Saltzman, W.M. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res. 1998, 58, 672–684. [Google Scholar]
- Anderson, J.M. Biocompatibility and the relationship to standards: Meaning and scope of biomaterials testing. In Comprehensive Biomaterials, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2011; pp. 7–26. [Google Scholar]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.K. Drug delivery systems—An overview. Methods Mol. Biol. 2008, 437, 1–50. [Google Scholar] [CrossRef]
- Sackett, C.K.; Narasimhan, B. Mathematical modeling of polymer erosion: Consequences for drug delivery. Int. J. Pharm. 2011, 418, 104–114. [Google Scholar] [CrossRef] [PubMed]
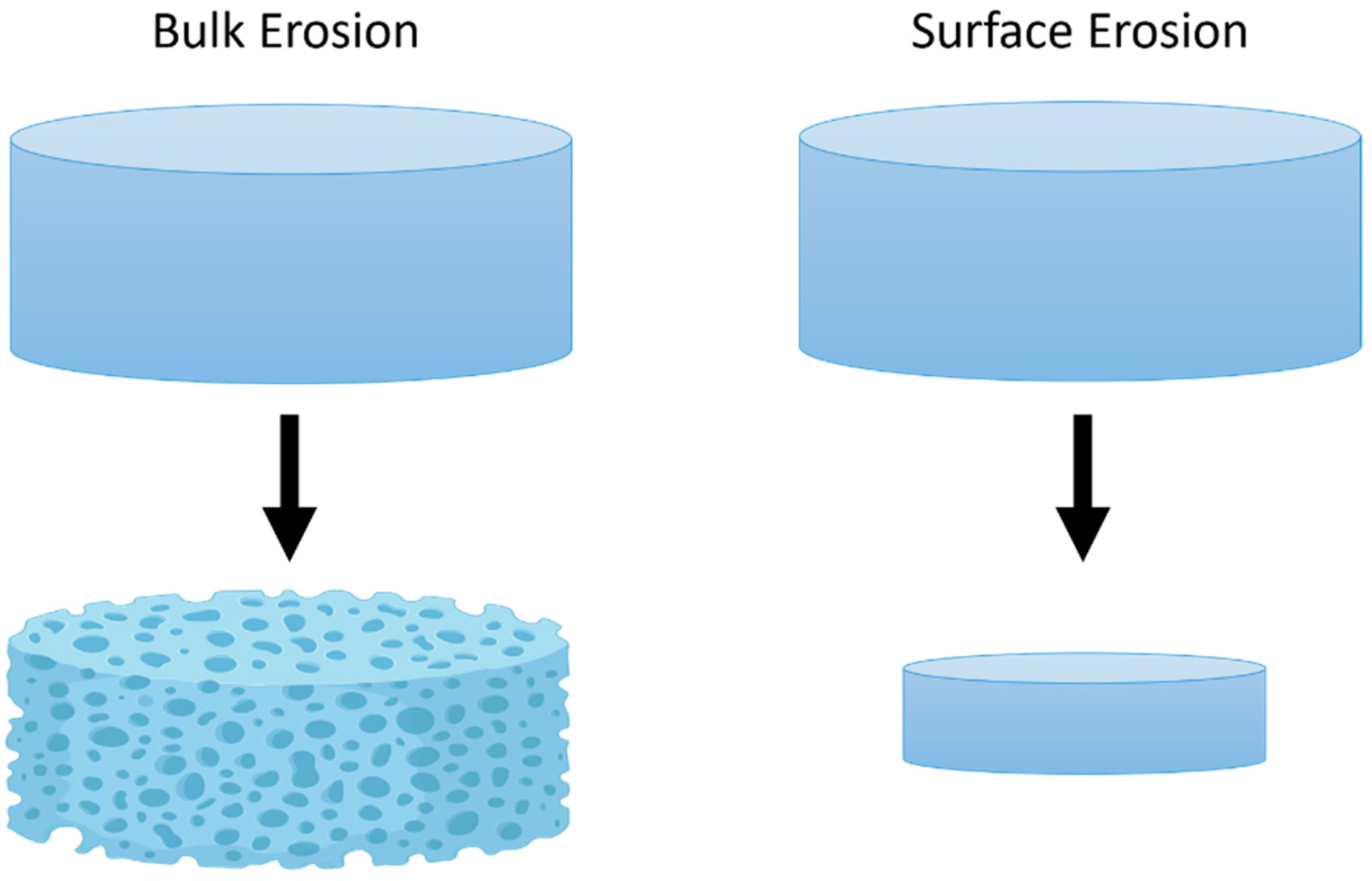
- Gopferich, A.; Langer, R. Modeling of polymer erosion. Macromolecules 1993, 26, 4105–4112. [Google Scholar] [CrossRef]
- Göpferich, A. Polymer bulk erosion. Macromolecules 1997, 30, 2598–2604. [Google Scholar] [CrossRef]
- von Burkersroda, F.; Schedl, L.; Göpferich, A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials 2002, 23, 4221–4231. [Google Scholar] [CrossRef]
- Wexler, P. (Ed.) Encyclopedia of Toxicology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Recinos, V.R.; Tyler, B.M.; Bekelis, K.; Sunshine, S.B.; Vellimana, A.; Li, K.W.; Brem, H. Combination of intracranial temozolomide with intracranial carmustine improves survival when compared with either treatment alone in a rodent glioma model. Neurosurgery 2010, 66, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Storm, P.B.; Moriarity, J.L.; Tyler, B.; Burger, P.C.; Brem, H.; Weingart, J. Polymer delivery of camptothecin against 9L gliosarcoma: Release, distribution, and efficacy. J. Neurooncol. 2002, 56, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, M.S.; Upadhyay, U.; Goodwin, R.; Tyler, B.; Brem, H. Local delivery of doxorubicin for the treatment of malignant brain tumors in rats. Anticancer Res. 2005, 25, 3825–3831. [Google Scholar]
- Legnani, F.G.; Pradilla, G.; Thai, Q.-A.; Fiorindi, A.; Recinos, P.F.; Tyler, B.M.; Gaini, S.M.; DiMeco, F.; Brem, H.; Olivi, A. Lactacystin exhibits potent anti-tumor activity in an animal model of malignant glioma when administered via controlled-release polymers. J. Neuro-Oncol. 2006, 77, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Yohay, K.; Tyler, B.; Weaver, K.D.; Pardo, A.C.; Gincel, D.; Blakeley, J.; Brem, H.; Rothstein, J.D. Efficacy of local polymer-based and systemic delivery of the anti-glutamatergic agents riluzole and memantine in rat glioma models. J. Neurosurg. 2014, 120, 854–863. [Google Scholar] [CrossRef] [Green Version]
- Pradilla, G.; Legnani, F.G.; Petrangolini, G.; Francescato, P.; Chillemi, F.; Tyler, B.M.; Gaini, S.M.; Brem, H.; Olivi, A.; DiMeco, F. Local delivery of a synthetic endostatin fragment for the treatment of experimental gliomas. Neurosurgery 2005, 57, 1032–1040. [Google Scholar] [CrossRef] [Green Version]
- Brem, S.; Tyler, B.; Li, K.; Pradilla, G.; Legnani, F.; Caplan, J.; Brem, H. Local delivery of temozolomide by biodegradable polymers is superior to oral administration in a rodent glioma model. Cancer Chemother. Pharmacol. 2007, 60, 643–650. [Google Scholar] [CrossRef]
- DiMeco, F.; Li, K.W.; Tyler, B.M.; Wolf, A.S.; Brem, H.; Olivi, A. Local delivery of mitoxantrone for the treatment of malignant brain tumors in rats. J. Neurosurg. 2002, 97, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Vogelhuber, W.; Spruss, T.; Bernhardt, G.; Buschauer, A.; Gopferich, A. Efficacy of BCNU and paclitaxel loaded subcutaneous implants in the interstitial chemotherapy of U-87 MG human glioblastoma xenografts. Int. J. Pharm. 2002, 238, 111–121. [Google Scholar] [CrossRef]
- Dang, H.; Wang, J.; Cheng, J.X.; Wang, P.Y.; Wang, Y.; Cheng, L.F.; Du, C.; Wang, X.J. Efficacy of local delivery of ardipusilloside I using biodegradable implants against cerebral tumor growth. Am. J. Cancer Res. 2015, 5, 243–254. [Google Scholar]
- Shapira-Furman, T.; Serra, R.; Gorelick, N.; Doglioli, M.; Tagliaferri, V.; Cecia, A.; Peters, M.; Kumar, A.; Rottenberg, Y.; Langer, R.; et al. Biodegradable wafers releasing Temozolomide and Carmustine for the treatment of brain cancer. J. Control Release 2019, 295, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; An, T.K.; Chae, G.S.; Jeong, J.K.; Cho, S.H.; Lee, H.B.; Khang, G. Evaluation of in vitro and in vivo antitumor activity of BCNU-loaded PLGA wafer against 9L gliosarcoma. Eur. J. Pharm. Biopharm. Off. J. Arb. Pharm. Verfahr. V 2005, 59, 169–175. [Google Scholar] [CrossRef]
- Zembko, I.; Ahmed, I.; Farooq, A.; Dail, J.; Tawari, P.; Wang, W.; McConville, C. Development of disulfiram-loaded poly(lactic-co-glycolic acid) wafers for the localised treatment of glioblastoma multiforme: A comparison of manufacturing techniques. J. Pharm. Sci. 2015, 104, 1076–1086. [Google Scholar] [CrossRef]
- Lee, L.Y.; Ranganath, S.H.; Fu, Y.; Zheng, J.L.; Lee, H.S.; Wang, C.-H.; Smith, K.A. Paclitaxel release from micro-porous PLGA disks. Chem. Eng. Sci. 2009, 64, 4341–4349. [Google Scholar] [CrossRef]
- Li, K.W.; Dang, W.; Tyler, B.M.; Troiano, G.; Tihan, T.; Brem, H.; Walter, K.A. Polilactofate microspheres for paclitaxel delivery to central nervous system malignancies. Clin. Cancer Res. 2003, 9, 3441–3447. [Google Scholar]
- Fu, K.; Pack, D.; Klibanov, A.; Langer, R. Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm. Res. 2000, 17, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Ding, A.G.; Shenderova, A.; Schwendeman, S.P. Prediction of microclimate pH in poly(lactic-co-glycolic acid) films. J. Am. Chem. Soc. 2006, 128, 5384–5390. [Google Scholar] [CrossRef]
- Sung, H.J.; Meredith, C.; Johnson, C.; Galis, Z.S. The effect of scaffold degradation rate on three-dimensional cell growth and angiogenesis. Biomaterials 2004, 25, 5735–5742. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.H.; Emmett, C.; Shen, P.; Aswani, S.; Iwamoto, T.; Vaghefi, F.; Cain, G.; Sanders, L. Bioerosion and biocompatibility of poly(d,l-lactic-co-glycolic acid) implants in brain. J. Control Release 1997, 43, 123–130. [Google Scholar] [CrossRef]
- Langer, R. New methods of drug delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Manome, Y.; Kobayashi, T.; Mori, M.; Suzuki, R.; Funamizu, N.; Akiyama, N.; Inoue, S.; Tabata, Y.; Watanabe, M. Local delivery of doxorubicin for malignant glioma by a biodegradable PLGA polymer sheet. Anticancer Res. 2006, 26, 3317–3326. [Google Scholar] [PubMed]
- Lam, C.X.; Savalani, M.M.; Teoh, S.H.; Hutmacher, D.W. Dynamics of in vitro polymer degradation of polycaprolactone-based scaffolds: Accelerated versus simulated physiological conditions. Biomed. Mater. 2008, 3, 034108. [Google Scholar] [CrossRef] [PubMed]
- Sailema-Palate, G.P.; Vidaurre, A.; Campillo-Fernández, A.J.; Castilla-Cortázar, I. A comparative study on poly(ε-caprolactone) film degradation at extreme pH values. Polym. Degrad. Stabil. 2016, 130, 118–125. [Google Scholar] [CrossRef]
- Chen, D.R.; Bei, J.Z.; Wang, S.G. Polycaprolactone microparticles and their biodegradation. Polym. Degrad. Stabil. 2000, 67, 455–459. [Google Scholar] [CrossRef]
- Irani, M.; Mir Mohamad Sadeghi, G.; Haririan, I. A novel biocompatible drug delivery system of chitosan/temozolomide nanoparticles loaded PCL-PU nanofibers for sustained delivery of temozolomide. Int. J. Biol. Macromol. 2017, 97, 744–751. [Google Scholar] [CrossRef]
- Irani, M.; Sadeghi, G.M.M.; Haririan, I. The sustained delivery of temozolomide from electrospun PCL-Diol-b-PU/gold nanocompsite nanofibers to treat glioblastoma tumors. Mater. Sci. Eng. C 2017, 75, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Fu, S.; Zhou, L.; Liang, H.; Fan, M.; Luo, F.; Qian, Z.; Wei, Y. Preparation of curcumin loaded poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone) nanofibers and their in vitro antitumor activity against Glioma 9L cells. Nanoscale 2011, 3, 3825–3832. [Google Scholar] [CrossRef]
- Bachelder, E.M.; Beaudette, T.T.; Broaders, K.E.; Dashe, J.; Fréchet, J.M.J. Acetal-Derivatized dextran: An acid-responsive biodegradable material for therapeutic applications. J. Am. Chem. Soc. 2008, 130, 10494–10495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, K.J.; Do, C.; Sharma, S.; Gallovic, M.D.; Bachelder, E.M.; Ainslie, K.M. Synthesis and characterization of acetalated dextran polymer and microparticles with ethanol as a degradation product. ACS Appl. Mater. Interfaces 2012, 4, 4149–4155. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Wang, C.H. Electrospun micro- and nanofibers for sustained delivery of paclitaxel to treat C6 glioma in vitro. Pharm. Res. 2006, 23, 1817–1826. [Google Scholar] [CrossRef]
- Zeng, J.; Yang, L.; Liang, Q.; Zhang, X.; Guan, H.; Xu, X.; Chen, X.; Jing, X. Influence of the drug compatibility with polymer solution on the release kinetics of electrospun fiber formulation. J. Control Release 2005, 105, 43–51. [Google Scholar] [CrossRef]
- Xue, J.; Wu, T.; Dai, Y.; Xia, Y. Electrospinning and electrospun nanofibers: Methods, materials, and applications. Chem. Rev. 2019, 119, 5298–5415. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Junnuthula, V.R.; Gowd, G.S.; Ashokan, A.; Thomas, J.; Peethambaran, R.; Thomas, A.; Unni, A.K.K.; Panikar, D.; Nair, S.V. Theranostic 3-Dimensional nano brain-implant for prolonged and localized treatment of recurrent glioma. Sci. Rep. 2017, 7, 43271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Lin, C.; Wen, X.; Gu, S.; Zhao, P. A potential nanofiber membrane device for filling surgical residual cavity to prevent glioma recurrence and improve local neural tissue reconstruction. PLoS ONE 2016, 11, e0161435. [Google Scholar] [CrossRef] [Green Version]
- Lian, H.; Meng, Z. Melt electrospinning of daunorubicin hydrochloride-loaded poly (ε-caprolactone) fibrous membrane for tumor therapy. Bioact. Mater. 2017, 2, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Sasaki, M.; Yoshino, H.; Kofuji, S.; Sasaki, A.T.; Steckl, A.J. In-Vitro evaluation of MPA-loaded electrospun coaxial fiber membranes for local treatment of glioblastoma tumor cells. J. Drug Deliv. Sci. Technol. 2017, 40, 45–50. [Google Scholar] [CrossRef]
- Zhu, X.; Ni, S.; Xia, T.; Yao, Q.; Li, H.; Wang, B.; Wang, J.; Li, X.; Su, W. Anti-Neoplastic cytotoxicity of SN-38-loaded PCL/Gelatin electrospun composite nanofiber scaffolds against human glioblastoma cells In Vitro. J. Pharm. Sci. 2015, 104, 4345–4354. [Google Scholar] [CrossRef]
- Xu, X.; Yang, L.; Xu, X.; Wang, X.; Chen, X.; Liang, Q.; Zeng, J.; Jing, X. Ultrafine medicated fibers electrospun from W/O emulsions. J. Control Release 2005, 108, 33–42. [Google Scholar] [CrossRef]
- Xu, X.; Chen, X.; Xu, X.; Lu, T.; Wang, X.; Yang, L.; Jing, X. BCNU-Loaded PEG–PLLA ultrafine fibers and their in vitro antitumor activity against Glioma C6 cells. J. Control Release 2006, 114, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, X.; Wang, Z.; Jing, X. Ultrafine PEG–PLA fibers loaded with both paclitaxel and doxorubicin hydrochloride and their in vitro cytotoxicity. Eur. J. Pharm. Biopharm. 2009, 72, 18–25. [Google Scholar] [CrossRef]
- Wang, B.; Li, H.; Yao, Q.; Zhang, Y.; Zhu, X.; Xia, T.; Wang, J.; Li, G.; Li, X.; Ni, S. Local in vitro delivery of rapamycin from electrospun PEO/PDLLA nanofibers for glioblastoma treatment. Biomed. Pharmacother. 2016, 83, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Su, C.H.; Yang, S.T.; Huang, Y.C.; Lee, W.H.; Wang, Y.C.; Liu, S.C.; Liu, S.J. Advanced interstitial chemotherapy combined with targeted treatment of malignant glioma in rats by using drug-loaded nanofibrous membranes. Oncotarget 2016, 7, 59902–59916. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Cui, Y.; Zheng, L.; Chow, P.K.-H.; Wang, C.-H. Development of a gene/drug dual delivery system for brain tumor therapy: Potent inhibition via RNA interference and synergistic effects. Biomaterials 2013, 34, 7483–7494. [Google Scholar] [CrossRef]
- Ni, S.; Fan, X.; Wang, J.; Qi, H.; Li, X. Biodegradable implants efficiently deliver combination of paclitaxel and temozolomide to glioma C6 cancer cells in vitro. Ann. Biomed. Eng. 2014, 42, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Steffens, L.; Morás, A.M.; Arantes, P.R.; Masterson, K.; Cao, Z.; Nugent, M.; Moura, D.J. Electrospun PVA-dacarbazine nanofibers as a novel nano brain-implant for treatment of glioblastoma: In Silico and In Vitro characterization. Eur. J. Pharm. Sci. 2020, 143, 105183. [Google Scholar] [CrossRef]
- Brudno, Y.; Pezone, M.J.; Snyder, T.K.; Uzun, O.; Moody, C.T.; Aizenberg, M.; Mooney, D.J. Replenishable drug depot to combat post-resection cancer recurrence. Biomaterials 2018, 178, 373–382. [Google Scholar] [CrossRef]
- Ranganath, S.H.; Kee, I.; Krantz, W.B.; Chow, P.K.-H.; Wang, C.-H. Hydrogel matrix entrapping PLGA-paclitaxel microspheres: Drug delivery with near zero-order release and implantability advantages for malignant brain tumour chemotherapy. Pharm. Res. 2009, 26, 2101–2114. [Google Scholar] [CrossRef]
- Zhao, M.; Danhier, F.; Bastiancich, C.; Joudiou, N.; Ganipineni, L.P.; Tsakiris, N.; Gallez, B.; Des Rieux, A.; Jankovski, A.; Bianco, J. Post-resection treatment of glioblastoma with an injectable nanomedicine-loaded photopolymerizable hydrogel induces long-term survival. Int. J. Pharm. 2018, 548, 522–529. [Google Scholar] [CrossRef]
- Zhao, M.; Bozzato, E.; Joudiou, N.; Ghiassinejad, S.; Danhier, F.; Gallez, B.; Préat, V. Codelivery of paclitaxel and temozolomide through a photopolymerizable hydrogel prevents glioblastoma recurrence after surgical resection. J. Control Release 2019, 309, 72–81. [Google Scholar] [CrossRef]
- Fourniols, T.; Randolph, L.D.; Staub, A.; Vanvarenberg, K.; Leprince, J.G.; Préat, V.; des Rieux, A.; Danhier, F. Temozolomide-Loaded photopolymerizable PEG-DMA-based hydrogel for the treatment of glioblastoma. J. Control Release 2015, 210, 95–104. [Google Scholar] [CrossRef]
- de la Puente, P.; Fettig, N.; Luderer, M.J.; Jin, A.A.; Shah, S.; Muz, B.; Kapoor, V.; Goddu, S.M.; Salama, N.N.; Tsien, C. Injectable hydrogels for localized chemotherapy and radiotherapy in brain tumors. J. Pharm. Sci. 2018, 107, 922–933. [Google Scholar] [CrossRef]
- Kim, S.; Nishimoto, S.K.; Bumgardner, J.D.; Haggard, W.O.; Gaber, M.W.; Yang, Y. A chitosan/β-glycerophosphate thermo-sensitive gel for the delivery of ellagic acid for the treatment of brain cancer. Biomaterials 2010, 31, 4157–4166. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-W.; Chen, P.-Y.; Wei, K.-C.; Huang, C.-Y.; Wang, C.-K.; Yang, H.-W. Rapid in situ MRI traceable gel-forming dual-drug delivery for synergistic therapy of brain tumor. Theranostics 2017, 7, 2524. [Google Scholar] [CrossRef] [PubMed]
- Bastiancich, C.; Vanvarenberg, K.; Ucakar, B.; Pitorre, M.; Bastiat, G.; Lagarce, F.; Préat, V.; Danhier, F. Lauroyl-gemcitabine-loaded lipid nanocapsule hydrogel for the treatment of glioblastoma. J. Control Release 2016, 225, 283–293. [Google Scholar] [CrossRef]
- Bastiancich, C.; Bianco, J.; Vanvarenberg, K.; Ucakar, B.; Joudiou, N.; Gallez, B.; Bastiat, G.; Lagarce, F.; Préat, V.; Danhier, F. Injectable nanomedicine hydrogel for local chemotherapy of glioblastoma after surgical resection. J. Control Release 2017, 264, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Vellimana, A.K.; Recinos, V.R.; Hwang, L.; Fowers, K.D.; Li, K.W.; Zhang, Y.; Okonma, S.; Eberhart, C.G.; Brem, H.; Tyler, B.M. Combination of paclitaxel thermal gel depot with temozolomide and radiotherapy significantly prolongs survival in an experimental rodent glioma model. J. Neuro-Oncol. 2013, 111, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastiancich, C.; Bozzato, E.; Luyten, U.; Danhier, F.; Bastiat, G.; Préat, V. Drug combination using an injectable nanomedicine hydrogel for glioblastoma treatment. Int. J. Pharm. 2019, 559, 220–227. [Google Scholar] [CrossRef]
- Kim, J.I.; Kim, B.; Chun, C.; Lee, S.H.; Song, S.-C. MRI-monitored long-term therapeutic hydrogel system for brain tumors without surgical resection. Biomaterials 2012, 33, 4836–4842. [Google Scholar] [CrossRef]
- Akbar, U.; Jones, T.; Winestone, J.; Michael, M.; Shukla, A.; Sun, Y.; Duntsch, C. Delivery of temozolomide to the tumor bed via biodegradable gel matrices in a novel model of intracranial glioma with resection. J. Neuro-Oncol. 2009, 94, 203–212. [Google Scholar] [CrossRef]
- Ozeki, T.; Kaneko, D.; Hashizawa, K.; Imai, Y.; Tagami, T.; Okada, H. Improvement of survival in C6 rat glioma model by a sustained drug release from localized PLGA microspheres in a thermoreversible hydrogel. Int. J. Pharm. 2012, 427, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, T.; Hashizawa, K.; Kaneko, D.; Imai, Y.; Okada, H. Treatment of rat brain tumors using sustained-release of camptothecin from poly (lactic-co-glycolic acid) microspheres in a thermoreversible hydrogel. Chem. Pharm. Bull. 2010, 58, 1142–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Benny, O.; Joki, T.; Menon, L.G.; Machluf, M.; Abe, T.; Carroll, R.S.; Black, P.M. Novel local drug delivery system using thermoreversible gel in combination with polymeric microspheres or liposomes. Anticancer Res. 2010, 30, 1057–1064. [Google Scholar]
- Arai, T.; Joki, T.; Akiyama, M.; Agawa, M.; Mori, Y.; Yoshioka, H.; Abe, T. Novel drug delivery system using thermoreversible gelation polymer for malignant glioma. J. Neuro-Oncol. 2006, 77, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Moreno Garcia, V.; Basu, B.; Molife, L.R.; Kaye, S.B. Combining antiangiogenics to overcome resistance: Rationale and clinical experience. Clin. Cancer Res. 2012, 18, 3750–3761. [Google Scholar] [CrossRef] [Green Version]
- Messaoudi, K.; Clavreul, A.; Lagarce, F. Toward an effective strategy in glioblastoma treatment. Part I: Resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov. Today 2015, 20, 899–905. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Evdokiou, A.; Bouralexis, S.; Atkins, G.J.; Chai, F.; Hay, S.; Clayer, M.; Findlay, D.M. Chemotherapeutic agents sensitize osteogenic sarcoma cells, but not normal human bone cells, to Apo2L/TRAIL-induced apoptosis. Int. J. Cancer 2002, 99, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.H.K.; Lee, T.; Wang, C.-H. Simulation of intratumoral release of etanidazole: Effects of the size of surgical opening. J. Pharm. Sci. 2003, 92, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, C.; Li, A.; Jing, W.; Sun, P.; Huang, X.; Liu, Y.; Zhang, S.; Du, W.; Zhang, R.; et al. Immunostimulant hydrogel for the inhibition of malignant glioma relapse post-resection. Nat. Nanotechnol. 2021, 16, 538–548. [Google Scholar] [CrossRef]
- Silakari, O.; Singh, P. ADMET Tools: Prediction and Assessment of Chemical ADMET Properties of NCEs. In Concepts and Experimental Protocols of Modelling and Informatics in Drug Design; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Mittal, B. Pharmacokinetics and Preformulation. In How to Develop Robust Solid Oral Dosage Forms from Conception to Post-Approval; Elsevier: Amsterdam, The Netherlands, 2017; pp. 17–37. [Google Scholar] [CrossRef]
- Natu, M.V.; de Sousa, H.C.; Gil, M.H. Effects of drug solubility, state and loading on controlled release in bicomponent electrospun fibers. Int. J. Pharm. 2010, 397, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Hardy, R.J.; Gu, X. Effect of drug solubility on polymer hydration and drug dissolution from polyethylene oxide (PEO) matrix tablets. AAPS PharmSciTech 2008, 9, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Carmustine|C5H9Cl2N3O2. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Carmustine (accessed on 31 May 2021).
- Hydroperoxycyclophosphamide|C7H15Cl2N2O4P. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/38347 (accessed on 31 May 2021).
- Paclitaxel|C47H51NO14. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/36314 (accessed on 31 May 2021).
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef]
- Kortagere, S.; Krasowski, M.D.; Ekins, S. The importance of discerning shape in molecular pharmacology. Trends Pharmacol. Sci. 2009, 30, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Quan, P.; Fang, L. Effect of drug physicochemical properties on drug release and their relationship with drug skin permeation behaviors in hydroxyl pressure sensitive adhesive. Eur. J. Pharm. Sci. 2016, 93, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, R.J. Properties of Organic Compounds, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Chaparro, F.J.; Presley, K.F.; Coutinho da Silva, M.A.; Mandan, N.; Colachis, M.L.; Posner, M.; Arnold, R.M.; Fan, F.; Moraes, C.R.; Lannutti, J.J. Sintered electrospun poly(ε-caprolactone)–poly(ethylene terephthalate) for drug delivery. J. Appl. Polym. Sci. 2019, 136, 47731. [Google Scholar] [CrossRef]
- Gilli, P.; Pretto, L.; Bertolasi, V.; Gilli, G. Predicting hydrogen-bond strengths from acid-base molecular properties. The pK(a) slide rule: Toward the solution of a long-lasting problem. ACC Chem. Res. 2009, 42, 33–44. [Google Scholar] [CrossRef] [PubMed]
- House, J.E. Inorganic Chemistry, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Hong, W.; Zhao, X.; Zhou, J.; Suo, Z. A theory of coupled diffusion and large deformation in polymeric gels. J. Mech. Phys. Solids 2008, 56, 1779–1793. [Google Scholar] [CrossRef]
- Omelczuk, M.O.; McGinity, J.W. The influence of polymer glass transition temperature and molecular weight on drug release from tablets containing poly(DL-lactic acid). Pharm. Res. 1992, 9, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Daviau, T.; Ying, P.; Zhao, Y.; Nowotnik, D.; Clow, C.S.; Tyler, B.; Brem, H. Effects of GLIADEL® wafer initial molecular weight on the erosion of wafer and release of BCNU. J. Control Release 1996, 42, 83–92. [Google Scholar] [CrossRef]
- Lassalle, V.; Ferreira, M.L. PLGA based drug delivery systems (DDS) for the sustained release of insulin: Insight into the protein/polyester interactions and the insulin release behavior. J. Chem. Technol. Biotechnol. 2010, 85, 1588–1596. [Google Scholar] [CrossRef]
- Chen, S.; Huang, X.; Cai, X.; Lu, J.; Yuan, J.; Shen, J. The influence of fiber diameter of electrospun poly (lactic acid) on drug delivery. Fibers Polym. 2012, 13, 1120–1125. [Google Scholar] [CrossRef]
- Zeng, J.; Haoqing, H.; Schaper, A.; Wendorff, J.H.; Greiner, A. Poly-L-Lactide nanofibers by electrospinning–Influence of solution viscosity and electrical conductivity on fiber diameter and fiber morphology. e-Polymers 2003, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Hu, G.; Ouyang, D. A numerical study of the distribution of chemotherapeutic drug carmustine in brain glioblastoma. Drug Deliv. Transl. Res. 2021. [Google Scholar] [CrossRef] [PubMed]





| Polymer | Drug(s) | Pre-Encapsulation Method |
|---|---|---|
| p(CPP:SA) | 4HC [55], BCNU [55,64], Camptothecin [65], DXR [66], Lactacystin [67], Memantine [68], PTX [31,55], Riluzole [68], Synthetic endostatin fragment [69], TMZ [64,70] | Solvent evaporation |
| p(CPP:SA) | Mitoxantrone [71] | Mix-melt |
| p(CPP:SA) and PLGA | BCNU [72], PTX [72] | Solvent evaporation |
| PLGA | ADS-I [73] | W/O/W double emulsion |
| BCNU [74,75] | Vortex mix [75], solvent evaporation [74] | |
| DSF [76] | Mortar and pestle | |
| PTX [30,77] | Spray dried microparticles [77], Supercritical CO2 foaming [30] | |
| TMZ [74] | Solvent evaporation | |
| PLGA and PEG | PTX [77] | Spray dried microparticles |
| p(DAPPG-EOP) | PTX [78] | In-line homogenizer to create microspheres |
| PCL-LA | TMZ [74] | Solvent evaporation |
| Hydrogel Matrix | Drug Carrier System | Drug | Crosslinking Method |
|---|---|---|---|
| Alginate | PLGA microparticles | PTX [28,112] | Ionic |
| Chitosan/glutaraldehyde | Alginate microparticles | TMZ [116], 131I [116] | Ionic |
| Chitosan/β-glycerophosphate | - | Ellagic acid [117] | Temperature |
| CMC-g-PNI PAAmMA/DTPAGd | BSA nanoparticles | EPI [118], PTX [118] | Temperature |
| Lipid nanocapsule | - | Gemcitabine [119,120,121], PTX [121] | Drug |
| P-CoFe2O4 NPs and PPZ | - | Irinotecan [115] | Temperature |
| PEG-DMA | PLGA nanoparticles | PTX [113,114], TMZ [114] | UV light |
| PEG-p(CL-co-TMC) micelles | TMZ [122] | UV light | |
| PLGA/ATEC/TEC | - | TMZ [123] | Plasticizer |
| PLGA/PEG | - | PTX [124] | Temperature |
| Thermoreversible gelation polymer | PLGA microparticles | CPT [125,126], DXR [127], VCR [125] | Temperature |
| Liposome | DXR [127] | Temperature | |
| - | DXR [127,128] | Temperature |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pena, E.S.; Graham-Gurysh, E.G.; Bachelder, E.M.; Ainslie, K.M. Design of Biopolymer-Based Interstitial Therapies for the Treatment of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 13160. https://doi.org/10.3390/ijms222313160
Pena ES, Graham-Gurysh EG, Bachelder EM, Ainslie KM. Design of Biopolymer-Based Interstitial Therapies for the Treatment of Glioblastoma. International Journal of Molecular Sciences. 2021; 22(23):13160. https://doi.org/10.3390/ijms222313160
Chicago/Turabian StylePena, Erik S., Elizabeth G. Graham-Gurysh, Eric M. Bachelder, and Kristy M. Ainslie. 2021. "Design of Biopolymer-Based Interstitial Therapies for the Treatment of Glioblastoma" International Journal of Molecular Sciences 22, no. 23: 13160. https://doi.org/10.3390/ijms222313160
APA StylePena, E. S., Graham-Gurysh, E. G., Bachelder, E. M., & Ainslie, K. M. (2021). Design of Biopolymer-Based Interstitial Therapies for the Treatment of Glioblastoma. International Journal of Molecular Sciences, 22(23), 13160. https://doi.org/10.3390/ijms222313160