
Fungal Secondary Metabolites as Inhibitors of the Ubiquitin–Proteasome System


Abstract
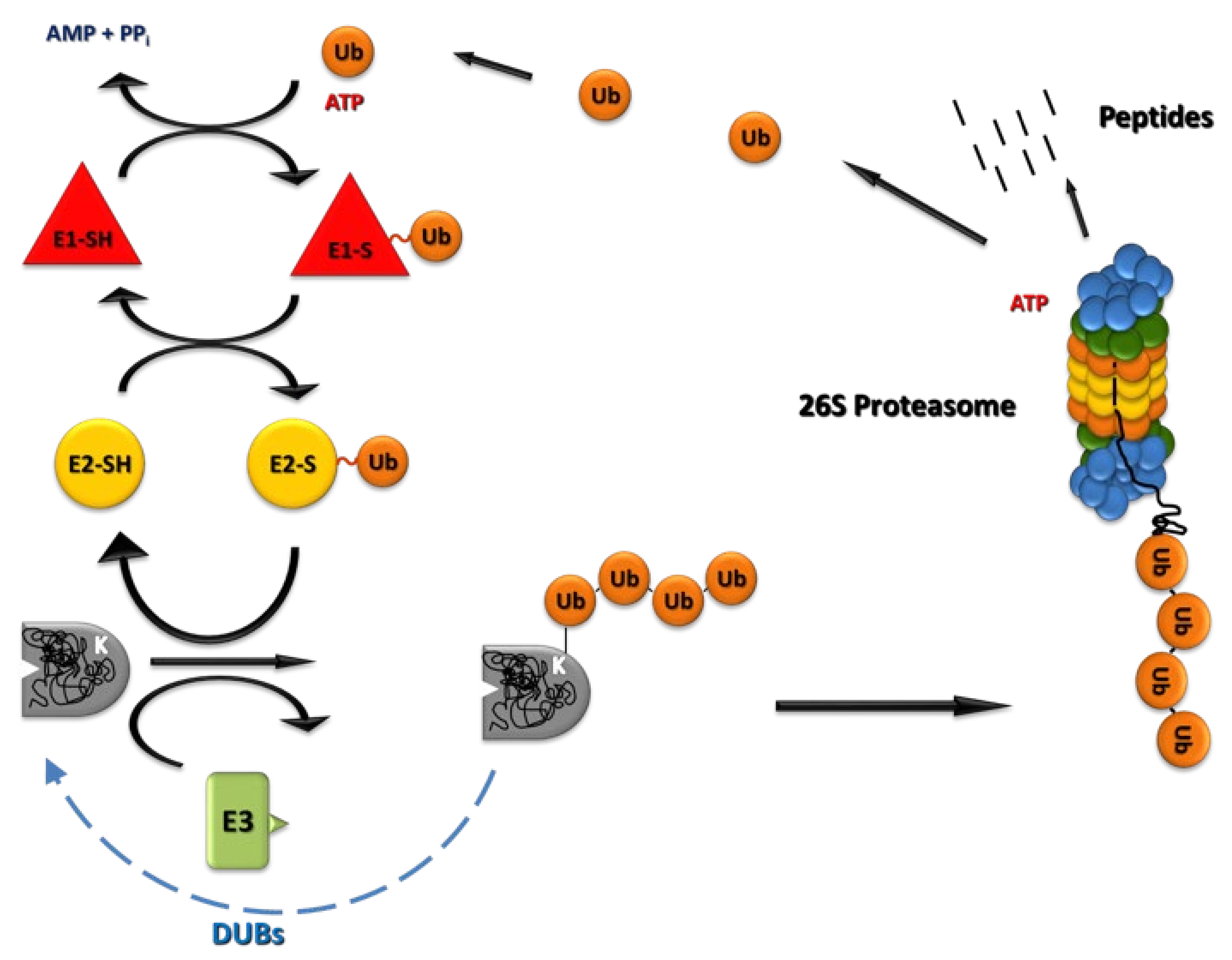
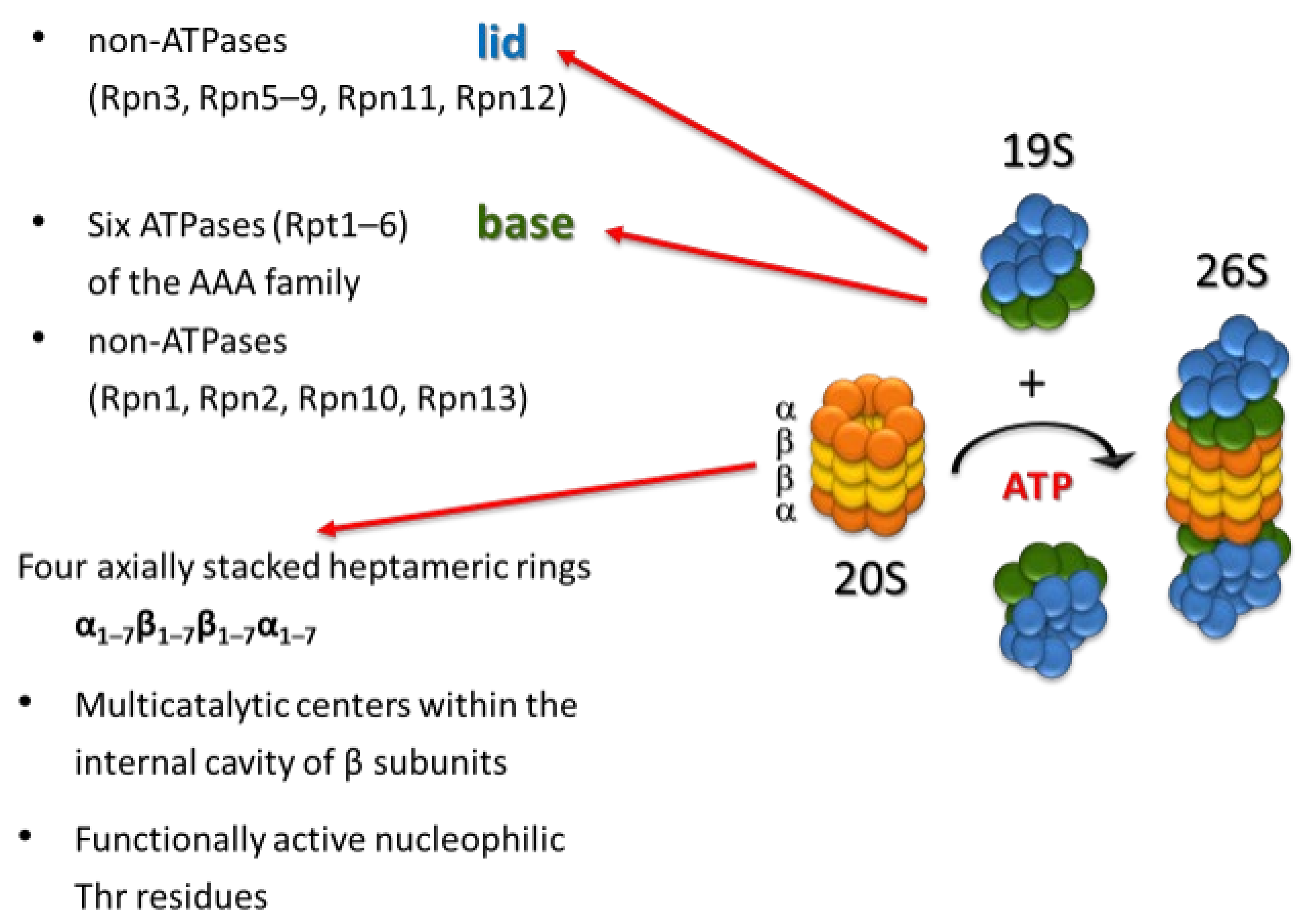
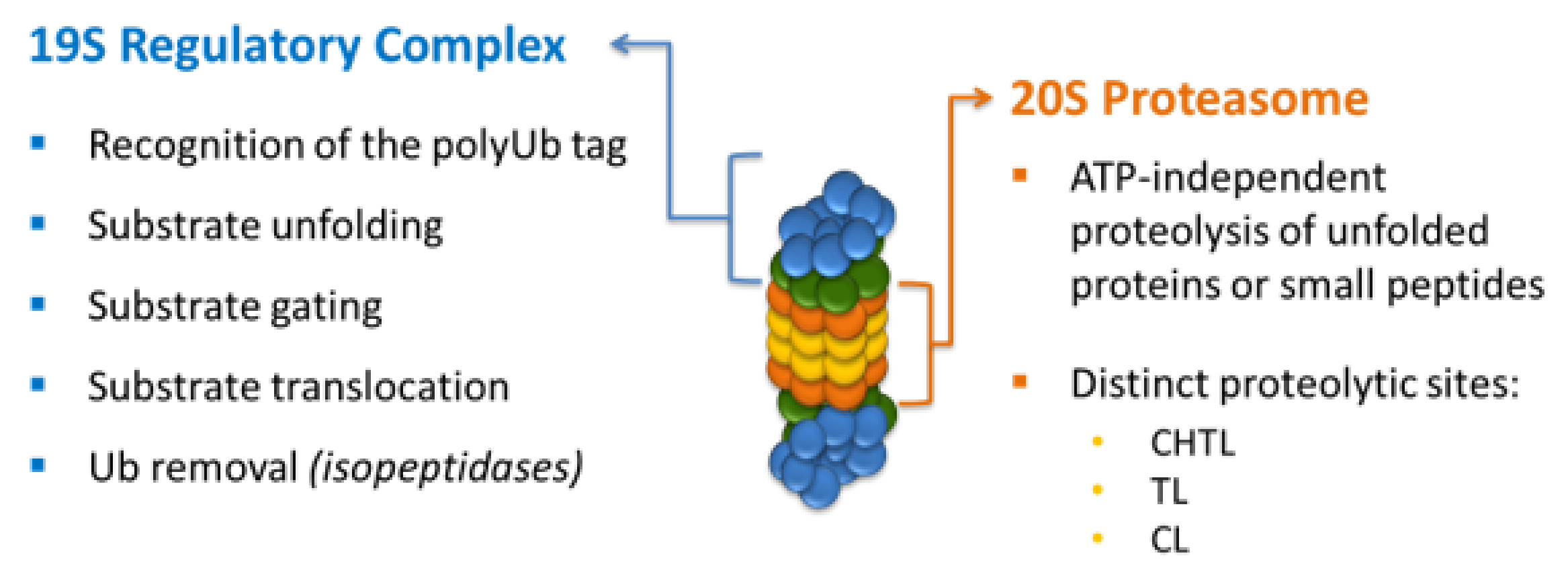
:1. Introduction
2. Proteasome Inhibitors
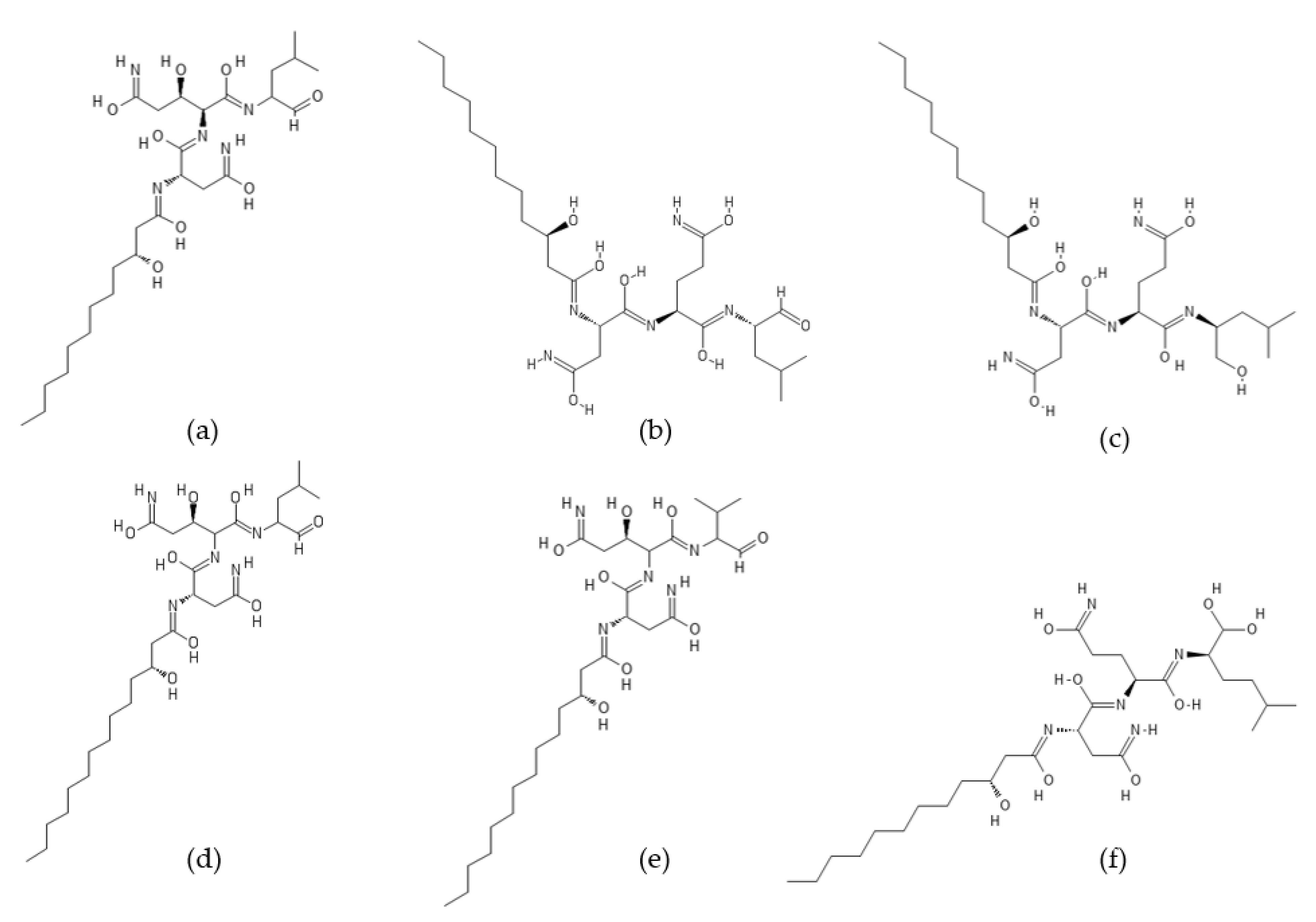
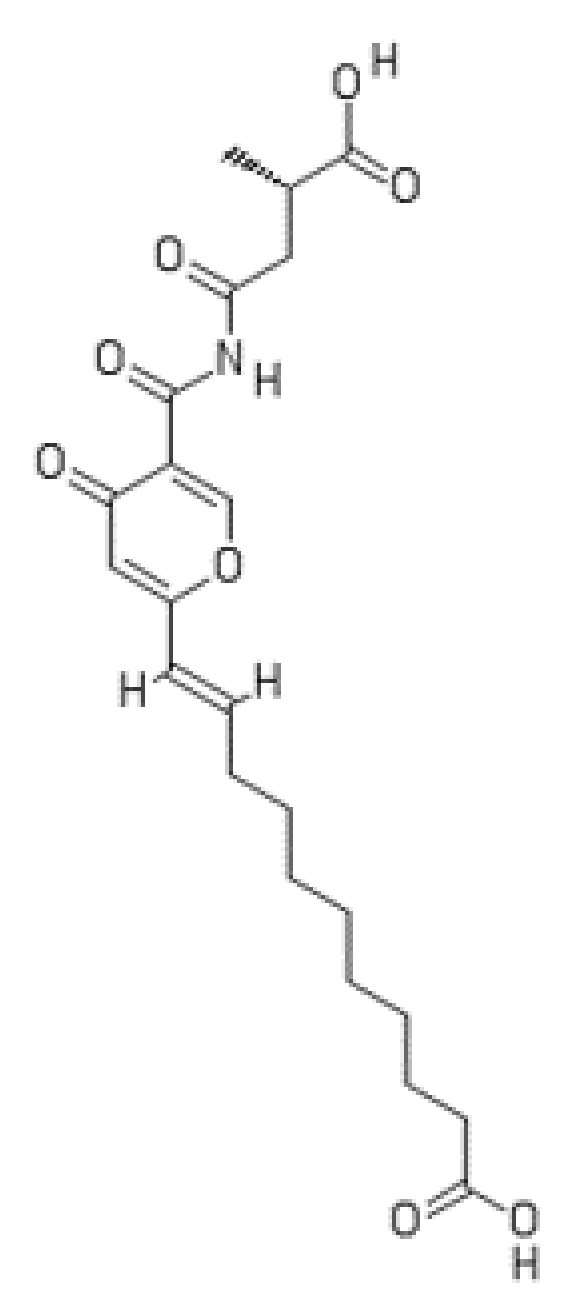
2.1. Fellutamides
2.2. TMC-95A-Related Peptides
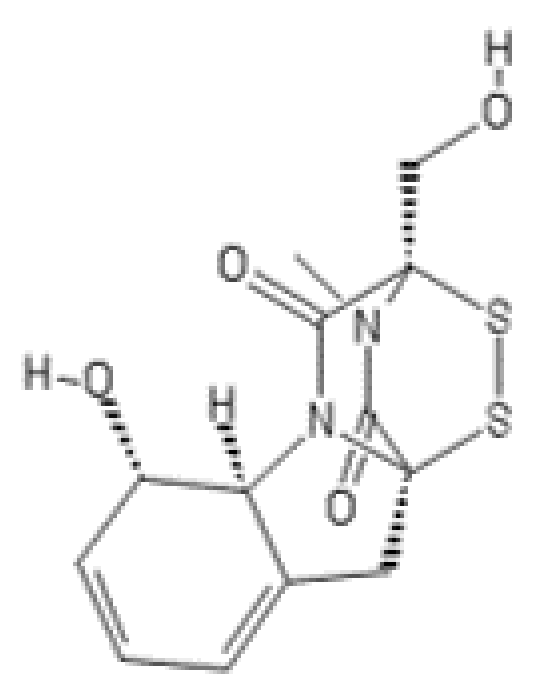
2.3. Gliotoxin
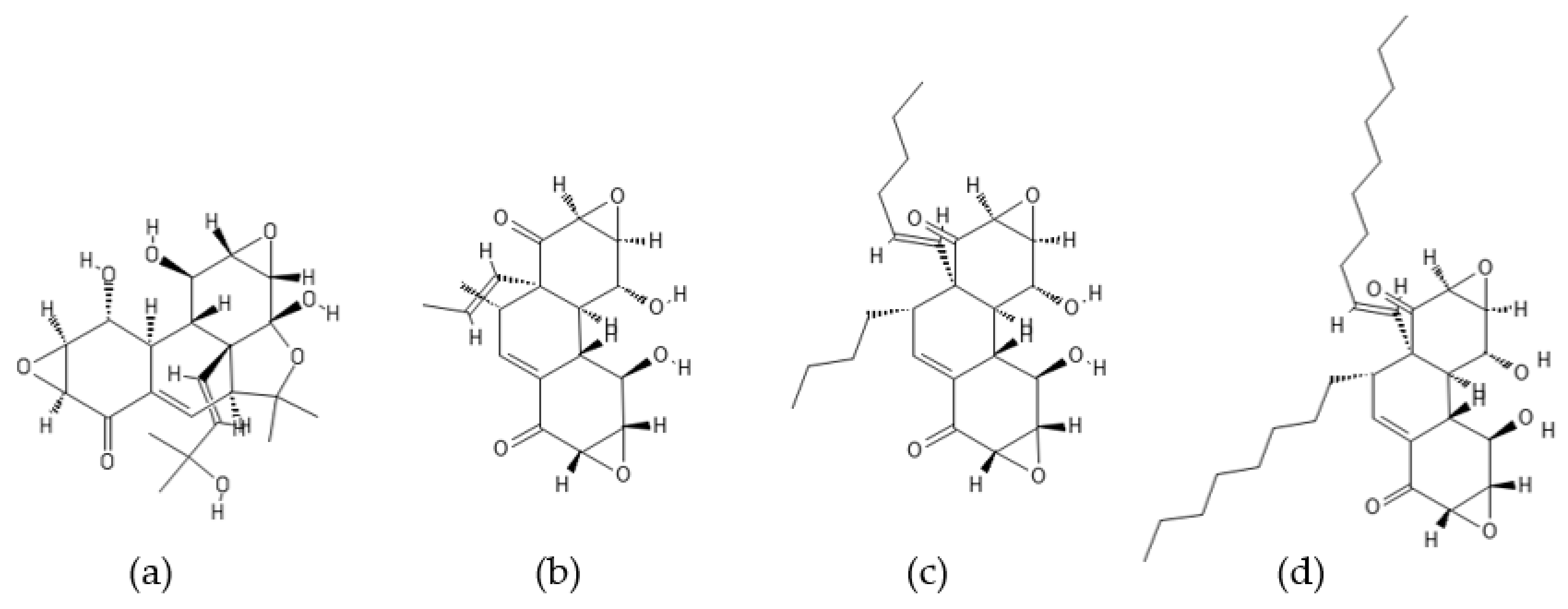
2.4. Epoxyphomalins
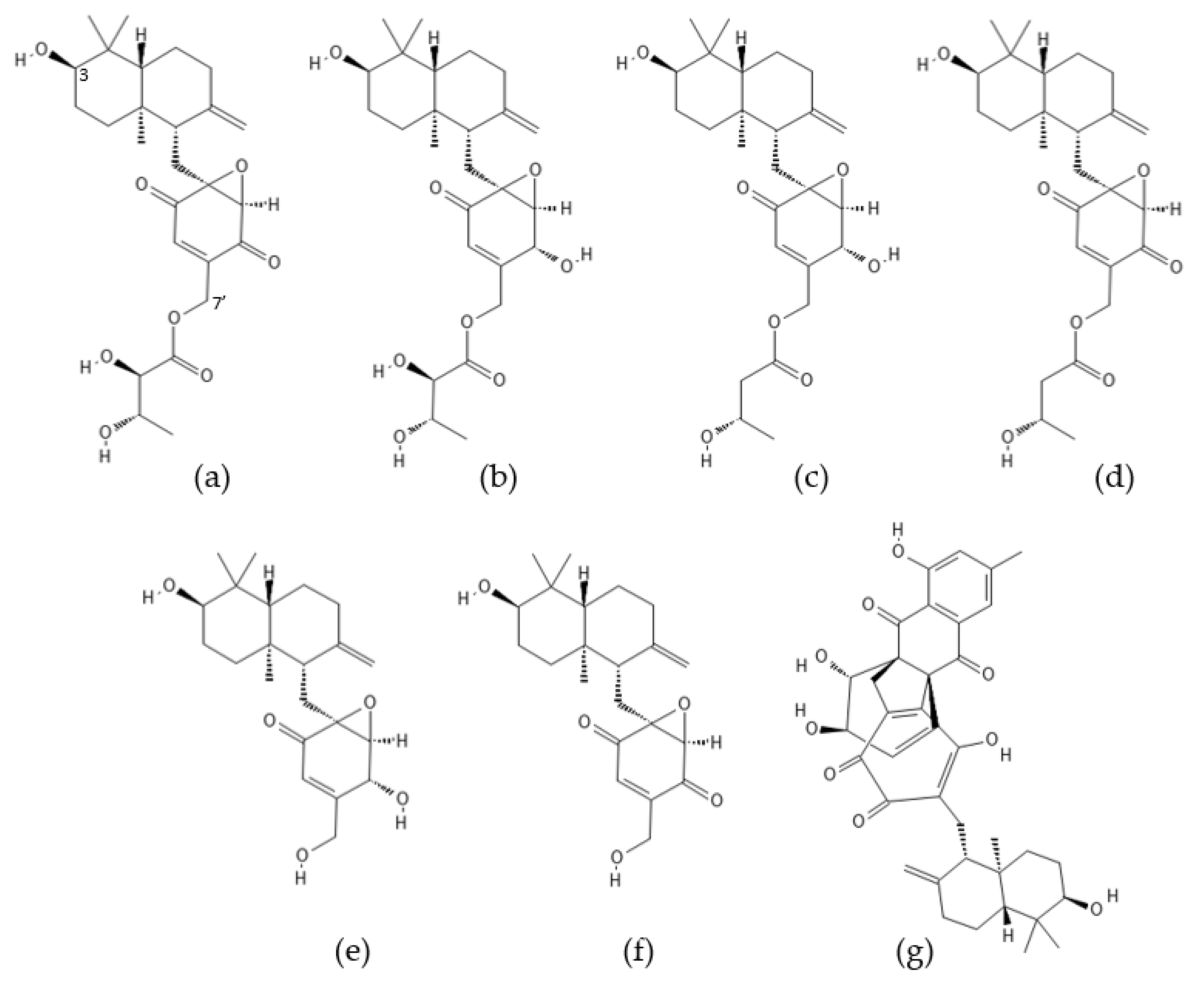
2.5. Neomacrophorins
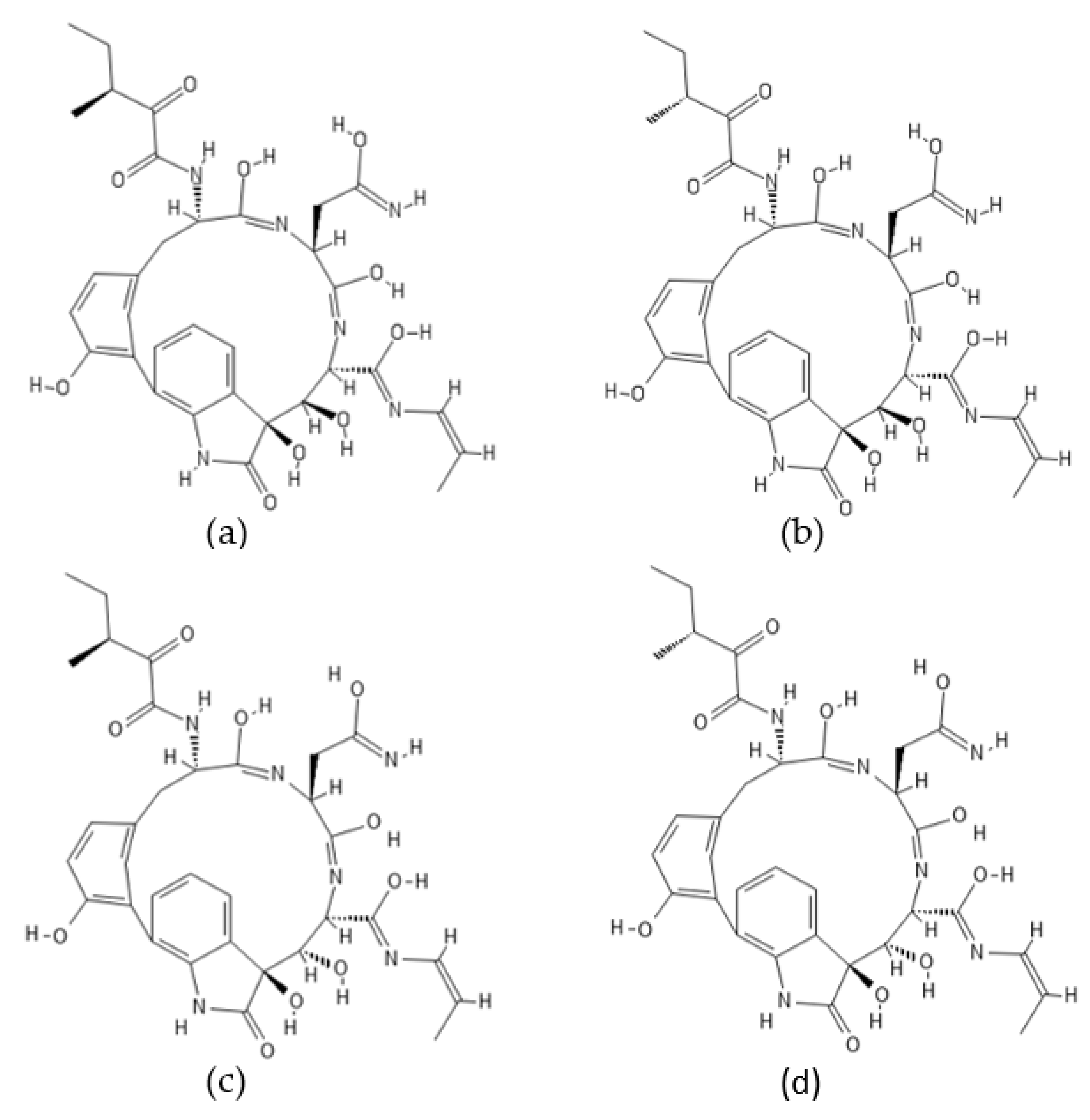
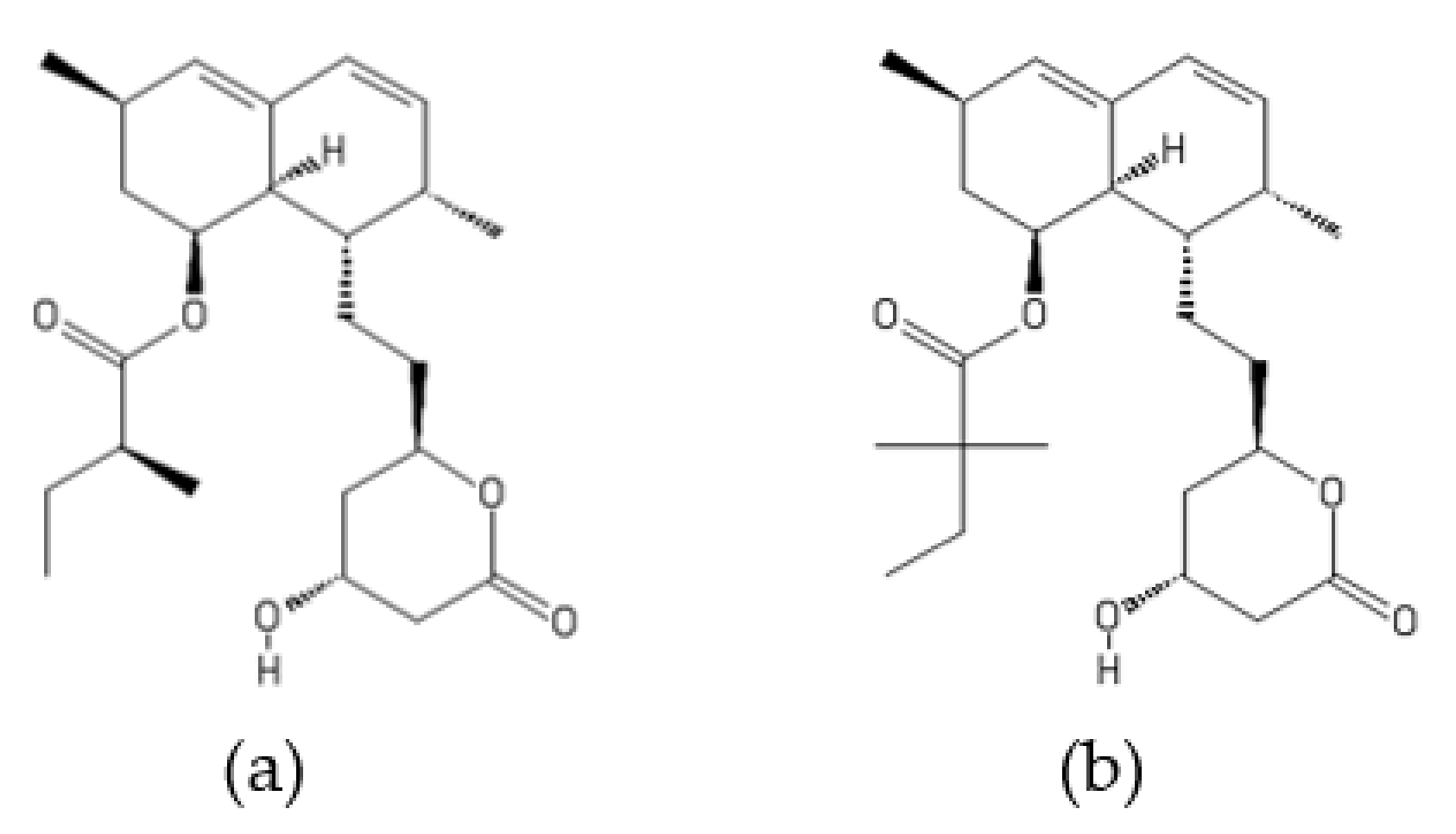
2.6. Trilongins
2.7. Terrein
2.8. Pyrrolizilactone
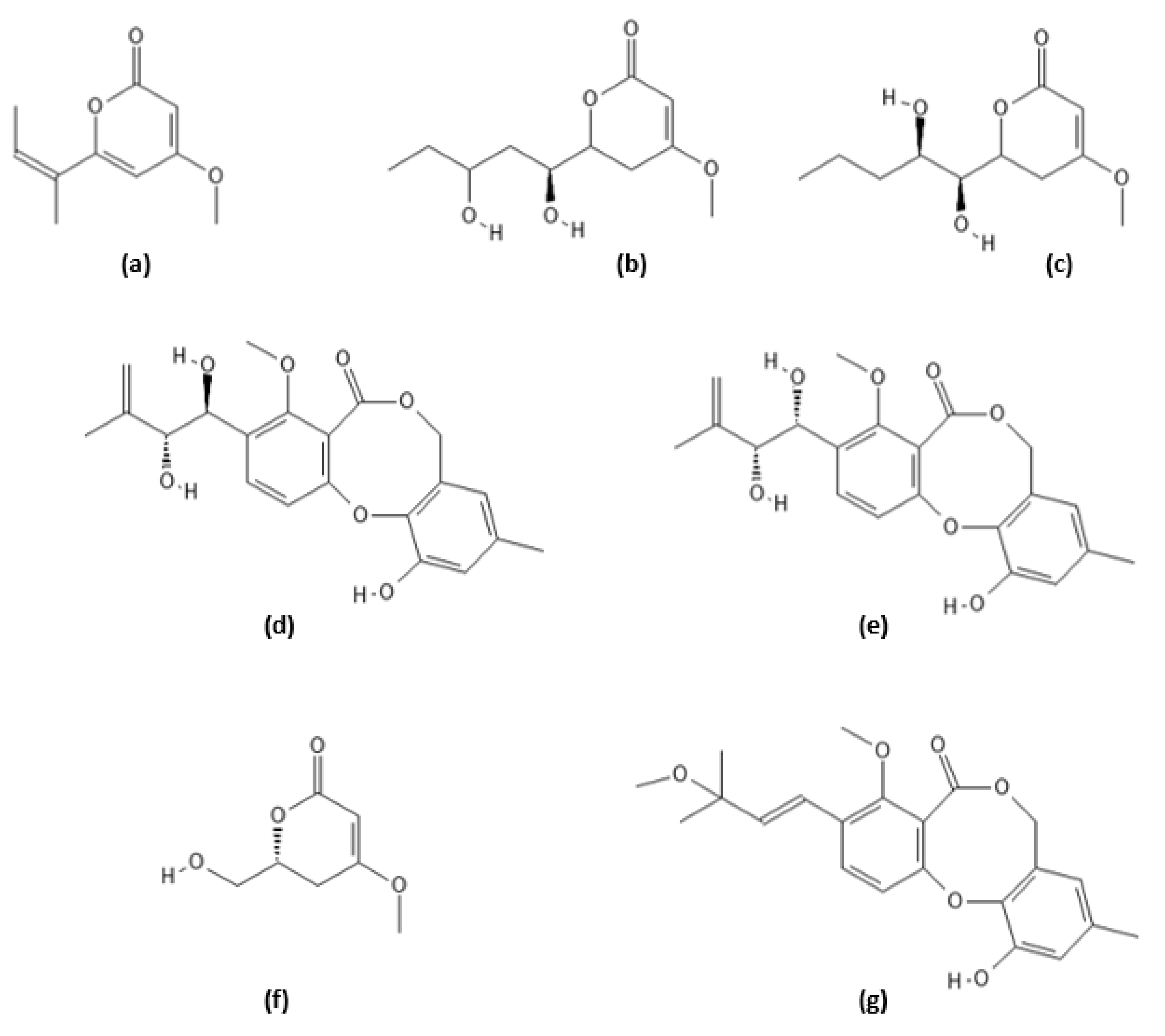
2.9. Secondary Metabolites of Pestalotiopsis sydowiana
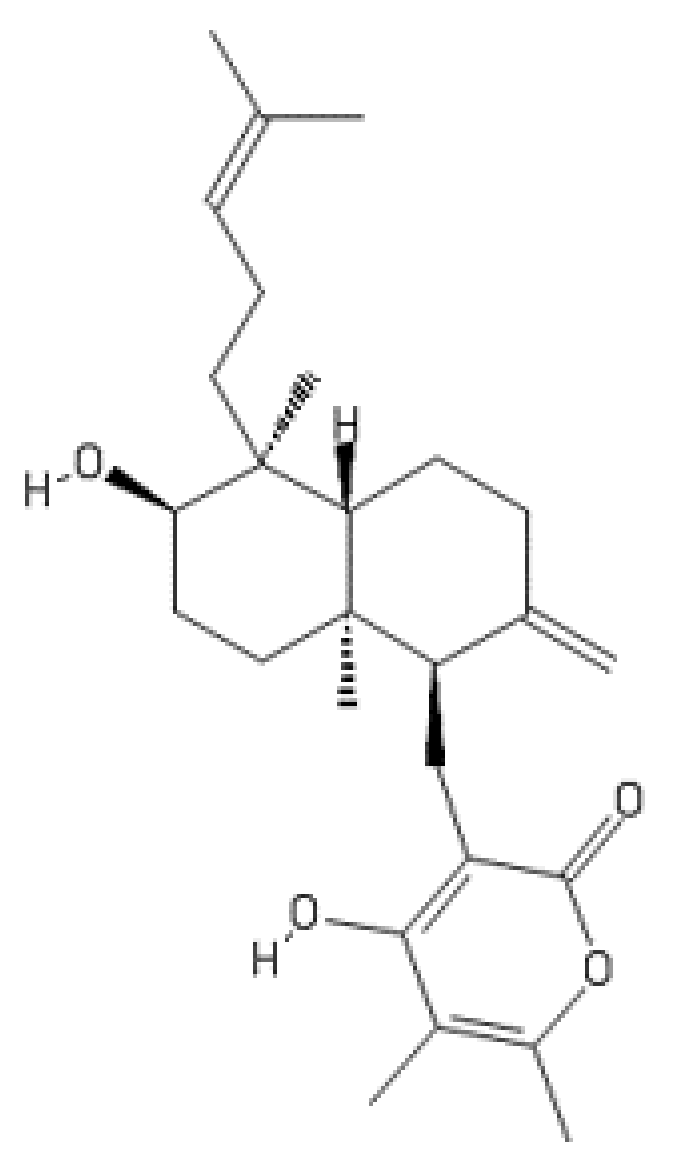
2.10. Higginsianin B
3. Inhibitors of the E1 Ubiquitin-Activating Enzyme
3.1. Panepophenanthrin
3.2. Himeic Acid A
4. Inhibitors of E3 Ligases
4.1. Chlorofusin
4.2. Hexylitaconic Acid
4.3. Potential Inhibitors of the MDM2–p53 Interaction
4.4. Statins as Inhibitors of the SCFSkp2 E3 Ligase
5. Inhibitors of Deubiquitinases
5.1. USP7 Inhibitors
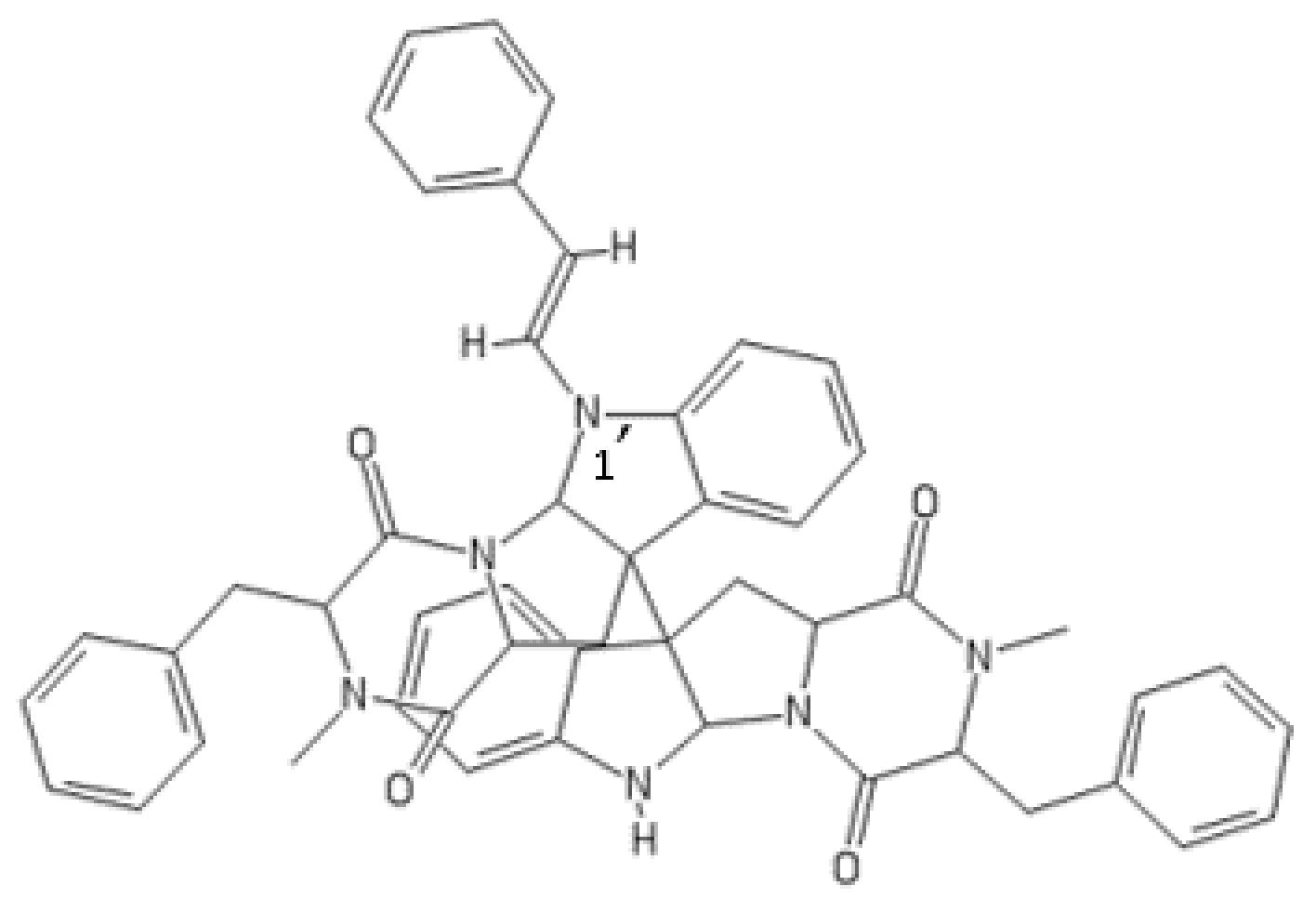
1′-(2-Phenylethylene)-ditryptophenaline
5.2. Rpn11 Inhibitors
Gliotoxin and Other Epidithiodiketopiperazines
5.3. USP5/USP4 Inhibitors
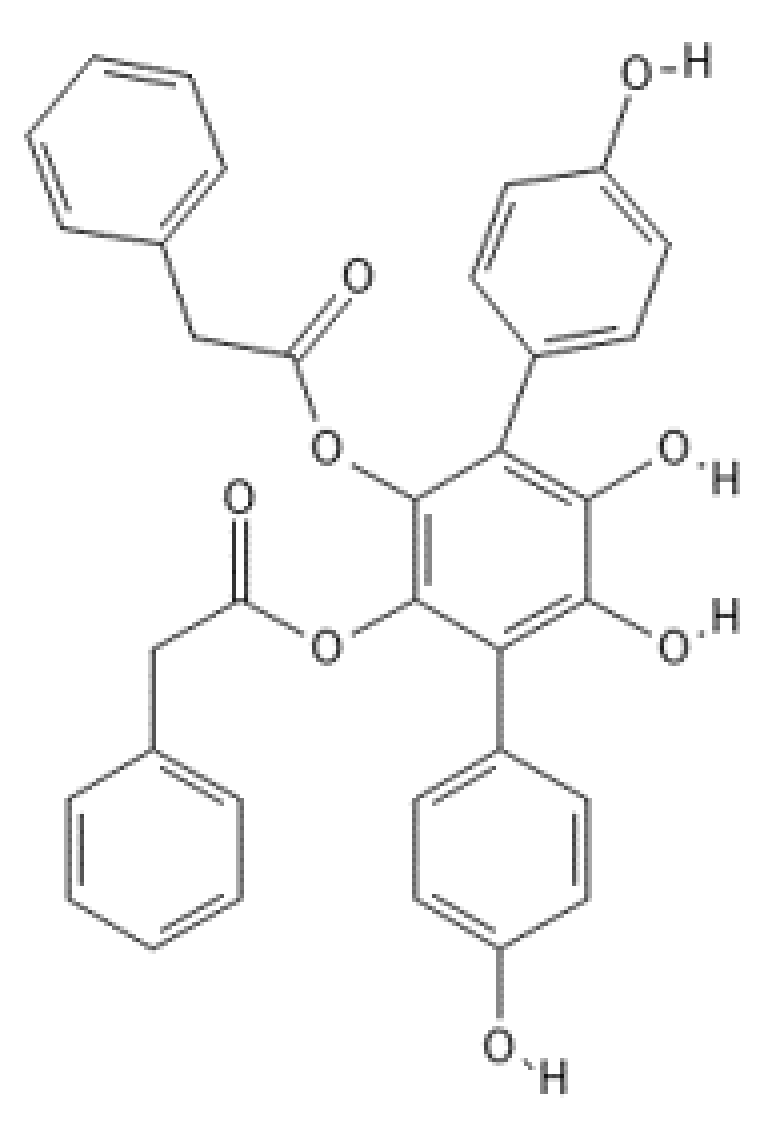
Vialinin A
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Ciechanover, A. Tracing the history of the ubiquitin proteolytic system: The pioneering article. Biochem. Biophys. Res. Commun. 2009, 387, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The unravelling of the ubiquitin system. Nat. Rev. Mol. Cell Biol. 2015, 16, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Greene, E.R.; Dong, K.C.; Martin, A. Understanding the 26S proteasome molecular machine from a structural and conformational dynamics perspective. Curr. Opin. Struct. Biol. 2020, 61, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y. Structure, dynamics and function of the 26S proteasome. In Macromolecular Protein Complexes III: Structure and Function; Harris, J.R., Marles-Wright, J., Eds.; Springer: Cham, Switzerland; Copenhagen, Denmark, 2020; pp. 1–151. ISBN 9783030589714. [Google Scholar]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Stanhill, A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Pickart, C.M.; Cohen, R.E. Proteasomes and their kin: Proteases in the machine age. Nat. Rev. Mol. Cell Biol. 2004, 5, 177–187. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Mykles, D.L. Intracellular proteinases of invertebrates: Calcium-dependent and proteasome/ubiquitin-dependent systems. Int. Rev. Cytol. 1998, 184, 157–289. [Google Scholar]
- Naujokat, C.; Šarić, T. Concise review: Role and function of the ubiquitin-proteasome system in mammalian stem and progenitor cells. Stem Cells 2007, 25, 2408–2418. [Google Scholar] [CrossRef]
- Kniepert, A.; Groettrup, M. The unique functions of tissue-specific proteasomes. Trends Biochem. Sci. 2014, 39, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vierstra, R.D. The ubiquitin/26S proteasome pathway, the complex last chapter in the life of many plant proteins. Trends Plant Sci. 2003, 8, 135–142. [Google Scholar] [CrossRef]
- Adams, E.H.G.; Spoel, S.H. The ubiquitin-proteasome system as a transcriptional regulator of plant immunity. J. Exp. Bot. 2018, 69, 4529–4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.; Hilt, W.; Richter-Ruoff, B.; Gonen, H.; Ciechanover, A.; Wolf, D.H. The 26S proteasome of the yeast Saccharomyces cerevisiae. FEBS Lett. 1994, 355, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Staszczak, M. The role of the ubiquitin–proteasome system in the response of the ligninolytic fungus Trametes versicolor to nitrogen deprivation. Fungal Genet. Biol. 2008, 45, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Staszczak, M. The 26S proteasome of the lignin-degrading Basidiomycete Phlebia radiata. Enzyme Microb. Technol. 2007, 40, 347–353. [Google Scholar] [CrossRef]
- Liu, F.; Walters, K.J. Multitasking with ubiquitin through multivalent interactions. Trends Biochem. Sci. 2010, 35, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Kannt, A.; Đikić, I. Expanding the arsenal of E3 ubiquitin ligases for proximity-induced protein degradation. Cell Chem. Biol. 2021, 28, 1014–1031. [Google Scholar] [CrossRef]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.; Spaller, B.L.; Matouschek, A. Mechanisms of substrate recognition by the 26S proteasome. Curr. Opin. Struct. Biol. 2021, 67, 161–169. [Google Scholar] [CrossRef]
- Sahu, I.; Glickman, M.H. Structural insights into substrate recognition and processing by the 20S proteasome. Biomolecules 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Wójcik, C.; DeMartino, G.N. Intracellular localization of proteasomes. Int. J. Biochem. Cell Biol. 2003, 35, 579–589. [Google Scholar] [CrossRef]
- Ehlinger, A.; Walters, K.J. Structural Insights into Proteasome Activation by the 19S Regulatory Particle. Biochemistry 2013, 52, 3618–3628. [Google Scholar] [CrossRef] [PubMed]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R.; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Songyang, Z.; Goldberg, A.L. Why does threonine, and not serine, function as the active site nucleophile in proteasomes? J. Biol. Chem. 2000, 275, 14831–14837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemüller, E.; Lupas, A.; Stock, D.; Löwe, J.; Huber, R.; Baumeister, W. Proteasome from Thermoplasma acidophilum: A Threonine Protease. Science 1995, 268, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A. Proteasome: A complex protease with a new fold and a distinct mechanism. Structure 1995, 3, 417–420. [Google Scholar] [CrossRef] [Green Version]
- Hewings, D.S.; Flygare, J.A.; Bogyo, M.; Wertz, I.E. Activity-based probes for the ubiquitin conjugation–deconjugation machinery: New chemistries, new tools, and new insights. FEBS J. 2017, 284, 1555–1576. [Google Scholar] [CrossRef] [Green Version]
- Lill, J.R.; Wertz, I.E. Toward understanding ubiquitin-modifying enzymes: From pharmacological targeting to proteomics. Trends Pharmacol. Sci. 2014, 35, 187–207. [Google Scholar] [CrossRef]
- Eletr, Z.M.; Wilkinson, K.D. Regulation of proteolysis by human deubiquitinating enzymes. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 114–128. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.Y.; Muniyappan, S.; Tran, N.N.; Park, H.; Lee, S.B.; Lee, B.H. Deubiquitination reactions on the proteasome for proteasome versatility. Int. J. Mol. Sci. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Aliabadi, F.; Sohrabi, B.; Mostafavi, E.; Pazoki-Toroudi, H.; Webster, T.J. Ubiquitin-proteasome system and the role of its inhibitors in cancer therapy. Open Biol. 2021, 11, 200390. [Google Scholar] [CrossRef]
- Laplante, G.; Zhang, W. Targeting the ubiquitin-proteasome system for cancer therapeutics by small-molecule inhibitors. Cancers 2021, 13, 1–43. [Google Scholar] [CrossRef]
- Fhu, C.W.; Ali, A. Dysregulation of the ubiquitin proteasome system in human malignancies: A window for therapeutic intervention. Cancers 2021, 13, 1513. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, N.; Bustamante, H.; Soza, A.; Burgos, P. Cellular responses to proteasome inhibition: Molecular mechanisms and beyond. Int. J. Mol. Sci. 2019, 20, 3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibaudeau, T.A.; Smith, D.M. A practical review of proteasome pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinjiyan, F.A.; Carbonneau, S.; Ross, N.T. Lead discovery and chemical biology approaches targeting the ubiquitin proteasome system. Bioorg. Med. Chem. Lett. 2017, 27, 4589–4596. [Google Scholar] [CrossRef] [PubMed]
- Staszczak, M. Ubiquitin-proteasome pathway as a target for therapeutic strategies. Postepy Biochem. 2017, 63, 287–303. [Google Scholar] [PubMed]
- Ciechanover, A. The ubiquitin proteolytic system: From an idea to the patient bed. Proc. Am. Thorac. Soc. 2006, 3, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Bibo-Verdugo, B.; Jiang, Z.; Caffrey, C.R.; O’Donoghue, A.J. Targeting proteasomes in infectious organisms to combat disease. FEBS J. 2017, 284, 1503–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longhitano, L.; Tibullo, D.; Giallongo, C.; Lazzarino, G.; Tartaglia, N.; Galimberti, S.; Li Volti, G.; Palumbo, G.A.; Liso, A. Proteasome inhibitors as a possible therapy for SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 1–11. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade®: U.S. FDA Approval for the Treatment of Multiple Myeloma Progressing on Prior Therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Wang, J.; Fang, Y.; Fan, R.A.; Kirk, C.J. Proteasome inhibitors and their pharmacokinetics, pharmacodynamics, and metabolism. Int. J. Mol. Sci. 2021, 22, 11595. [Google Scholar] [CrossRef]
- Guedes, R.A.; Aniceto, N.; Andrade, M.A.P.; Salvador, J.A.R.; Guedes, R.C. Chemical patterns of proteasome inhibitors: Lessons learned from two decades of drug design. Int. J. Mol. Sci. 2019, 20, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Sherman, D.J.; Li, J. Proteasome inhibitors: Harnessing proteostasis to combat disease. Molecules 2020, 25, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antao, A.M.; Tyagi, A.; Kim, K.; Ramakrishna, S. Advances in Deubiquitinating Enzyme Inhibition. Cancers 2020, 1–34. [Google Scholar]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of ubiquitin-specific proteases as a novel anticancer therapeutic strategy. Front. Pharmacol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wan, L.; Dai, X.; Sun, Y.; Wei, W. Functional characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2014, 1845, 277–293. [Google Scholar] [CrossRef] [Green Version]
- Mattern, M.R.; Wu, J.; Nicholson, B. Ubiquitin-based anticancer therapy: Carpet bombing with proteasome inhibitors vs surgical strikes with E1, E2, E3, or DUB inhibitors. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 2014–2021. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Liu, Y.; Zhou, H. Advances in the development ubiquitin-specific peptidase (USP) inhibitors. Int. J. Mol. Sci. 2021, 22, 4546. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin–proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm. Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, G.E.; Tepe, J.J. Natural product scaffolds as inspiration for the design and synthesis of 20S human proteasome inhibitors. RSC Chem. Biol. 2020, 1, 305–332. [Google Scholar] [CrossRef]
- Sengupta, S.; Mehta, G. Non-peptidic natural products as ubiquitin-proteasome inhibitors. Tetrahedron 2019, 75, 817–853. [Google Scholar] [CrossRef]
- Della Sala, G.; Agriesti, F.; Mazzoccoli, C.; Tataranni, T.; Costantino, V.; Piccoli, C. Clogging the ubiquitin-proteasome machinery with marine natural products: Last decade update. Mar. Drugs 2018, 16, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, W.; Mackeen, M.M.; Kramer, H.B. Natural Product Inhibitors of Ubiquitin Conjugation and Deconjugation. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2016; Volume 49, pp. 207–242. ISBN 9780444636010. [Google Scholar]
- Tsukamoto, S. Search for inhibitors of the ubiquitin–proteasome system from natural sources for cancer therapy. Chem. Pharm. Bull. 2016, 64, 112–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momose, I.; Kawada, M. The therapeutic potential of microbial proteasome inhibitors. Int. Immunopharmacol. 2016, 37, 23–30. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Atanasov, A.G.; Khan, H.; Barreca, D.; Trombetta, D.; Testai, L.; Sureda, A.; Tejada, S.; Vacca, R.A.; Pittalà, V.; et al. Targeting ubiquitin-proteasome pathway by natural, in particular polyphenols, anticancer agents: Lessons learned from clinical trials. Cancer Lett. 2018, 434, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Bonfili, L.; Cecarini, V.; Amici, M.; Cuccioloni, M.; Angeletti, M.; Keller, J.N.; Eleuteri, A.M. Natural polyphenols as proteasome modulators and their role as anti-cancer compounds. FEBS J. 2008, 275, 5512–5526. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.L.; Groll, M. Applied techniques for mining natural proteasome inhibitors. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, R.; Kaur, T.; Guleria, G.; Rana, K.L.; Kour, D.; Yadav, N.; Yadav, A.N.; Saxena, K.A. Fungal secondary metabolites and their biotechnological applications for human health. In New and Future Developments in Microbial Biotechnology and Bioengineering; Rastegari, A.A., Yadav, A.N., Yadav, N., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2020; pp. 147–161. [Google Scholar]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism—From biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 4. [Google Scholar] [CrossRef]
- Greco, C.; Keller, N.P.; Rokas, A. Unearthing fungal chemodiversity and prospects for drug discovery. Curr. Opin. Microbiol. 2019, 51, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Skellam, E. Strategies for engineering natural product biosynthesis in fungi. Trends Biotechnol. 2019, 37, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Robey, M.T.; Caesar, L.K.; Drott, M.T.; Keller, N.P.; Kelleher, N.L. An interpreted atlas of biosynthetic gene clusters from 1,000 fungal genomes. Proc. Natl. Acad. Sci. USA 2021, 118, 19. [Google Scholar] [CrossRef]
- Hautbergue, T.; Jamin, E.L.; Debrauwer, L.; Puel, O.; Oswald, I.P. From genomics to metabolomics, moving toward an integrated strategy for the discovery of fungal secondary metabolites. Nat. Prod. Rep. 2018, 35, 147–173. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.C.; Nielsen, J. Development of fungal cell factories for the production of secondary metabolites: Linking genomics and metabolism. Synth. Syst. Biotechnol. 2017, 2, 5–12. [Google Scholar] [CrossRef]
- Yeh, H.-H.; Ahuja, M.; Chiang, Y.-M.; Oakley, C.E.; Moore, S.; Yoon, O.; Hajovsky, H.; Bok, J.-W.; Keller, N.P.; Wang, C.C.C.; et al. Resistance gene-guided genome mining: Serial promoter exchanges in Aspergillus nidulans reveal the biosynthetic pathway for fellutamide B, a proteasome inhibitor. ACS Chem. Biol. 2016, 11, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Shigemori, H.; Wakuri, S.; Yazawa, K.; Nakamura, T.; Sasaki, T.; Kobayashi, J. Fellutamides A and B, cytotoxic peptides from a marine fish-possessing fungus. Tetrahedron 1991, 47, 8529–8534. [Google Scholar] [CrossRef]
- Lee, Y.M.; Dang, H.T.; Hong, J.; Lee, C.O.; Bae, K.S.; Kim, D.K.; Jung, J.H. A cytotoxic lipopeptide from the sponge-derived fungus Aspergillus versicolor. Bull. Korean Chem. Soc. 2010, 31, 205–208. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.M.; Dang, H.T.; Li, J.; Zhang, P.; Hong, J.; Lee, C.O.; Jung, J.H. A cytotoxic fellutamide analogue from the sponge-derived fungus Aspergillus versicolor. Bull. Korean Chem. Soc. 2011, 32, 3817–3820. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Ondeyka, J.; Harris, G.H.; Zink, D.; Kahn, J.N.; Wang, H.; Bills, G.; Platas, G.; Wang, W.; Szewczak, A.A.; et al. Isolation, structure, and biological activities of fellutamides C and D from an undescribed Metulocladosporiella (Chaetothyriales) using the genome-wide Candida albicans fitness test. J. Nat. Prod. 2011, 74, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-J.; Li, C.-W.; Cui, C.-B. Seven new and two known lipopeptides as well as five known polyketides: The activated production of silent metabolites in a marine-derived fungus by chemical mutagenesis strategy using diethyl sulphate. Mar. Drugs 2014, 12, 1815–1838. [Google Scholar] [CrossRef] [Green Version]
- Pirrung, M.C.; Zhang, F.; Ambadi, S.; Gangadhara Rao, Y. Total synthesis of fellutamides, lipopeptide proteasome inhibitors. More sustainable peptide bond formation. Org. Biomol. Chem. 2016, 14, 8367–8375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hines, J.; Groll, M.; Fahnestock, M.; Crews, C.M. Proteasome inhibition by fellutamide B induces nerve growth factor synthesis. Chem. Biol. 2008, 15, 501–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, E.M.; Groll, M. Inhibitors for the immuno- and constitutive proteasome: Current and future trends in drug development. Angew. Chem. Int. Ed. 2012, 51, 8708–8720. [Google Scholar] [CrossRef]
- De Bettignies, G.; Coux, O. Proteasome inhibitors: Dozens of molecules and still counting. Biochimie 2010, 92, 1530–1545. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Li, D.; Chidawanyika, T.; Nathan, C.; Li, H. Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome. Arch. Biochem. Biophys. 2010, 501, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneekloth, J.S.; Sanders, J.L.; Hines, J.; Crews, C.M. Neurotrophic peptide aldehydes: Solid phase synthesis of fellutamide B and a simplified analog. Bioorg. Med. Chem. Lett. 2006, 16, 3855–3858. [Google Scholar] [CrossRef] [Green Version]
- Koguchi, Y.; Kohno, J.; Nishio, M.; Takahashi, K.; Okuda, T.; Ohnuki, T.; Komatsubara, S. TMC-95A, B, C, and D, novel proteasome inhibitors produced by Apiospora montagnei Sacc. TC 1093. Taxonomy, production, isolation, and biological activities. J. Antibiot. 2000, 53, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohno, J.; Koguchi, Y.; Nishio, M.; Nakao, K.; Kuroda, M.; Shimizu, R.; Ohnuki, T.; Komatsubara, S. Structures of TMC-95A-D: Novel proteasome inhibitors from Apiospora montagnei Sacc. TC 1093. J. Org. Chem. 2000, 65, 990–995. [Google Scholar] [CrossRef]
- Groll, M.; Koguchi, Y.; Huber, R.; Kohno, J. Crystal structure of the 20 S proteasome: TMC-95A complex: A non-covalent proteasome inhibitor. J. Mol. Biol. 2001, 311, 543–548. [Google Scholar] [CrossRef]
- Berthelot, A.; Piguel, S.; Le Dour, G.; Vidal, J. Synthesis of macrocyclic peptide analogues of proteasome inhibitor TMC-95A. J. Org. Chem. 2003, 68, 9835–9838. [Google Scholar] [CrossRef]
- Kaiser, M.; Groll, M.; Siciliano, C.; Assfalg-Machleidt, I.; Weyher, E.; Kohno, J.; Milbradt, A.G.; Renner, C.; Huber, R.; Moroder, L. Binding mode of TMC-95A analogues to eukaryotic 20S proteasome. ChemBioChem 2004, 5, 1256–1266. [Google Scholar] [CrossRef]
- Groll, M.; Götz, M.; Kaiser, M.; Weyher, E.; Moroder, L. TMC-95-based inhibitor design provides evidence for the catalytic versatility of the proteasome. Chem. Biol. 2006, 13, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Basse, N.; Piguel, S.; Papapostolou, D.; Ferrier-Berthelot, A.; Richy, N.; Pagano, M.; Sarthou, P.; Sobczak-Thépot, J.; Reboud-Ravaux, M.; Vidal, J. Linear TMC-95-based proteasome inhibitors. J. Med. Chem. 2007, 50, 2842–2850. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Gallastegui, N.; Maréchal, X.; Le Ravalec, V.; Basse, N.; Richy, N.; Genin, E.; Huber, R.; Moroder, L.; Vidal, J.; et al. 20S proteasome inhibition: Designing noncovalent linear peptide mimics of the natural product TMC-95A. ChemMedChem 2010, 5, 1701–1705. [Google Scholar] [CrossRef]
- Desvergne, A.; Genin, E.; Maréchal, X.; Gallastegui, N.; Dufau, L.; Richy, N.; Groll, M.; Vidal, J.; Reboud-Ravaux, M. Dimerized linear mimics of a natural cyclopeptide (TMC-95A) are potent noncovalent inhibitors of the eukaryotic 20S proteasome. J. Med. Chem. 2013, 56, 3367–3378. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Wang, K.; Yang, Y.; Lai, F.F.; Chen, X.G.; Xiao, Z.Y. Design, synthesis and biological evaluation of dipeptides as novel non-covalent 20S proteasome inhibitors. J. Asian Nat. Prod. Res. 2021, 23, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Scharf, D.H.; Brakhage, A.A.; Mukherjee, P.K. Gliotoxin-bane or boon? Environ. Microbiol. 2016, 18, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Welch, T.R.; Williams, R.M. Epidithiodioxopiperazines. Occurrence, synthesis and biogenesis. Nat. Prod. Rep. 2014, 31, 1376–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardiner, D.M.; Waring, P.; Howlett, B.J. The epipolythiodioxopiperazine (ETP) class of fungal toxins: Distribution, mode of action, functions and biosynthesis. Microbiology 2005, 151, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Knowles, S.L.; Mead, M.E.; Pereira Silva, L.; Raja, H.A.; Steenwyk, J.L.; Goldman, G.H.; Oberlies, N.H.; Rokas, A. Gliotoxin, a known virulence factor in the major human pathogen Aspergillus fumigatus, is also biosynthesized by its nonpathogenic relative Aspergillus fischeri. MBio 2020, 11, e03361-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahl, H.L.; Krauss, B.; Schulze-Osthoff, K.; Decker, T.; Traenckner, E.B.; Vogt, M.; Myers, C.; Parks, T.; Warring, P.; Mühlbacher, A.; et al. The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-κB. J. Exp. Med. 1996, 183, 1829–1840. [Google Scholar] [CrossRef] [PubMed]
- Kroll, M.; Arenzana-Seisdedos, F.; Bachelerie, F.; Thomas, D.; Friguet, B.; Conconi, M. The secondary fungal metabolite gliotoxin targets proteolytic activities of the proteasome. Chem. Biol. 1999, 6, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, H.; Egashira, S.; Saito, N.; Kirisako, T.; Miller, S.; Sasaki, Y.; Matsumoto, T.; Shimonishi, M.; Komatsu, T.; Terai, T.; et al. Gliotoxin suppresses NF-κB activation by selectively inhibiting linear ubiquitin chain assembly complex (LUBAC). ACS Chem. Biol. 2015, 10, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K. Discovery of linear ubiquitination, a crucial regulator for immune signaling and cell death. FEBS J. 2021, 288, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Paugam, A.; Creuzet, C.; Dupouy-Camet, J.; Roisin, M. In vitro effects of gliotoxin, a natural proteasome inhibitor, on the infectivity and proteolytic activity of Toxoplasma gondii. Parasitol. Res. 2002, 88, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Hatabu, T.; Hagiwara, M.; Taguchi, N.; Kiyozawa, M.; Suzuki, M.; Kano, S.; Sato, K. Plasmodium falciparum: The fungal metabolite gliotoxin inhibits proteasome proteolytic activity and exerts a plasmodicidal effect on P. falciparum. Exp. Parasitol. 2006, 112, 179–183. [Google Scholar] [CrossRef]
- Pereira-Neves, A.; Menna-Barreto, R.F.S.; Benchimol, M. The fungal metabolite gliotoxin inhibits proteasome proteolytic activity and induces an irreversible pseudocystic transformation and cell death in Tritrichomonas foetus. Parasitol. Res. 2016, 115, 3057–3069. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y.; Da Silva Sil Dos Santos, B.; Wang, F.; Ma, Y.; Perez, C.; Yang, Y.; Peng, J.; Cohen, S.M.; Chou, T.F.; et al. Epidithiodiketopiperazines inhibit protein degradation by targeting proteasome deubiquitinase Rpn11. Cell Chem. Biol. 2018, 25, 1350–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Wu, Z.; Liu, L.; Chen, S. The chemistry and biology of fungal meroterpenoids (2009–2019). Org. Biomol. Chem. 2021, 19, 1644–1704. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.E.; Gross, H.; Pontius, A.; Kehraus, S.; Krick, A.; Kelter, G.; Maier, A.; Fiebig, H.H.; König, G.M. Epoxyphomalin A and B, Prenylated Polyketides with Potent Cytotoxicity from the Marine-Derived Fungus Phoma sp. Org. Lett. 2009, 11, 5014–5017. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.E.; Kehraus, S.; Krick, A.; König, G.M.; Kelter, G.; Maier, A.; Fiebig, H.H.; Kalesse, M.; Malek, N.P.; Gross, H. Mode of Action of Epoxyphomalins A and B and Characterization of Related Metabolites from The Marine-Derived Fungus Paraconiothyrium sp. J. Nat. Prod. 2010, 73, 2053–2056. [Google Scholar] [CrossRef] [PubMed]
- Li, C.S.; Ren, G.; Yang, B.J.; Miklossy, G.; Turkson, J.; Fei, P.; Ding, Y.; Walker, L.A.; Cao, S. Meroterpenoids with Antiproliferative Activity from a Hawaiian-Plant Associated Fungus Peyronellaea coffeae-arabicae FT238. Org. Lett. 2016, 18, 2335–2338. [Google Scholar] [CrossRef] [PubMed]
- Hirose, A.; Maeda, H.; Tonouchi, A.; Nehira, T.; Hashimoto, M. Neomacrophorin I, II, and III, novel drimenyl cyclohexanes with hydroxylated butanoates from Trichoderma sp. 1212-03. Tetrahedron 2014, 70, 1458–1463. [Google Scholar] [CrossRef]
- Kusakabe, K.; Honmura, Y.; Uesugi, S.; Tonouchi, A.; Maeda, H.; Kimura, K.I.; Koshino, H.; Hashimoto, M. Neomacrophorin X, a [4.4.3]Propellane-Type Meroterpenoid from Trichoderma sp. 1212-03. J. Nat. Prod. 2017, 80, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, S.; Honmura, Y.; Nishiyama, M.; Kusakabe, K.; Tonouchi, A.; Yamashita, T.; Hashimoto, M.; Kimura, K. Identification of neomacrophorins isolated from Trichoderma sp. 1212-03 as proteasome inhibitors. Bioorganic Med. Chem. 2019, 27, 115161. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Thaochan, N.; Hu, Q. Diversity of linear non-ribosomal peptide in biocontrol fungi. J. Fungi 2020, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, R.; Andersson, M.A.; Kredics, L.; Grigoriev, P.A.; Sundell, N.; Salkinoja-Salonen, M.S. 20-Residue and 11-residue peptaibols from the fungus Trichoderma longibrachiatum are synergistic in forming Na+/K+- permeable channels and adverse action towards mammalian cells. FEBS J. 2012, 279, 4172–4190. [Google Scholar] [CrossRef]
- Grigoletto, D.F.; Trivella, D.B.B.; Tempone, A.G.; Rodrigues, A.; Correia, A.M.L.; Lira, S.P. Antifungal compounds with anticancer potential from Trichoderma sp. P8BDA1F1, an endophytic fungus from Begonia venosa. Braz. J. Microbiol. 2020, 51, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Tamandegani, P.R.; Marik, T.; Zafari, D.; Balázs, D.; Vágvölgyi, C.; Szekeres, A.; Kredics, L. Changes in peptaibol production of Trichoderma species during in vitro antagonistic interactions with fungal plant pathogens. Biomolecules 2020, 10, 730. [Google Scholar] [CrossRef] [PubMed]
- Raistrick, H.; Smith, G. Studies in the biochemistry of micro-organisms: The metabolic products of Aspergillus terreus Thom. A new mould metabolic product-terrein. Biochem. J. 1935, 29, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarintanan, F.; Jongrungruangchok, S.; Uthaisang-Tanechpongtamb, W. Terrein from Aspergillus terreus induced cytotoxic and nuclear changes on human colon cancer COLO205 cells. J. Pharm. Drug Deliv. Res. 2019, 8, 2. [Google Scholar]
- Asfour, H.Z.; Awan, Z.A.; Bagalagel, A.A.; Elfaky, M.A.; Abdelhameed, R.F.A.; Elhady, S.S. Large-Scale Production of Bioactive Terrein by Aspergillus Terreus Strain S020 Isolated from the Saudi Coast of the Red Sea. Biomolecules 2019, 9, 480. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Wang, S.Y.; Shen, H.; Yao, X.F.; Zhang, F.L.; Lai, D. The marine-derived fungal metabolite, terrein, inhibits cell proliferation and induces cell cycle arrest in human ovarian cancer cells. Int. J. Mol. Med. 2014, 34, 1591–1598. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Ibaragi, S.; Mandai, H.; Tsumura, T.; Kishimoto, K.; Okui, T.; Hassan, N.M.M.; Shimo, T.; Omori, K.; Hu, G.F.; et al. Synthetic terrein inhibits progression of head and neck cancer by suppressing angiogenin production. Anticancer Res. 2016, 36, 2161–2168. [Google Scholar] [PubMed]
- Demasi, M.; Felicio, A.L.; Pacheco, A.O.; Leite, H.G.; Lima, C.; Andrade, L.H. Studies on terrein as a new class of proteasome inhibitors. J. Braz. Chem. Soc. 2010, 21, 299–305. [Google Scholar] [CrossRef]
- Nogawa, T.; Kawatani, M.; Uramoto, M.; Okano, A.; Aono, H.; Futamura, Y.; Koshino, H.; Takahashi, S.; Osada, H. Pyrrolizilactone, a new pyrrolizidinone metabolite produced by a fungus. J. Antibiot. 2013, 66, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Futamura, Y.; Kawatani, M.; Muroi, M.; Aono, H.; Nogawa, T.; Osada, H. Identification of a molecular target of a novel fungal metabolite, pyrrolizilactone, by phenotypic profiling systems. ChemBioChem 2013, 14, 2456–2463. [Google Scholar] [CrossRef]
- Deshmukh, S.K.; Prakash, V.; Ranjan, N. Recent advances in the discovery of bioactive metabolites from Pestalotiopsis. Phytochem. Rev. 2017, 16, 883–920. [Google Scholar] [CrossRef]
- Nishad, J.H.; Singh, A.; Gautam, V.S.; Kumar, D.; Kumar, J.; Kharwar, R.N. Endophytic fungi: A cryptic fountainhead for biodiversity, functional metabolites, host stress tolerance, and myco-mediated nanoparticles (Nps) synthesis. In Endophytes and Secondary Metabolites; Reference Series in Phytochemistry; Jha, S., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–29. ISBN 9783319769004. Reference Series in Phytochemistry. [Google Scholar]
- Xia, X.; Kim, S.; Liu, C.; Shim, S.H. Secondary Metabolites Produced by an Endophytic Fungus Pestalotiopsis Sydowiana and Their 20S Proteasome Inhibitory Activities. Molecules 2016, 21, 944. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, H.; Kaneko, T.; Koshino, H.; Esumi, Y.; Uzawa, J.; Sugawara, F. Penicillides from Penicillium sp. isolated from Taxus cuspidata. Nat. Prod. Lett. 2000, 14, 477–484. [Google Scholar] [CrossRef]
- Xu, J.; Aly, A.H.; Wray, V.; Proksch, P. Polyketide derivatives of endophytic fungus Pestalotiopsis sp. isolated from the Chinese mangrove plant Rhizophora mucronata. Tetrahedron Lett. 2011, 52, 21–25. [Google Scholar] [CrossRef]
- Mcgahren, W.J.; Ellestad, G.A.; Morton, G.; Kunstmann, M.P. A new fungal lactone, LL-P88Oβ, and a new pyrone, LL-P880x, from a Penicillium sp. J. Org. Chem. 1973, 38, 3542–3544. [Google Scholar] [CrossRef]
- Ding, G.; Qi, Y.; Liu, S.; Guo, L.; Chen, X. Photipyrones A and B, new pyrone derivatives from the plant endophytic fungus Pestalotiopsis photiniae. J. Antibiot. 2012, 65, 271–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, A.; Mathieu, V.; Masi, M.; Baroncelli, R.; Boari, A.; Pescitelli, G.; Ferderin, M.; Lisy, R.; Evidente, M.; Tuzi, A.; et al. Higginsianins A and B, two diterpenoid α-pyrones produced by Colletotrichum higginsianum, with in vitro cytostatic activity. J. Nat. Prod. 2016, 79, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallery, J.F.; Zimmer, M.; Halder, V.; Suliman, M.; Pigné, S.; Le Goff, G.; Gianniou, D.D.; Trougakos, I.P.; Ouazzani, J.; Gasperini, D.; et al. Inhibition of jasmonate-mediated plant defences by the fungal metabolite higginsianin B. J. Exp. Bot. 2020, 71, 2910–2921. [Google Scholar] [CrossRef] [Green Version]
- Sekizawa, R.; Ikeno, S.; Nakamura, H.; Naganawa, H.; Matsui, S.; Iinuma, H.; Takeuchi, T. Panepophenanthrin, from a mushroom strain, a novel inhibitor of the ubiquitin-activating enzyme. J. Nat. Prod. 2002, 65, 1491–1493. [Google Scholar] [CrossRef]
- Matsuzawa, M.; Kakeya, H.; Yamaguchi, J.; Shoji, M.; Onose, R.; Osada, H.; Hayashi, Y. Enantio- and diastereoselective total synthesis of (+)-panepophenanthrin, a ubiquitin-activating enzyme inhibitor, and biological properties of its new derivatives. Chem. Asian J. 2006, 1, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Moses, J.E.; Commeiras, L.; Baldwin, J.E.; Adlington, R.M. Total synthesis of panepophenanthrin. Org. Lett. 2003, 5, 2987–2988. [Google Scholar] [CrossRef]
- Mehta, G.; Sengupta, S. Progress in the total synthesis of epoxyquinone natural products: An update. Tetrahedron 2017, 73, 6223–6247. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Hirota, H.; Imachi, M.; Fujimuro, M.; Onuki, H.; Ohta, T.; Yokosawa, H. Himeic acid A: A new ubiquitin-activating enzyme inhibitor isolated from a marine-derived fungus, Aspergillus sp. Bioorg. Med. Chem. Lett. 2005, 15, 191–194. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kato, H.; Katsuki, A.; Tsukamoto, S.; Fujii, I. Identification of the biosynthetic gene cluster for himeic acid A: A ubiquitin-activating enzyme (E1) inhibitor in Aspergillus japonicus MF275. ChemBioChem 2018, 19, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Kim, M.J.; Li, H.; Zhang, P.; Bao, B.; Lee, K.J.; Jung, J.H. Marine-derived Aspergillus Species as a Source of Bioactive Secondary Metabolites. Mar. Biotechnol. 2013, 15, 499–519. [Google Scholar] [CrossRef]
- Katsuki, A.; Kato, H.; Tahara, Y.; Hashimoto, M.; Fujii, I.; Tsukamoto, S. pH-dependent production of himeic acid A and its non-enzymatic conversions to himeic acids B and C. Bioorg. Med. Chem. 2018, 26, 1869–1874. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.; Pavletich, N. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos, A.; Leão, M.; Soares, J.; Palmeira, A.; Pinto, M.; Saraiva, L.; Sousa, M.E. Medicinal chemistry strategies to disrupt the p53-MDM2/MDMX interaction. Med. Res. Rev. 2016, 36, 789–844. [Google Scholar] [CrossRef] [PubMed]
- Sane, S.; Rezvani, K. Essential roles of E3 ubiquitin ligases in p53 regulation. Int. J. Mol. Sci. 2017, 18, 442. [Google Scholar] [CrossRef]
- Duncan, S.J.; Grüschow, S.; Williams, D.H.; McNicholas, C.; Purewal, R.; Hajek, M.; Gerlitz, M.; Martin, S.; Wrigley, S.K.; Moore, M. Isolation and structure elucidation of chlorofusin, a novel p53-MDM2 antagonist from a Fusarium sp. J. Am. Chem. Soc. 2001, 123, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.J.; Cooper, M.A.; Williams, D.H. Binding of an inhibitor of the p53/MDM2 Interaction to MDM2. Chem. Commun. 2003, 3, 316–317. [Google Scholar] [CrossRef]
- Qian, W.J.; Wei, W.G.; Zhang, Y.X.; Yao, Z.J. Total synthesis, assignment of absolute stereochemistry, and structural revision of chlorofusin. J. Am. Chem. Soc. 2007, 129, 6400–6401. [Google Scholar] [CrossRef]
- Clark, R.C.; Lee, S.Y.; Searcey, M.; Boger, D.L. The isolation, total synthesis and structure elucidation of chlorofusin, a natural product inhibitor of the p53-MDM2 protein-protein interaction. Nat. Prod. Rep. 2009, 26, 465–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, H.B.; Chen, X.Y.; Li, Q.; Qian, W.J.; Yu, S.M.; Tang, G.L.; Yao, Z.J. Unified flexible total synthesis of chlorofusin and artificial Click mimics as antagonists against p53-HDM2 interactions. Tetrahedron Lett. 2014, 55, 6055–6059. [Google Scholar] [CrossRef]
- Qiu, H.B.; Qian, W.J.; Yu, S.M.; Yao, Z.J. Stereodivergent total synthesis of chlorofusin and its all seven chromophore diastereomers. Tetrahedron 2015, 71, 370–380. [Google Scholar] [CrossRef]
- Cominetti, M.M.D.; Goffin, S.A.; Raffel, E.; Turner, K.D.; Ramoutar, J.C.; O’Connell, M.A.; Howell, L.A.; Searcey, M. Identification of a new p53/MDM2 inhibitor motif inspired by studies of chlorofusin. Bioorg. Med. Chem. Lett. 2015, 25, 4878–4880. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, S.; Yoshida, T.; Hosono, H.; Ohta, T.; Yokosawa, H. Hexylitaconic acid: A new inhibitor of p53-HDM2 interaction isolated from a marine-derived fungus, Arthrinium sp. Bioorg. Med. Chem. Lett. 2006, 16, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Isogai, A.; Washizu, M.; Kondo, K.; Murakoshi, S.; Suzuki, A. Isolation and identification of (+)-hexylitaconic acid as a plant growth regulator. Agric. Biol. Chem. 1984, 48, 2607–2609. [Google Scholar] [CrossRef] [Green Version]
- Klemke, C.; Kehraus, S.; Wright, A.D.; König, G.M. New secondary metabolites from the marine endophytic fungus Apiospora montagnei. J. Nat. Prod. 2004, 67, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Yang, N.; Wei, S.; Jia, J.; Lin, R.; Li, J.; Bi, H.; Song, F.; Xu, X. Dimeric hexylitaconic acids from the marine-derived fungus Aspergillus welwitschiae CUGBMF180262. Nat. Prod. Res. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nakahashi, A.; Miura, N.; Monde, K.; Tsukamoto, S. Stereochemical studies of hexylitaconic acid, an inhibitor of p53-HDM2 interaction. Bioorg. Med. Chem. Lett. 2009, 19, 3027–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froufe, H.J.C.; Abreu, R.M.V.; Ferreira, I.C.F.R. Virtual screening of low molecular weight mushrooms compounds as potential Mdm2 inhibitors. J. Enzyme Inhib. Med. Chem. 2013, 28, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandi, S.; Sikder, R.; Acharya, K. Secondary Metabolites of Mushrooms: A potential source for anticancer therapeutics with translational opportunities. In Advancing Frontiers in Mycology and Mycotechnology; Satyanarayana, T., Deshmukh, S.K., Deshpande, M.V., Eds.; Springer: Singapore, 2019; pp. 563–598. [Google Scholar]
- Li, C.H.; Chen, P.Y.; Chang, U.M.; Kan, L.S.; Fang, W.H.; Tsai, K.S.; Lin, S. Bin Ganoderic acid X, a lanostanoid triterpene, inhibits topoisomerases and induces apoptosis of cancer cells. Life Sci. 2005, 77, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Bok, J.W.; Lermer, L.; Chilton, J.; Klingeman, H.G.; Towers, G.H.N. Antitumor sterols from the mycelia of Cordyceps sinensis. Phytochemistry 1999, 51, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Pleszczyńska, M.; Wiater, A.; Siwulski, M.; Lemieszek, M.K.; Kunaszewska, J.; Kaczor, J.; Rzeski, W.; Janusz, G.; Szczodrak, J. Cultivation and utility of Piptoporus betulinus fruiting bodies as a source of anticancer agents. World J. Microbiol. Biotechnol. 2016, 32, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Endo, A. The origin of the statins. Int. Congr. Ser. 2004, 1262, 3–8. [Google Scholar] [CrossRef]
- Manzoni, M.; Rollini, M. Biosynthesis and biotechnological production of statins by filamentous fungi and application of these cholesterol-lowering drugs. Appl. Microbiol. Biotechnol. 2002, 58, 555–564. [Google Scholar] [PubMed]
- Manzoni, M.; Bergomi, S.; Rollini, M.; Cavazzoni, V. Production of statins by filamentous fungi. Biotechnol. Lett. 1999, 21, 253–257. [Google Scholar] [CrossRef]
- Subhan, M.; Faryal, R.; Macreadie, I. Production of statins by fungal fermentation. Microbiol. Aust. 2017, 38, 70–72. [Google Scholar] [CrossRef] [Green Version]
- Kała, K.; Kryczyk-Poprawa, A.; Rzewińska, A.; Muszyńska, B. Fruiting bodies of selected edible mushrooms as a potential source of lovastatin. Eur. Food Res. Technol. 2020, 246, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Wójcik, C.; Bury, M.; Stoklosa, T.; Giermasz, A.; Feleszko, W.; Mlynarczuk, I.; Pleban, E.; Basak, G.; Omura, S.; Jakóbisiak, M. Lovastatin and simvastatin are modulators of the proteasome. Int. J. Biochem. Cell Biol. 2000, 32, 957–965. [Google Scholar] [CrossRef]
- Murray, S.S.; Tu, K.N.; Young, K.L.; Brochmann Murray, E.J. The effects of lovastatin on proteasome activities in highly purified rabbit 20 S proteasome preparations and mouse MC3T3-E1 osteoblastic cells. Metabolism 2002, 51, 1153–1160. [Google Scholar] [CrossRef]
- Rao, S.; Porter, D.C.; Chen, X.; Herliczek, T.; Lowe, M.; Keyomarsi, K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc. Natl. Acad. Sci. USA 1999, 96, 7797–7802. [Google Scholar] [CrossRef] [Green Version]
- Efuet, E.T.; Keyomarsi, K. Curcumin and simvastatin mediate growth arrest in breast cancer cells by targeting the proteasome. Proc. Amer. Assoc. Cancer Res. 2006, 47, 1092. [Google Scholar]
- Park, I.H.; Kim, J.Y.; Choi, J.Y.; Han, J.Y. Simvastatin enhances irinotecan-induced apoptosis in human non-small cell lung cancer cells by inhibition of proteasome activity. Invest. New Drugs 2011, 29, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Barbalata, C.I.; Tefas, L.R.; Achim, M.; Tomuta, I.; Porfire, A.S. Statins in risk-reduction and treatment of cancer. World J. Clin. Oncol. 2020, 11, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Vosper, J.; Masuccio, A.; Kullmann, M.; Ploner, C.; Geley, S.; Hengst, L. Statin-induced depletion of geranylgeranyl pyrophosphate inhibits cell proliferation by a novel pathway of Skp2 degradation. Oncotarget 2015, 6, 2889–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.T.; Ho, H.J.; Lin, J.T.; Shieh, J.J.; Wu, C.Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017, 8, e2626-e2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system—implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Colland, F. The therapeutic potential of deubiquitinating enzyme inhibitors. Biochem. Soc. Trans. 2010, 38, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.; Muniyappan, S.; Lee, S.-B.; Lee, B.-H. Small-molecule inhibitors targeting proteasome-associated deubiquitinases. Int. J. Mol. Sci. 2021, 22, 6213. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, H.M. Recent advances in the development of ubiquitin-specific-processing protease 7 (USP7) inhibitors. Eur. J. Med. Chem. 2020, 191, 112107. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Crews, C.M. Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew. Chem. Int. Ed. 2014, 53, 2312–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.L.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, D.R.; et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486. [Google Scholar] [CrossRef]
- Barrow, C.J.; Sedlock, D.M. 1’-(2-phenyl-ethylene)-ditryptophenaline, a new dimeric diketopiperazine from Aspergillus flavus. J. Nat. Prod. 1994, 57, 1239–1244. [Google Scholar] [CrossRef]
- Uka, V.; Cary, J.W.; Lebar, M.D.; Puel, O.; De Saeger, S.; Diana Di Mavungu, J. Chemical repertoire and biosynthetic machinery of the Aspergillus flavus secondary metabolome: A review. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2797–2842. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.G.M.; Pereira, R.B.; Andrade, P.B.; Valentão, P. Double the chemistry, double the fun: Structural diversity and biological activity of marine-derived diketopiperazine dimers. Mar. Drugs 2019, 17, 551. [Google Scholar] [CrossRef] [Green Version]
- Tadano, S.; Sugimachi, Y.; Sumimoto, M.; Tsukamoto, S.; Ishikawa, H. Collective synthesis and biological evaluation of tryptophan-based dimeric diketopiperazine alkaloids. Chem. Eur. J. 2016, 22, 1277–1291. [Google Scholar] [CrossRef] [PubMed]
- Ning, F.; Xin, H.; Liu, J.; Lv, C.; Xu, X.; Wang, M.; Wang, Y.; Zhang, W.; Zhang, X. Structure and function of USP5: Insight into physiological and pathophysiological roles. Pharmacol. Res. 2020, 157, 104557. [Google Scholar] [CrossRef] [PubMed]
- Kaistha, B.P.; Krattenmacher, A.; Fredebohm, J.; Schmidt, H.; Behrens, D.; Widder, M.; Hackert, T.; Strobel, O.; Hoheisel, J.D.; Gress, T.M.; et al. The deubiquitinating enzyme USP5 promotes pancreatic cancer via modulating cell cycle regulators. Oncotarget 2017, 8, 66215–66225. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Zhang, D.; Zhao, K.; Wang, Y.; Pei, L.; Fu, Q.; Ma, X. Spotlight on USP4: Structure, Function, and Regulation. Front. Cell Dev. Biol. 2021, 9, 1–15. [Google Scholar] [CrossRef]
- Li, F.; Hu, Q.; He, T.; Xu, J.; Yi, Y.; Xie, S.; Ding, L.; Fu, M.; Guo, R.; Xiao, Z.X.J.; et al. The deubiquitinase usp4 stabilizes twist1 protein to promote lung cancer cell stemness. Cancers 2020, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, L.; Lu, J.; Jiang, B.; Liu, C.; Guo, J. USP4 function and multifaceted roles in cancer: A possible and potential therapeutic target. Cancer Cell Int. 2020, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Koshino, H.; Esumi, Y.; Takahashi, S.; Yoshikawa, K.; Abe, N. Vialinin A, a novel 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenger from an edible mushroom in China. Biosci. Biotechnol. Biochem. 2005, 69, 2326–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norikura, T.; Fujiwara, K.; Narita, T.; Yamaguchi, S.; Morinaga, Y.; Iwai, K.; Matsue, H. Anticancer activities of thelephantin O and vialinin A isolated from Thelephora aurantiotincta. J. Agric. Food Chem. 2011, 59, 6974–6979. [Google Scholar] [CrossRef]
- Onose, J.I.; Xie, C.; Ye, Y.Q.; Sugaya, K.; Takahashi, S.; Koshino, H.; Yasunaga, K.; Abe, N.; Yoshikawa, K. Vialinin A, a novel potent inhibitor of TNF-α production from RBL-2H3 cells. Biol. Pharm. Bull. 2008, 31, 831–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, K.; Ye, Y.Q.; Taniguchi, K.; Yoshida, A.; Akiyama, T.; Yoshioka, Y.; Onose, J.I.; Koshino, H.; Takahashi, S.; Yajima, A.; et al. Vialinin A is a ubiquitin-specific peptidase inhibitor. Bioorg. Med. Chem. Lett. 2013, 23, 4328–4331. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Ye, Y.Q.; Okada, K.; Taniguchi, K.; Yoshida, A.; Sugaya, K.; Onose, J.I.; Koshino, H.; Takahashi, S.; Yajima, A.; et al. Ubiquitin-specific peptidase 5, a target molecule of vialinin A, is a key molecule of TNF-α production in RBL-2H3 cells. PLoS ONE 2013, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Originally Derived from | Active Sites Targeted |
|---|---|---|
| Fellutamide B | Penicillium fellutanum | CHTL >> TL~CL |
| Fellutamide D | Metulocladosporiella sp. | CHTL1 |
| Fellutamide E | Metulocladosporiella sp. | CHTL1 |
| TMC-95A, TMC-95B | Apiospora montagnei Sacc. TC 1093 | CHTL > CL > TL |
| TMC-95C | Apiospora montagnei Sacc. TC 1093 | CHTL~ CL > TL |
| TMC-95D | Apiospora montagnei Sacc. TC 1093 | CHTL > TL~CL |
| Gliotoxin | Gliocladium fimbriatum | CHTL > TL ≥ CL (Rpn11)2 |
| Epoxyphomalin A | Phoma sp. (revised to Paraconiothyrium cf sporulosum) | CHTL~TL~CL |
| Epoxyphomalin B | Phoma sp. (revised to Paraconiothyrium cf sporulosum) | CHTL >> TL~CL |
| Neomacrophorins I, IV, VI, X | Trichoderma sp. 1212-03 | CHTL~CL > TL |
| Trilongins BI–BIV | Trichoderma longibrachiatum | CHTL1 |
| Terrein | Aspergillus terreus Thom | CHTL~TL1 |
| Pyrrolizilactone | Uncharacterized fungus | TL >> CHTL > CL |
| Pestalotiopyrone G | Pestalotiopsis sydowiana | CHTL1 |
| Photipyrone B | Pestalotiopsis sydowiana | CHTL1 |
| LL-P880β lactone | Pestalotiopsis sydowiana | CHTL1 |
| Pestalotiollides A, B | Pestalotiopsis sydowiana | CHTL1 |
| 6-hydroxymethyl-4-methoxy-5,6-dihydro-2H-pyran-2-one | Pestalotiopsis sydowiana | CHTL1 |
| 3′-O-methyldehydroisopenicillide | Pestalotiopsis sydowiana | CHTL1 |
| Higginsianin B | Colletotrichum higginsianum | CHTL~CL1 |
| Compound Name | Originally Derived from | Mechanism of Action |
|---|---|---|
| Panepophenanthrin | Panus rudis Fr. IFO 8994 | Inhibits formation of the E1–ubiquitin thioester intermediate |
| Himeic acid A | Aspergillus japonicus MF275 | Specific inhibitor of the E1 enzyme; inhibits formation of the E1–ubiquitin thioester intermediate |
| Compound Name | Originally Derived from | Mechanism of Action |
|---|---|---|
| Chlorofusin | Fusarium sp. 22026 (corrected as Microdochium caespitosum) | Antagonizes the p53–MDM2 interaction by direct binding to the N-terminal domain of MDM2 |
| (–)-Hexylitaconic acid | Apiospora montagnei | Inhibits the p53–HDM2 interaction by direct binding to HDM2, but not to the p53 protein |
| (+)-Hexylitaconic acid | Aspergillus niger strain K-88 | Inhibits the p53–HDM2 interaction |
| Ganoderic acids X, Y, F | Ganoderma lucidum | Potential inhibitors of the p53–MDM2 interaction |
| EMCD1 | Cordyceps sinensis | Potential inhibitor of the p53–MDM2 interaction |
| Polyporenic acid C | Piptoporus betulinus | Potential inhibitor of the p53–MDM2 interaction |
| Lovastatin and simvastatin2 | Aspergillus terreus | Induce the degradation of Skp2, a subunit of the SCFSkp2 ubiquitin ligase that targets p27Kip1 and p21Cip1 for proteasomal destruction |
| Compound Name | Originally Derived from | Cellular Target |
|---|---|---|
| 1′-(2-phenylethylene)-ditryptophenaline | Aspergillus flavus | USP7 |
| Gliotoxin and other ETPs1 | Gliocladium fimbriatum | Rpn11 (inhibition by chelating the catalytic Zn2+ ion) |
| Vialinin A | Thelephora vialis | USP5/IsoT, USP4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staszczak, M. Fungal Secondary Metabolites as Inhibitors of the Ubiquitin–Proteasome System. Int. J. Mol. Sci. 2021, 22, 13309. https://doi.org/10.3390/ijms222413309
Staszczak M. Fungal Secondary Metabolites as Inhibitors of the Ubiquitin–Proteasome System. International Journal of Molecular Sciences. 2021; 22(24):13309. https://doi.org/10.3390/ijms222413309
Chicago/Turabian StyleStaszczak, Magdalena. 2021. "Fungal Secondary Metabolites as Inhibitors of the Ubiquitin–Proteasome System" International Journal of Molecular Sciences 22, no. 24: 13309. https://doi.org/10.3390/ijms222413309
APA StyleStaszczak, M. (2021). Fungal Secondary Metabolites as Inhibitors of the Ubiquitin–Proteasome System. International Journal of Molecular Sciences, 22(24), 13309. https://doi.org/10.3390/ijms222413309