
Gene Expression Profile in Primary Tumor Is Associated with Brain-Tropism of Metastasis from Lung Adenocarcinoma

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Basic Clinical and Pathological Characteristics
2.2. No significant Genomic Difference Was Identified between Brain-Metastasizing and Non-Brain-Metastasizing Lung Adenocarcinomas by Targeted Next-Generation Sequencing
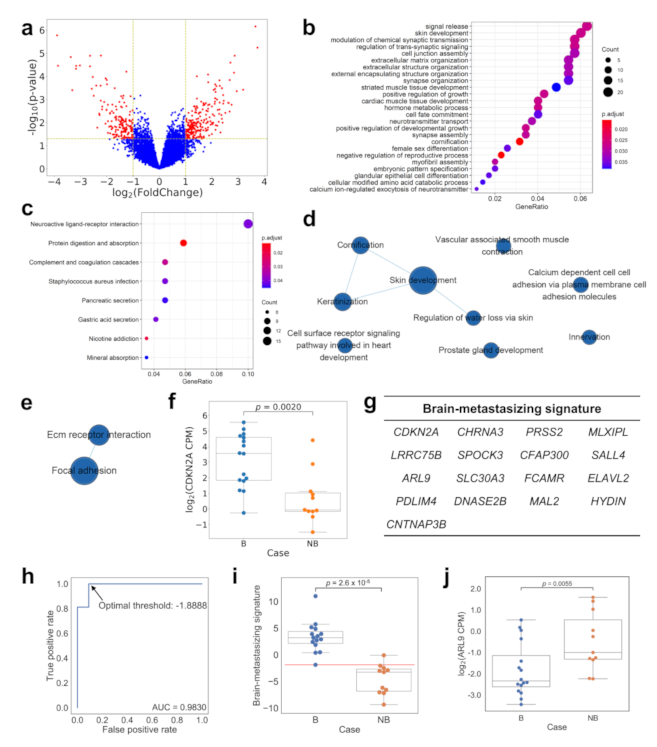
2.3. The mRNA Expression Profile, including CDKN2A, Is Significantly Different between Brain-Metastasizing and Non-Brain-Metastasizing Lung Adenocarcinomas
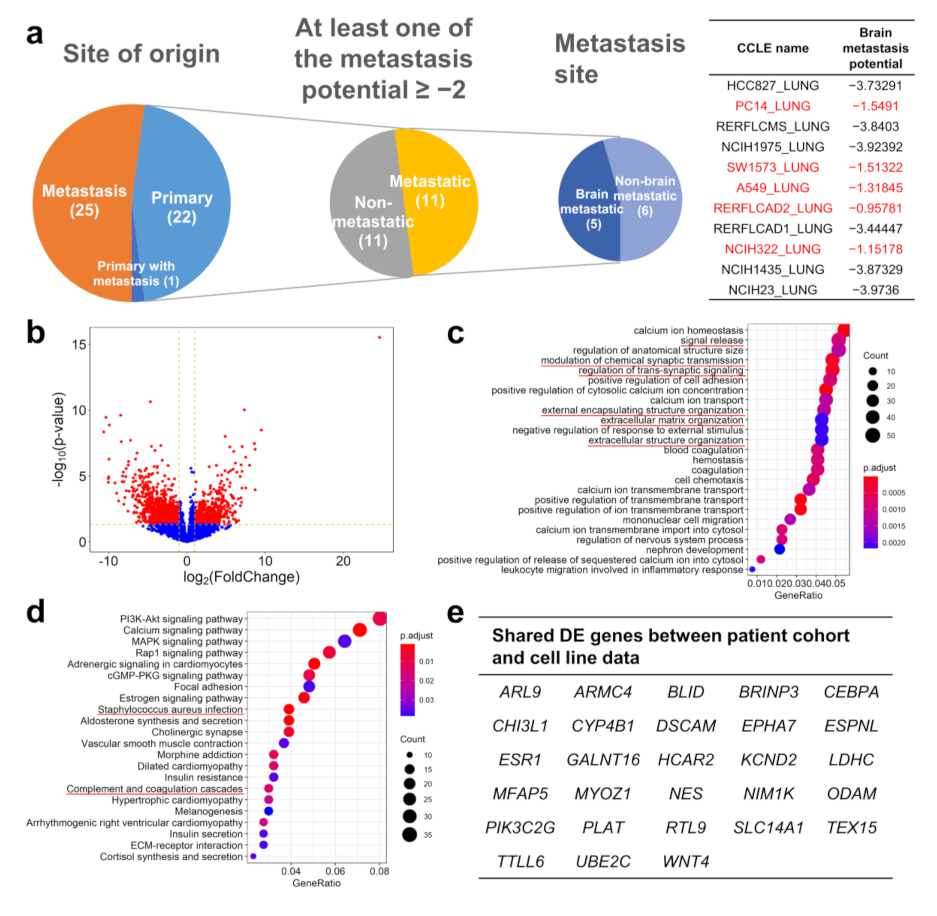
2.4. Comparing the Gene Expression Pattern between Brain-Metastasizing Patient Tumors and Brain-Tropic Lung Adenocarcinoma Cell Lines Showed a Small Set of Shared Differentially Expressed Genes
3. Discussion
4. Materials and Methods
4.1. Patient Selection
4.2. Targeted DNA Next-Generation Sequencing to Detect Genomic Alterations
4.3. Transcriptome Analysis and Identification of Differentially Expressed Genes
4.4. Immunohistochemistry
4.5. Comparison of Gene Expression Profile between Brain-Tropic Lung Adenocarcinoma Cell Lines and Patients with Brain-Metastasizing Lung Adenocarcinoma
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef] [Green Version]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [Green Version]
- Cagney, D.N.; Martin, A.M.; Catalano, P.J.; Redig, A.J.; Lin, N.U.; Lee, E.Q.; Wen, P.Y.; Dunn, I.F.; Bi, W.L.; Weiss, S.E.; et al. Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: A population-based study. Neuro-Oncol. 2017, 19, 1511–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnholtz-Sloan, J.S.; Sloan, A.E.; Davis, F.G.; Vigneau, F.D.; Lai, P.; Sawaya, R.E. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J. Clin. Oncol. 2004, 22, 2865–2872. [Google Scholar] [CrossRef] [PubMed]
- Owen, S.; Souhami, L. The management of brain metastases in non-small cell lung cancer. Front. Oncol. 2014, 4, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, D.J.H.; Nayyar, N.; Bihun, I.; Dagogo-Jack, I.; Gill, C.M.; Aquilanti, E.; Bertalan, M.; Kaplan, A.; D’Andrea, M.R.; Chukwueke, U.; et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat. Genet. 2020, 52, 371–377. [Google Scholar] [CrossRef]
- Wang, H.; Ou, Q.; Li, D.; Qin, T.; Bao, H.; Hou, X.; Wang, K.; Wang, F.; Deng, Q.; Liang, J.; et al. Genes associated with increased brain metastasis risk in non-small cell lung cancer: Comprehensive genomic profiling of 61 resected brain metastases versus primary non-small cell lung cancer (Guangdong Association Study of Thoracic Oncology 1036). Cancer 2019, 125, 3535–3544. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Ding, X.; Bi, N.; Wang, Z.; Wu, L.; Zhou, W.; Zhao, Z.; Wang, J.; Zhang, W.; Fan, J.; et al. Molecular predictors of brain metastasis-related microRNAs in lung adenocarcinoma. PLoS Genet. 2019, 15, e1007888. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Lin, Z.; Peng, W.; Hu, Z. Identification of potential biomarkers of lung adenocarcinoma brain metastases via microarray analysis of cDNA expression profiles. Oncol. Lett. 2019, 17, 2228–2236. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Jin, X.; Demere, Z.; Nair, K.; Ali, A.; Ferraro, G.B.; Natoli, T.; Deik, A.; Petronio, L.; Tang, A.A.; Zhu, C.; et al. A metastasis map of human cancer cell lines. Nature 2020, 588, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, F.; Zhu, C.Q.; Brambilla, E.; Graziano, S.L.; Olaussen, K.; Le-Chevalier, T.; Pignon, J.P.; Kratzke, R.; Soria, J.C.; Shepherd, F.A.; et al. Genome-wide copy number analyses of samples from LACE-Bio project identify novel prognostic and predictive markers in early stage non-small cell lung cancer. Transl. Lung Cancer Res. 2018, 7, 416–427. [Google Scholar] [CrossRef]
- Bradly, D.P.; Gattuso, P.; Pool, M.; Basu, S.; Liptay, M.; Bonomi, P.; Buckingham, L. CDKN2A (p16) promoter hypermethylation influences the outcome in young lung cancer patients. Diagn Mol. Pathol. 2012, 21, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Bian, C.; Li, Z.; Xu, Y.; Wang, J.; Xu, L.; Shen, H. Clinical outcome and expression of mutant P53, P16, and Smad4 in lung adenocarcinoma: A prospective study. World J. Surg. Oncol. 2015, 13, 128. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Sugita, H.; Sima, C.S.; Zauderer, M.; Rudin, C.M.; Kris, M.G.; Rusch, V.W.; Azzoli, C.G. A prospective study of tumor suppressor gene methylation as a prognostic biomarker in surgically resected stage I to IIIA non-small-cell lung cancers. J. Thorac. Oncol. 2014, 9, 1272–1277. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Sun, X.; Cheng, H.; Zhao, D.; Ma, J.; Zhen, Q.; Cao, Y.; Zhu, H.; Bai, J. Expression of p16 in non-small cell lung cancer and its prognostic significance: A meta-analysis of published literatures. Lung Cancer 2011, 74, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Kohno, M.; Ito, K.; Takada, K.; Katsura, M.; Morodomi, Y.; Toyokawa, G.; Shoji, F.; Maehara, Y. Clinical Significance of DNA Damage Response Factors and Chromosomal Instability in Primary Lung Adenocarcinoma. Anticancer Res. 2017, 37, 1729–1735. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.L.; Hung, J.Y.; Lee, Y.L.; Chen, F.W.; Chang, K.F.; Chang, W.A.; Tsai, Y.M.; Chong, I.W.; Kuo, P.L. Identification of novel gene expression signature in lung adenocarcinoma by using next-generation sequencing data and bioinformatics analysis. Oncotarget 2017, 8, 104831–104854. [Google Scholar] [CrossRef] [Green Version]
- Fabian, K.; Nemeth, Z.; Furak, J.; Tiszlavicz, L.; Papay, J.; Krenacs, T.; Timar, J.; Moldvay, J. Protein expression differences between lung adenocarcinoma and squamous cell carcinoma with brain metastasis. Anticancer Res. 2014, 34, 5593–5597. [Google Scholar]
- Dok, R.; Glorieux, M.; Holacka, K.; Bamps, M.; Nuyts, S. Dual role for p16 in the metastasis process of HPV positive head and neck cancers. Mol. Cancer 2017, 16, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Solimando, A.G.; Summa, S.; Vacca, A.; Ribatti, D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers 2020, 12, 3380. [Google Scholar] [CrossRef]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef]
- Li, J.; Poi, M.J.; Tsai, M.D. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef] [Green Version]
- Ko, E.; Kim, Y.; Lee, B.B.; Han, J.; Song, S.Y.; Shim, Y.M.; Park, J.; Kim, D.H. Relationship of phospho-pRb (Ser-807/811) level to exposure to tobacco smoke in primary non-small cell lung cancer. Cancer Lett. 2009, 274, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.A.; Jaing, C.J.; Pierce Campbell, C.; Magliocco, A.; Xiong, Y.; Magliocco, G.; Thissen, J.B.; Antonia, S. Molecular evidence of viral DNA in non-small cell lung cancer and non-neoplastic lung. Br. J. Cancer 2016, 115, 497–504. [Google Scholar] [CrossRef]
- Baba, M.; Castillo, A.; Koriyama, C.; Yanagi, M.; Matsumoto, H.; Natsugoe, S.; Shuyama, K.Y.; Khan, N.; Higashi, M.; Itoh, T.; et al. Human papillomavirus is frequently detected in gefitinib-responsive lung adenocarcinomas. Oncol. Rep. 2010, 23, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.F.; Cheng, Y.W.; Lai, J.C.; Hsu, M.C.; Chen, J.T.; Liu, W.S.; Chiou, M.C.; Chen, C.Y.; Lee, H. Frequent p16INK4a promoter hypermethylation in human papillomavirus-infected female lung cancer in Taiwan. Int. J. Cancer 2005, 113, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; Keeney, M.; Law, M.; Donovan, J.; Aubry, M.C.; Garcia, J. Detection of human papillomavirus in non-small cell carcinoma of the lung. Hum. Pathol. 2015, 46, 1592–1597. [Google Scholar] [CrossRef]
- van Boerdonk, R.A.; Daniels, J.M.; Bloemena, E.; Krijgsman, O.; Steenbergen, R.D.; Brakenhoff, R.H.; Grunberg, K.; Ylstra, B.; Meijer, C.J.; Smit, E.F.; et al. High-risk human papillomavirus-positive lung cancer: Molecular evidence for a pattern of pulmonary metastasis. J. Thorac. Oncol. 2013, 8, 711–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Barton, L.F.; Chi, Y.; Clurman, B.E.; Roberts, J.M. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol. Cell 2007, 26, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Coryell, P.R.; Goraya, S.K.; Griffin, K.A.; Redick, M.A.; Sisk, S.R.; Purvis, J.E. Autophagy regulates the localization and degradation of p16(INK4a). Aging Cell 2020, 19, e13171. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Kim, M.; Jung, H.S.; Kim, Y.; Jeoung, D. Targeting Autophagy for Overcoming Resistance to Anti-EGFR Treatments. Cancers 2019, 11, 1374. [Google Scholar] [CrossRef] [Green Version]
- Stachowicz, K. The role of DSCAM in the regulation of synaptic plasticity: Possible involvement in neuropsychiatric disorders. Acta Neurobiol. Exp. 2018, 78, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. USA 2014, 111, 984–989. [Google Scholar] [CrossRef] [Green Version]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; El-Naggar, A.K.; Kim, E.S.; Kurie, J.M.; Lozano, G. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene 2007, 26, 6896–6904. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | Brain Metastasizing | Non-Brain Metastasizing | p Value | |
|---|---|---|---|---|
| n | 16 | 11 | ||
| Mean age (range) | 62 (45–78) | 67 (46–77) | 0.19 | |
| Male sex (%) | 6 (37.5) | 8 (72.7) | 0.12 | |
| Smoking history (%) | 7 (43.8) | 7 (63.6) | 0.44 | |
| Mean tumor size (S.D.) | 2.9 (1.1) | 3.4 (1.9) | 0.40 | |
| Received adjuvant chemotherapy (%) | 11 (68.8) | 8 (72.7) | 1 | |
| Predominant growth pattern in primary tumor | Acinar (%) | 7 (43.7) | 5 (45.4) | 0.55 |
| Papillary (%) | 1 (6.3) | 1 (9.1) | ||
| Micropapillary (%) | 5 (31.3) | 1 (9.1) | ||
| Solid (%) | 3 (18.7) | 4 (36.4) | ||
| T stage (%) | T1a | 1 (6.3) | 1 (9.1) | 0.28 |
| T1b | 3 (18.7) | 3 (27.3) | ||
| T2a | 11 (68.7) | 4 (36.3) | ||
| T2b | 1 (6.3) | 1 (9.1) | ||
| T3 | 0 (0) | 2 (18.2) | ||
| N stage (%) | N0 | 8 (50.0) | 5 (45.5) | 1 |
| N1 | 3 (18.7) | 3 (27.3) | ||
| N2 | 5 (31.3) | 3 (27.3) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-Y.; Wang, Y.-C.; Yeh, D.-W.; Hung, C.-Y.; Yeh, Y.-C.; Ho, H.-L.; Mon, H.-C.; Chen, M.-Y.; Wu, Y.-C.; Chou, T.-Y. Gene Expression Profile in Primary Tumor Is Associated with Brain-Tropism of Metastasis from Lung Adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 13374. https://doi.org/10.3390/ijms222413374
Lin Y-Y, Wang Y-C, Yeh D-W, Hung C-Y, Yeh Y-C, Ho H-L, Mon H-C, Chen M-Y, Wu Y-C, Chou T-Y. Gene Expression Profile in Primary Tumor Is Associated with Brain-Tropism of Metastasis from Lung Adenocarcinoma. International Journal of Molecular Sciences. 2021; 22(24):13374. https://doi.org/10.3390/ijms222413374
Chicago/Turabian StyleLin, Yen-Yu, Yu-Chao Wang, Da-Wei Yeh, Chen-Yu Hung, Yi-Chen Yeh, Hsiang-Ling Ho, Hsiang-Chen Mon, Mei-Yu Chen, Yu-Chung Wu, and Teh-Ying Chou. 2021. "Gene Expression Profile in Primary Tumor Is Associated with Brain-Tropism of Metastasis from Lung Adenocarcinoma" International Journal of Molecular Sciences 22, no. 24: 13374. https://doi.org/10.3390/ijms222413374