
Decreased Podocyte Vesicle Transcytosis and Albuminuria in APC C-Terminal Deficiency Mice with Puromycin-Induced Nephrotic Syndrome

 ,
,
Abstract
:1. Introduction
2. Results
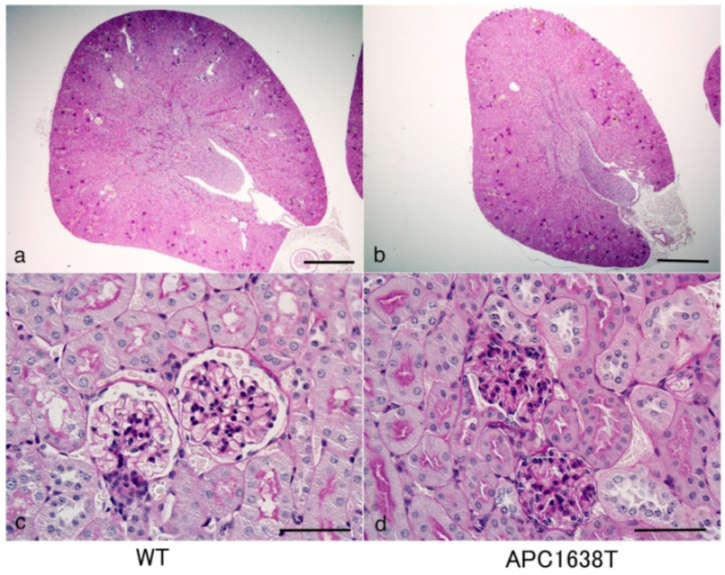
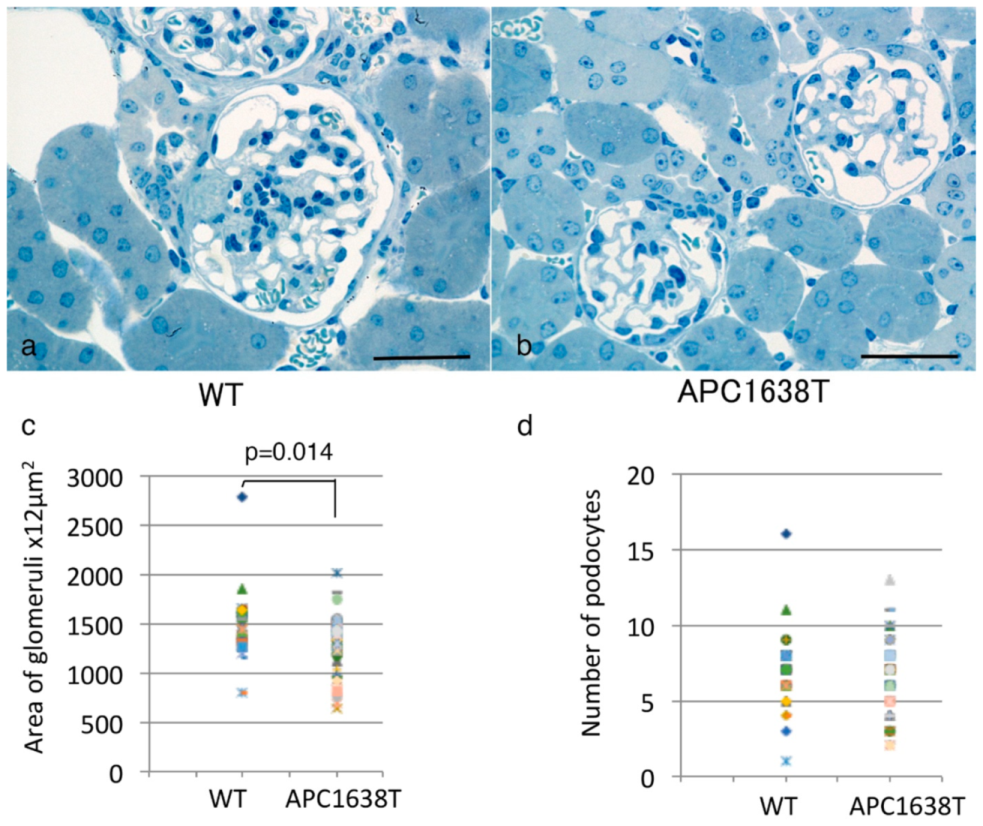
2.1. Kidney Size, Glomerular Volume, and Podocyte Number and Structure in APC Mutant Mice
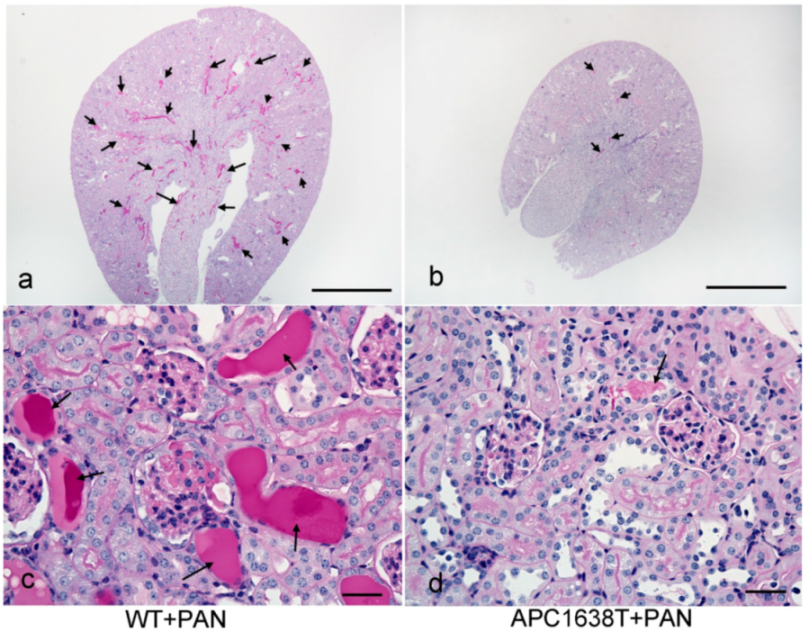
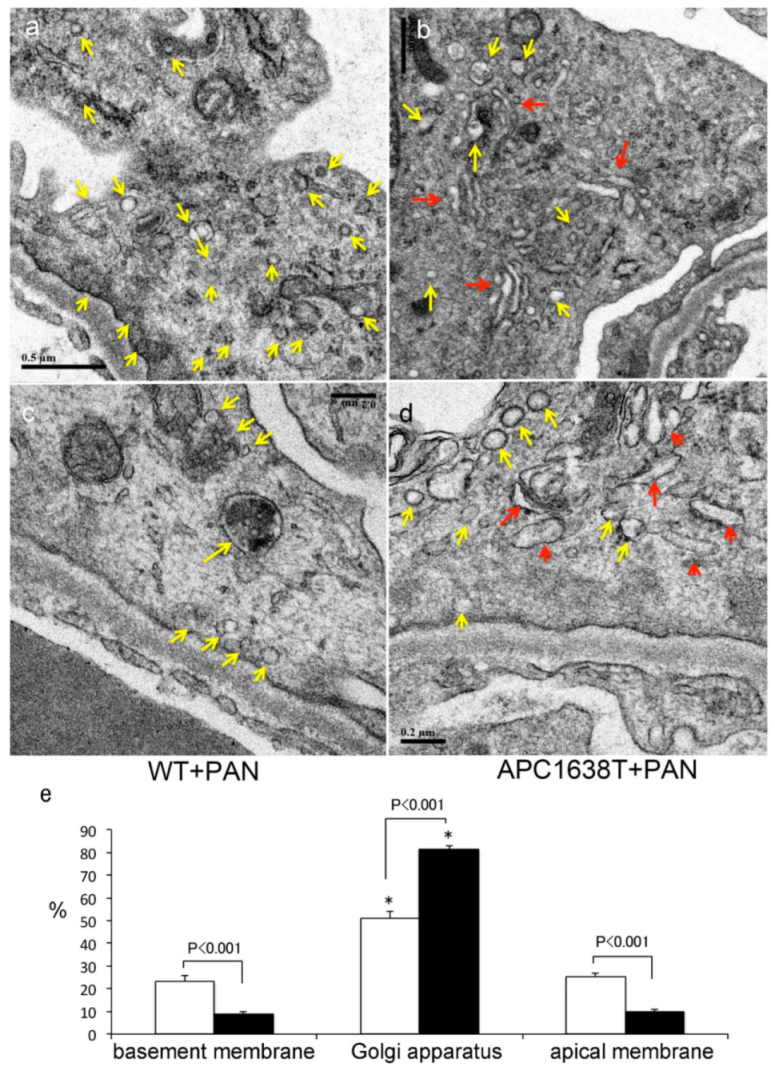
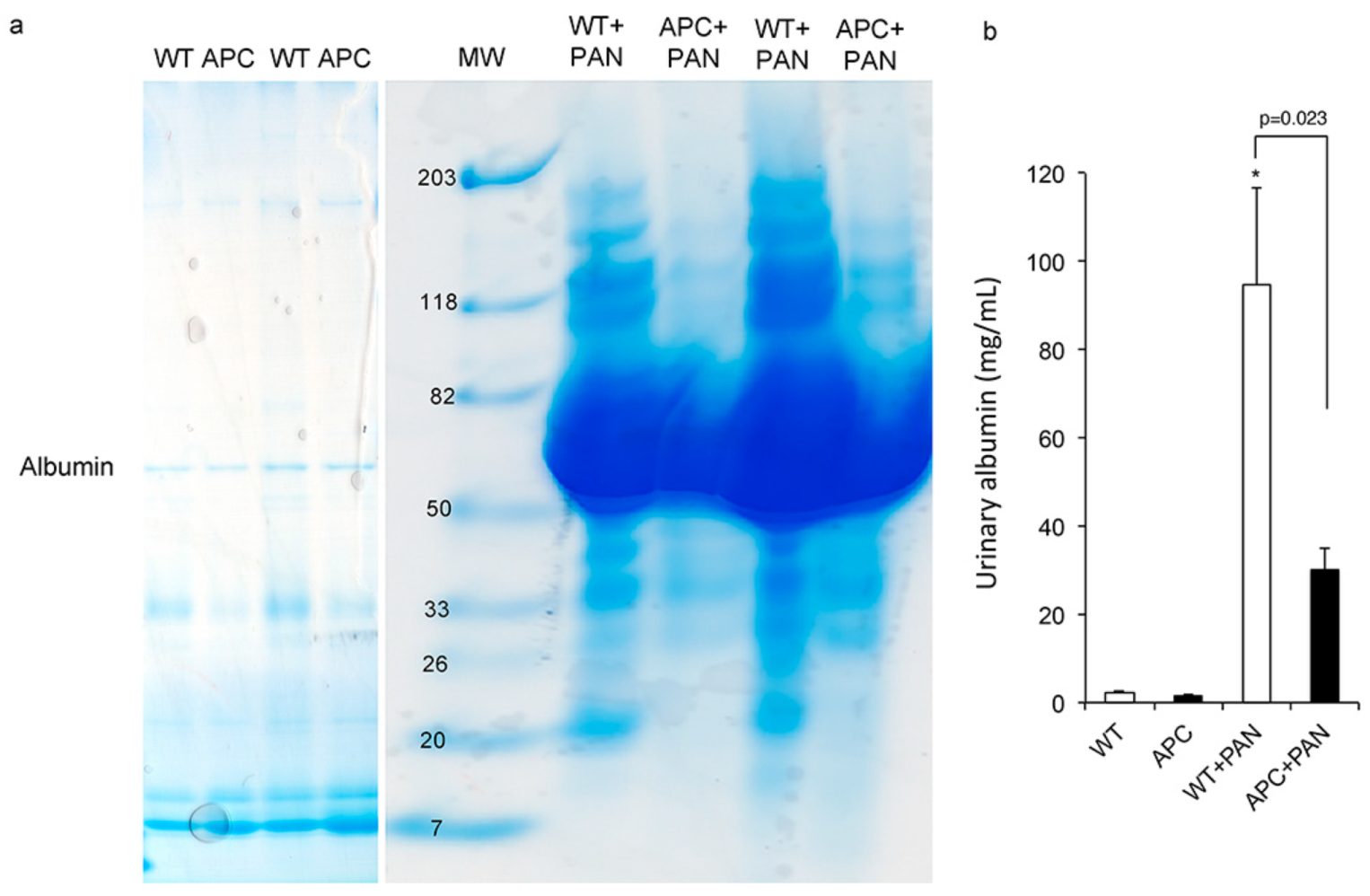
2.2. Kidneys of APC Mutant Mice with PAN-Induced Nephrotic Syndrome
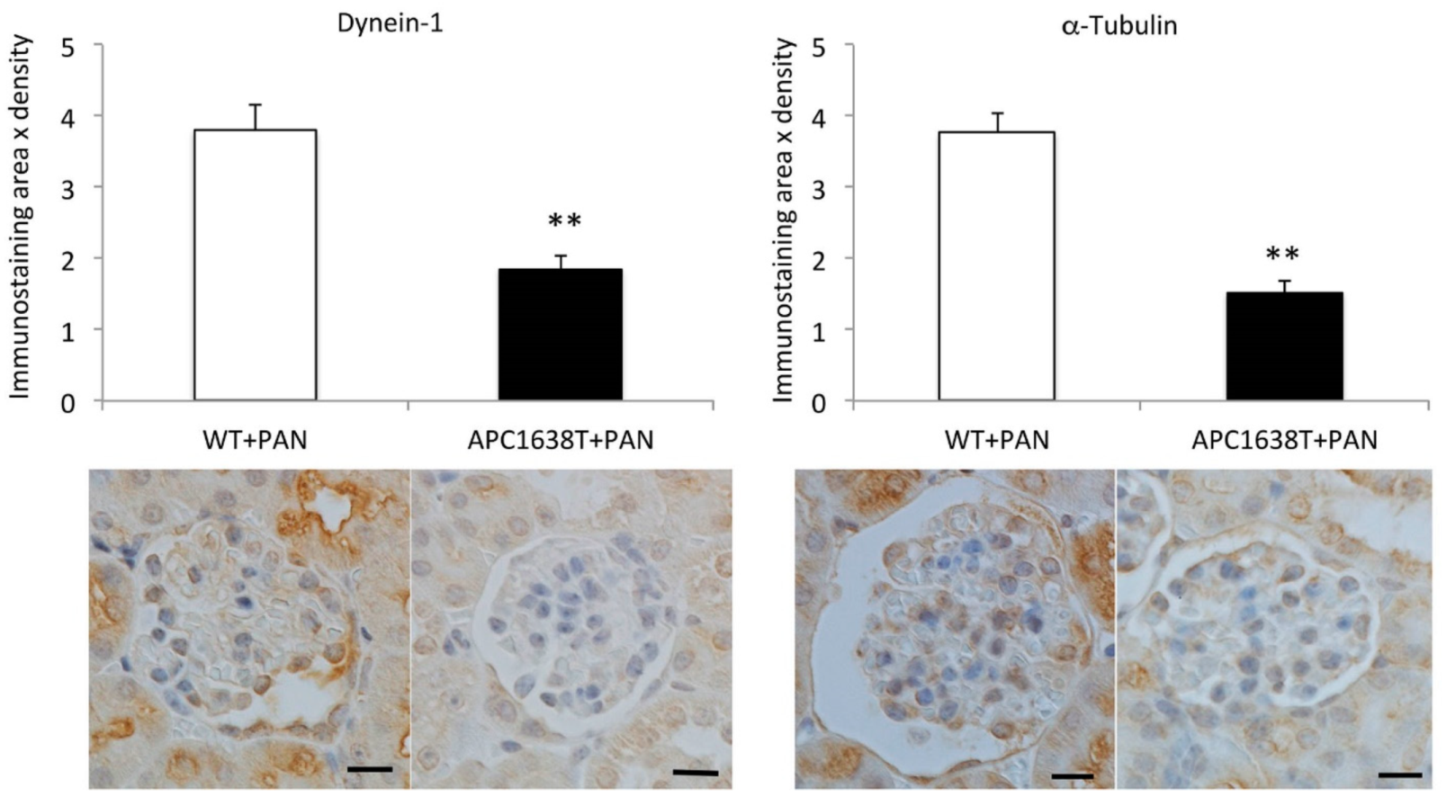
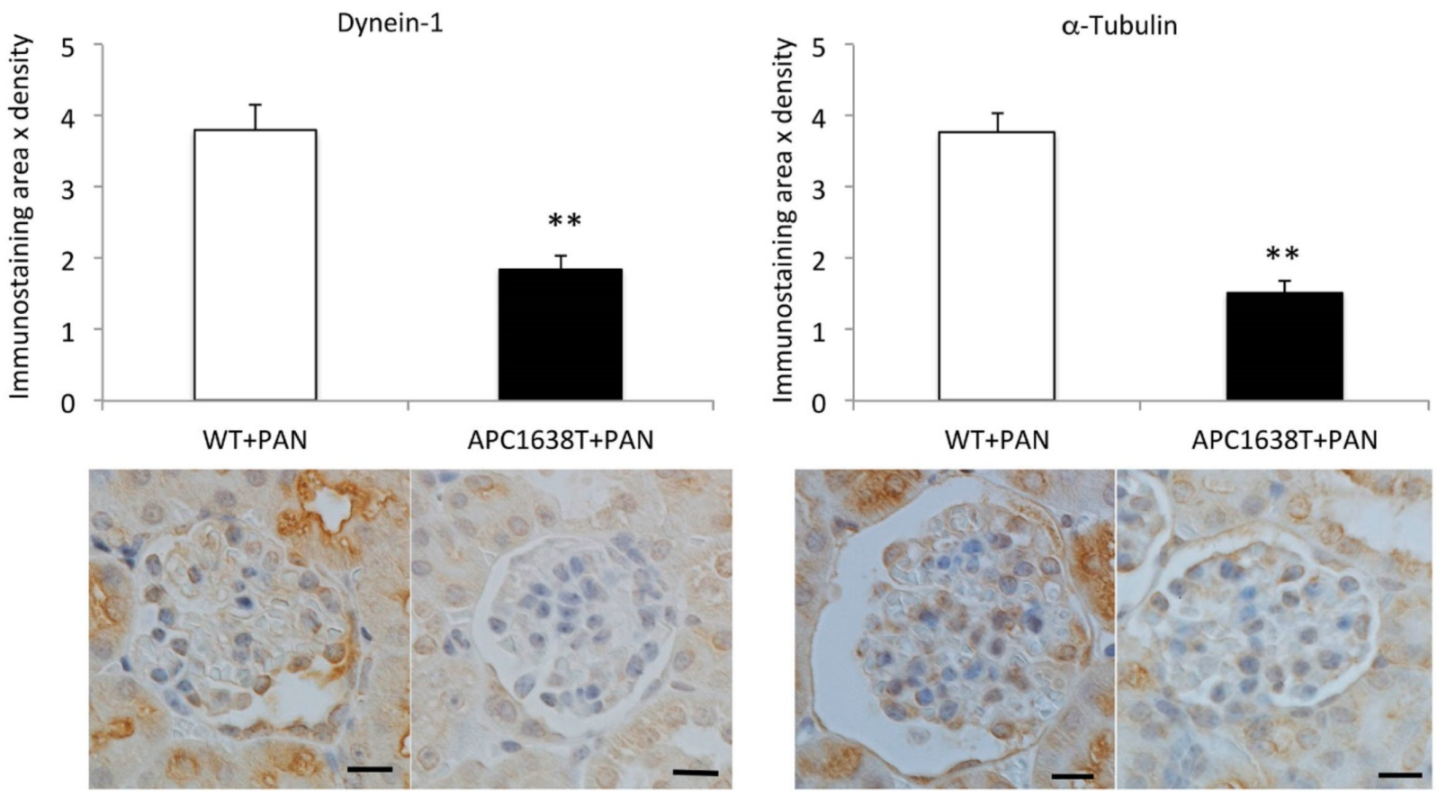
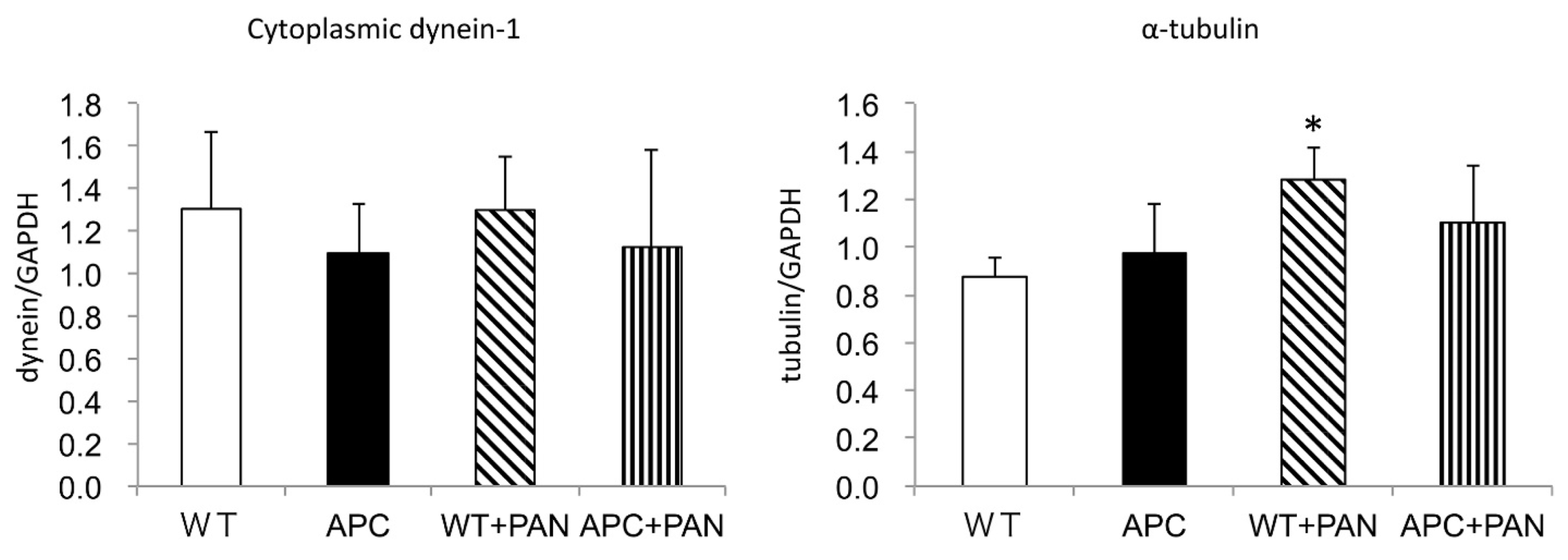
2.3. Cytoplasmic Dynein and α–Tubulin Expression in the Podocyte and Albuminuria in APC Mutant Mice with Nephrotic Syndrome
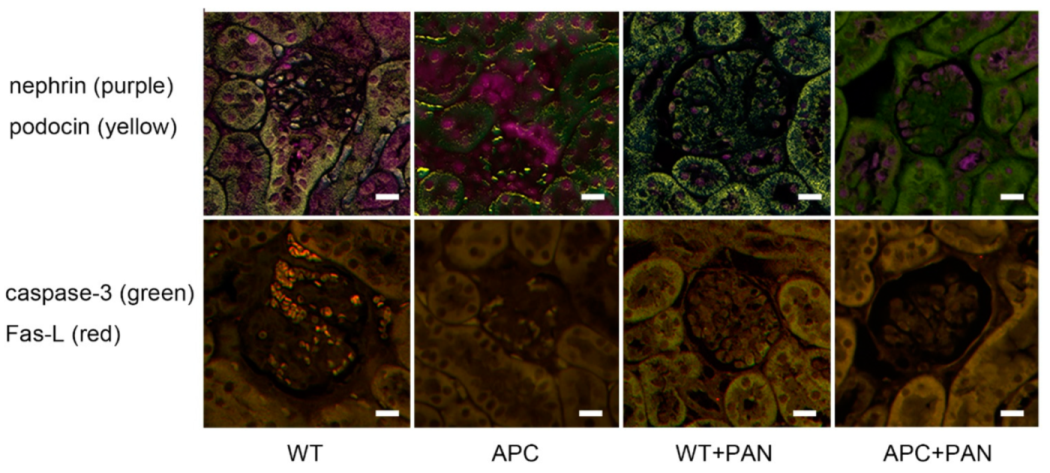
2.4. Immunofluorescence of Nephrin and Podocin Expression
3. Discussion
3.1. Renal Morphological Change in APC Mutant Mice under Control Conditions
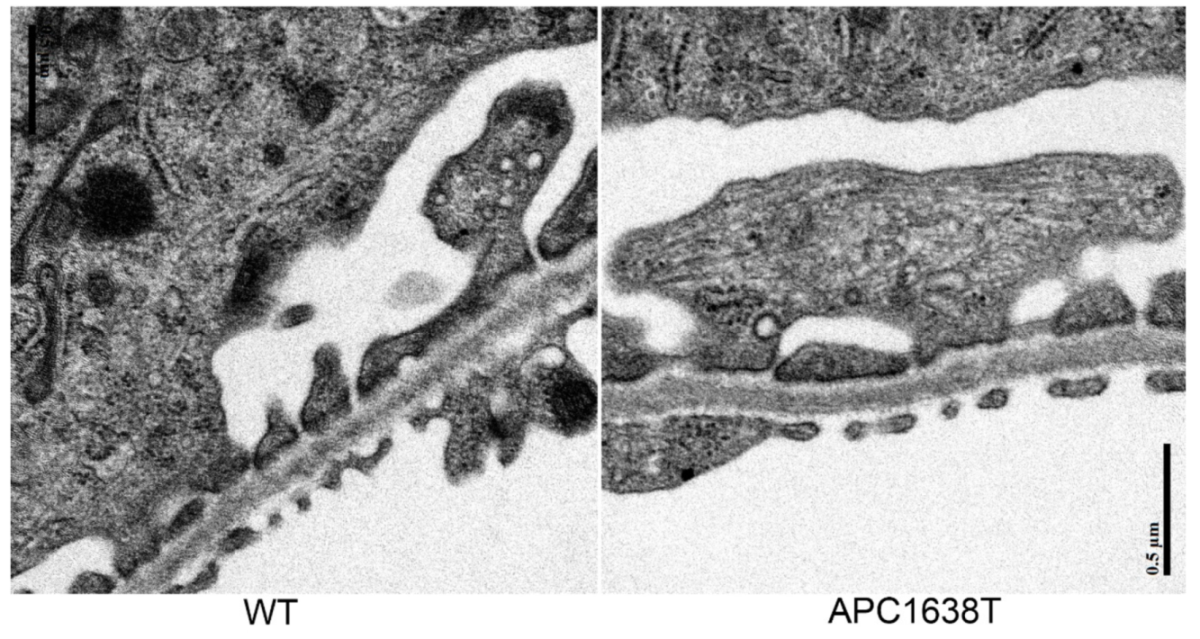
3.2. Morphological Characteristics of Podocyte of APC Mutant Mice with PAN-Induced Nephrotic Syndrome
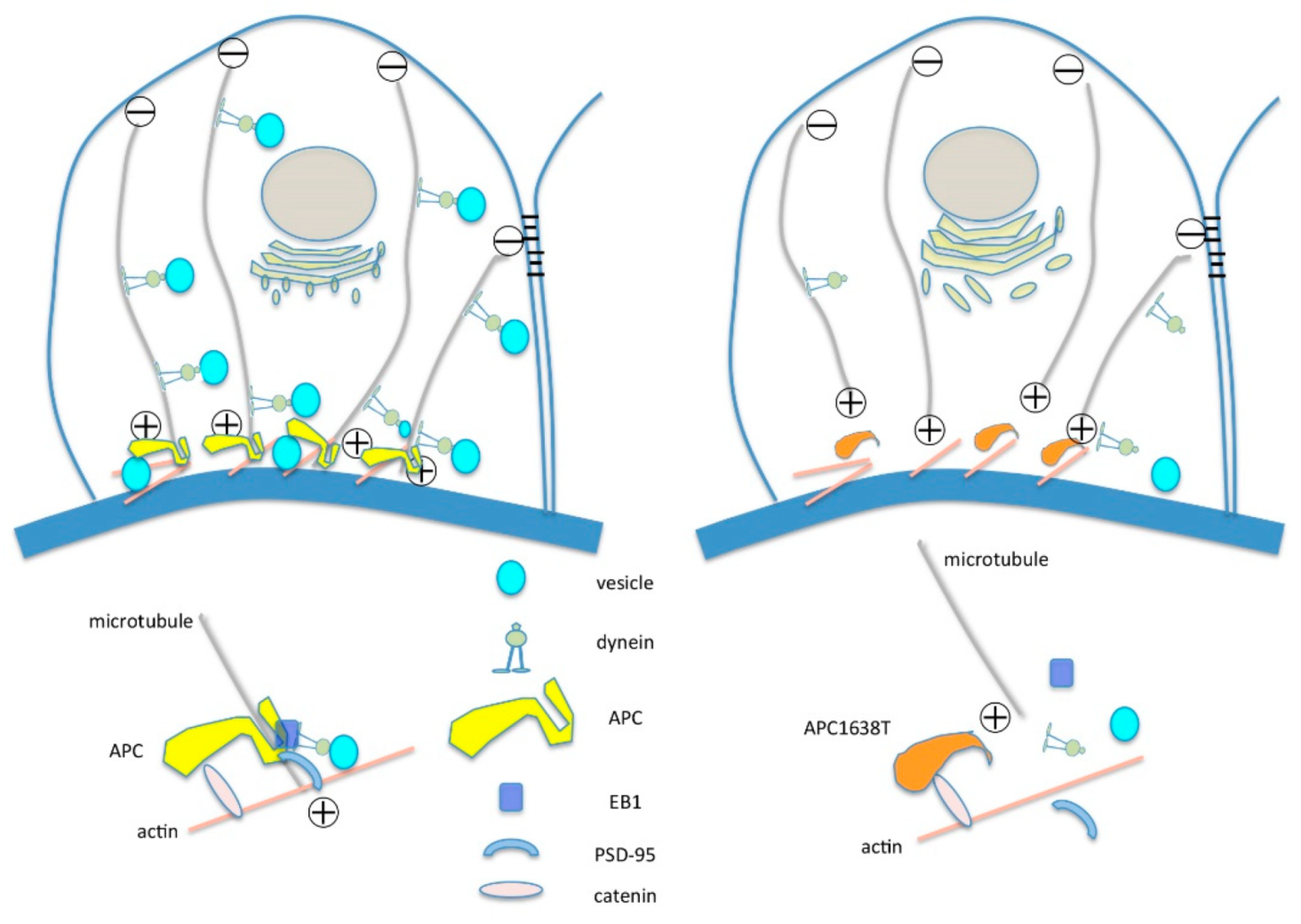
3.3. APC as a Therapeutic Target of Selective Microalbuminuria
4. Materials and Methods
4.1. Animal Experiments
4.2. Periodic Acid Schiff (PAS) Staining, Immunohistochemistry and Image Analyses
4.3. Electron Microscopy (EM)
4.4. Quantitative Real-Time PCR
4.5. Statistical Analyses
5. Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamada, E. The fine structure of the renal glomerulus of the mouse. J. Biophys. Biochem. Cytol. 1955, 1, 551–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinehart, J.F. Fine structure of renal glomerulus as revealed by electron microscopy. AMA Arch. Pathol. 1955, 59, 439–448. [Google Scholar] [PubMed]
- Farquhar, M.G.; Wissig, S.L.; Palade, G.E. Glomerular permeability. I. Ferritin transfer across the normal glomerular capillary wall. J. Exp. Med. 1961, 113, 47–66. [Google Scholar] [CrossRef] [Green Version]
- Rodewald, R.; Karnovsky, M.J. Porous substructure of the glomerular slit diaphragm in the rat and mouse. J. Cell Biol. 1974, 60, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruotsalainen, V.; Ljungberg, P.; Wartiovaara, J.; Lenkkeri, U.; Kestila, M.; Jalanko, H.; Holmberg, C.; Tryggvason, K. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 7962–7967. [Google Scholar] [CrossRef] [Green Version]
- Tryggvason, K.; Wartiovaara, J. Molecular basis of glomerular permselectivity. Curr. Opin. Nephrol. Hypertens. 2001, 10, 543–549. [Google Scholar] [CrossRef]
- Wartiovaara, J.; Ofverstedt, L.G.; Khoshnoodi, J.; Zhang, J.J.; Makela, E.; Sandin, S.; Ruotsalainen, V.; Cheng, R.H.; Jalanko, H.; Skoglund, U.; et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J. Clin. Investig. 2004, 114, 1475–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarad, G.; Cunningham, J.; Shaw, A.S.; Miner, J.H. Proteinuria precedes podocyte abnormalities in Lamb2(-/-) mice, implicating the glomerular basement membrane as an albumin barrier. J. Clin. Investig. 2006, 116, 2272–2279. [Google Scholar] [CrossRef] [Green Version]
- Farquhar, M.G. The glomerular basement membrane: Not gone, just forgotten. J. Clin. Investig. 2006, 116, 2090–2093. [Google Scholar] [CrossRef]
- Smithies, O. Why the kidney glomerulus does not clog: A gel permeation/diffusion hypothesis of renal function. Proc. Natl. Acad. Sci. USA 2003, 100, 4108–4113. [Google Scholar] [CrossRef] [Green Version]
- Tojo, A. The role of the kidney in protein metabolism: The capacity of tubular lysosomal proteolysis in nephrotic syndrome. Kidney Int. 2013, 84, 861–863. [Google Scholar] [CrossRef] [Green Version]
- Moeller, M.J.; Tenten, V. Renal albumin filtration: Alternative models to the standard physical barriers. Nat. Rev. Nephrol. 2013, 9, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Tojo, A.; Onozato, M.L.; Kitiyakara, C.; Kinugasa, S.; Fukuda, S.; Sakai, T.; Fujita, T. Glomerular albumin filtration through podocyte cell body in puromycin aminonucleoside nephrotic rat. Med. Mol. Morphol. 2008, 41, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, S.; Tojo, A.; Sakai, T.; Tsumura, H.; Takahashi, M.; Hirata, Y.; Fujita, T. Selective albuminuria via podocyte albumin transport in puromycin nephrotic rats is attenuated by an inhibitor of NADPH oxidase. Kidney Int. 2011, 80, 1328–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, K.; Balkin, D.M.; Ferguson, S.M.; Paradise, S.; Milosevic, I.; Giovedi, S.; Volpicelli-Daley, L.; Tian, X.; Wu, Y.; Ma, H.; et al. Role of dynamin, synaptojanin, and endophilin in podocyte foot processes. J. Clin. Investig. 2012, 122, 4401–4411. [Google Scholar] [CrossRef] [Green Version]
- Schiessl, I.M.; Hammer, A.; Kattler, V.; Gess, B.; Theilig, F.; Witzgall, R.; Castrop, H. Intravital Imaging Reveals Angiotensin II-Induced Transcytosis of Albumin by Podocytes. J. Am. Soc. Nephrol. 2016, 27, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Dobrinskikh, E.; Okamura, K.; Kopp, J.B.; Doctor, R.B.; Blaine, J. Human podocytes perform polarized, caveolae-dependent albumin endocytosis. Am. J. Physiol. Renal Physiol. 2014, 306, F941–F951. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.J.; Huber, T.B.; Godel, M.; Jarad, G.; Hartleben, B.; Kwoh, C.; Keil, A.; Karpitskiy, A.; Hu, J.; Huh, C.J.; et al. Albumin-associated free fatty acids induce macropinocytosis in podocytes. J. Clin. Investig. 2015, 125, 2307–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tojo, A. Mechanism Underlying Selective Albuminuria in Minimal Change Nephrotic Syndrome. Int. J. Nephrol. 2019, 2019, 5859102. [Google Scholar] [CrossRef]
- Farquhar, M.G.; Vernier, R.L.; Good, R.A. Studies on familial nephrosis. II. Glomerular changes observed with the electron microscope. Am. J. Pathol. 1957, 33, 791–817. [Google Scholar] [PubMed]
- Farquhar, M.G.; Vernier, R.L.; Good, R.A. An electron microscope study of the glomerulus in nephrosis, glomerulonephritis, and lupus erythematosus. J. Exp. Med. 1957, 106, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Tojo, A.; Hatakeyama, S.; Kinugasa, S.; Fukuda, S.; Sakai, T. Enhanced podocyte vesicle transport in the nephrotic rat. Med. Mol. Morphol. 2017, 50, 86–93. [Google Scholar] [CrossRef]
- Rishal, I.; Fainzilber, M. Axon-soma communication in neuronal injury. Nat. Rev. Neurosci. 2014, 15, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, K.L.; Greensmith, L.; Schiavo, G. Regulation of Axonal Transport by Protein Kinases. Trends Biochem. Sci. 2015, 40, 597–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, S.M.; Zilz, N.; Beazerbarclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. Apc Mutations Occur Early during Colorectal Tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Brakeman, J.S.F.; Gu, S.H.; Wang, X.B.; Dolin, G.; Baraban, J.M. Neuronal localization of the Adenomatous polyposis coli tumor suppressor protein. Neuroscience 1999, 91, 661–672. [Google Scholar] [CrossRef]
- Temburni, M.K.; Rosenberg, M.M.; Pathak, N.; McConnell, R.; Jacob, M.H. Neuronal nicotinic synapse assembly requires the adenomatous polyposis coli tumor suppressor protein. J. Neurosci. 2004, 24, 6776–6784. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, M.M.; Tucker, J.B.; Mackie, J.B.; Prescott, A.R.; Nathke, I.S. The adenomatous polyposis coli protein unambiguously localizes to microtubule plus ends and is involved in establishing parallel arrays of microtubule bundles in highly polarized epithelial cells. J. Cell Biol. 2002, 157, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Smits, R.; Kielman, M.F.; Breukel, C.; Zurcher, C.; Neufeld, K.; Jagmohan-Changur, S.; Hofland, N.; van Dijk, J.; White, R.; Edelmann, W.; et al. Apc1638T: A mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev. 1999, 13, 1309–1321. [Google Scholar] [CrossRef]
- Kakinuma, N.; Nishimura, Y.; Akiyama, T.; Senda, T. APC is colocalized with beta-catenin and hDLG in Henle‘s loop of the mouse kidney. Acta Histochem. Cytochem. 2000, 33, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Senda, T.; Iizuka-Kogo, A.; Onouchi, T.; Shimomura, A. Adenomatous polyposis coli (APC) plays multiple roles in the intestinal and colorectal epithelia. Med. Mol. Morphol. 2007, 40, 68–81. [Google Scholar] [CrossRef]
- Li, C.G.; Onouchi, T.; Hirayama, M.; Sakai, K.; Matsuda, S.; Yamada, N.O.; Senda, T. Morphological and functional abnormalities of hippocampus inAPC(1638T/1638T)mice. Med. Mol. Morphol. 2021, 54, 31–40. [Google Scholar] [CrossRef]
- Tojo, A.; Bredt, D.S.; Wilcox, C.S. Distribution of postsynaptic density proteins in rat kidney: Relationship to neuronal nitric oxide synthase. Kidney Int. 1999, 55, 1384–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Kasama, Y.; Lee, J.S.; Jim, B.; Marin, M.; Ziyadeh, F.N. Podocyte-derived vascular endothelial growth factor mediates the stimulation of alpha 3(IV) collagen production by transforming growth factor-beta 1 in mouse podocytes. Diabetes 2004, 53, 2939–2949. [Google Scholar] [CrossRef] [Green Version]
- Alsabban, A.H.; Morikawa, M.; Tanaka, Y.; Takei, Y.; Hirokawa, N. Kinesin Kif3b mutation reduces NMDAR subunit NR2A trafficking and causes schizophrenia-like phenotypes in mice. EMBO J. 2020, 39, e101090. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Onouchi, T.; Yamada, N.O.; Matsuda, S.; Senda, T. A disturbance of intestinal epithelial cell population and kinetics in APC1638T mice. Med. Mol. Morphol. 2017, 50, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Nathke, I.S.; Adams, C.L.; Polakis, P.; Sellin, J.H.; Nelson, W.J. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J. Cell Biol. 1996, 134, 165–179. [Google Scholar] [CrossRef]
- Mogensen, M.M.; Malik, A.; Piel, M.; Bouckson-Castaing, V.; Bornens, M. Microtubule minus-end anchorage at centrosomal and non-centrosomal sites: The role of ninein. J. Cell Sci. 2000, 113, 3013–3023. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Villarin, J.M.; McCurdy, E.P.; Martinez, J.C.; Hengst, U. Local synthesis of dynein cofactors matches retrograde transport to acutely changing demands. Nat. Commun. 2016, 7, 13865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraldsson, B.; Nystrom, J.; Deen, W.M. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol. Rev. 2008, 88, 451–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, N.O.; Wenduerma, S.; Matsuda, S.; Senda, T. Validation and application of a novel APC antibody in western blotting, immunoprecipitation, and immunohistochemistry. Med. Mol. Morphol. 2018, 51, 227–236. [Google Scholar] [CrossRef]
- Tojo, A.; Asaba, K.; Kinugasa, S.; Ikeda, Y.; Shintani, Y.; Fukayama, M.; Nangaku, M. The reduced expression of proximal tubular transporters in acquired Fanconi syndrome with kappa light chain deposition. Med. Mol. Morphol. 2016, 49, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Hanamura, K.; Tojo, A.; Fujita, T. Urinary and glomerular podocytes in patients with chronic kidney diseases. Clin. Exp. Nephrol. 2014, 18, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Satonaka, H.; Nagata, D.; Takahashi, M.; Kiyosue, A.; Myojo, M.; Fujita, D.; Ishimitsu, T.; Nagano, T.; Nagai, R.; Hirata, Y. Involvement of P2Y(12) receptor in vascular smooth muscle inflammatory changes via MCP-1 upregulation and monocyte adhesion. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H853–H861. [Google Scholar] [CrossRef] [Green Version]
- Asano-Matsuda, K.; Ibrahim, S.; Takano, T.; Matsuda, J. Role of Rho GTPase Interacting Proteins in Subcellular Compartments of Podocytes. Int. J. Mol. Sci. 2021, 22, 3656. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequences 5′ to 3′ |
|---|---|
| Dync1h | Forward AGTCACAGGTCTGAAGCTCC |
| Reverse ACTGTGGAGATGGCATTGGA | |
| tubulinAlpha1a | Forward AGCGGCTCTCTGTGGATTAC |
| Reverse CAACCACAGCAGTGGAAACC | |
| GAPDH | Forward ACCCAGAAGACTGTGGATGG |
| Reverse GGATGCAGGGATGATGTTCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatakeyama, S.; Tojo, A.; Satonaka, H.; Yamada, N.O.; Senda, T.; Ishimitsu, T. Decreased Podocyte Vesicle Transcytosis and Albuminuria in APC C-Terminal Deficiency Mice with Puromycin-Induced Nephrotic Syndrome. Int. J. Mol. Sci. 2021, 22, 13412. https://doi.org/10.3390/ijms222413412
Hatakeyama S, Tojo A, Satonaka H, Yamada NO, Senda T, Ishimitsu T. Decreased Podocyte Vesicle Transcytosis and Albuminuria in APC C-Terminal Deficiency Mice with Puromycin-Induced Nephrotic Syndrome. International Journal of Molecular Sciences. 2021; 22(24):13412. https://doi.org/10.3390/ijms222413412
Chicago/Turabian StyleHatakeyama, Saaya, Akihiro Tojo, Hiroshi Satonaka, Nami O. Yamada, Takao Senda, and Toshihiko Ishimitsu. 2021. "Decreased Podocyte Vesicle Transcytosis and Albuminuria in APC C-Terminal Deficiency Mice with Puromycin-Induced Nephrotic Syndrome" International Journal of Molecular Sciences 22, no. 24: 13412. https://doi.org/10.3390/ijms222413412
APA StyleHatakeyama, S., Tojo, A., Satonaka, H., Yamada, N. O., Senda, T., & Ishimitsu, T. (2021). Decreased Podocyte Vesicle Transcytosis and Albuminuria in APC C-Terminal Deficiency Mice with Puromycin-Induced Nephrotic Syndrome. International Journal of Molecular Sciences, 22(24), 13412. https://doi.org/10.3390/ijms222413412