
C-Terminal Domain of Aquaporin-5 Is Required to Pass Its Protein Quality Control and Ensure Its Trafficking to Plasma Membrane

Abstract
:1. Introduction
2. Results
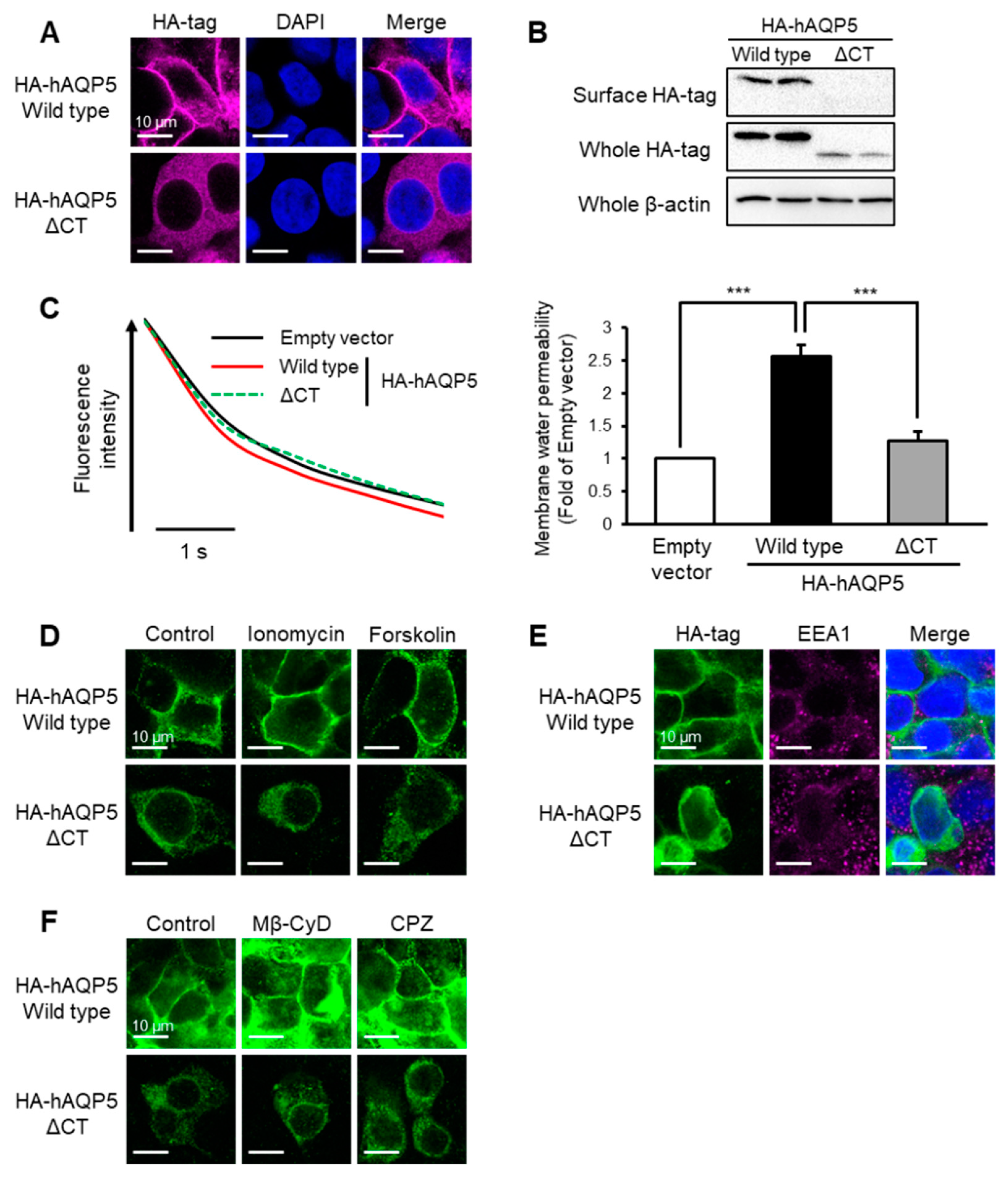
2.1. C-Terminal Domain Is Required for Plasma Membrane Localization of AQP5
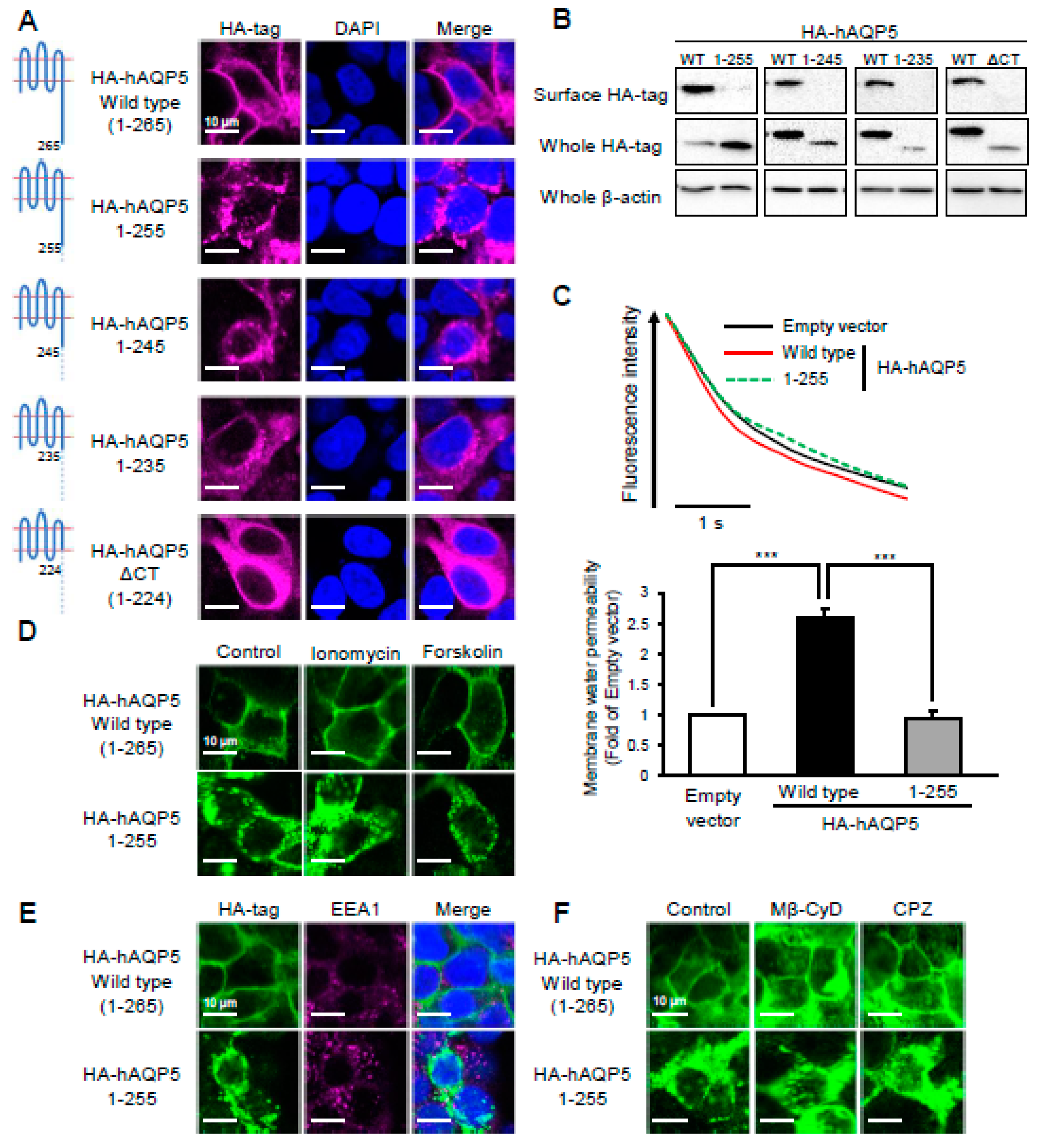
2.2. Ten Amino Acid Residues between Arg256 and Arg265 Are Required for Plasma Membrane Localization of AQP5
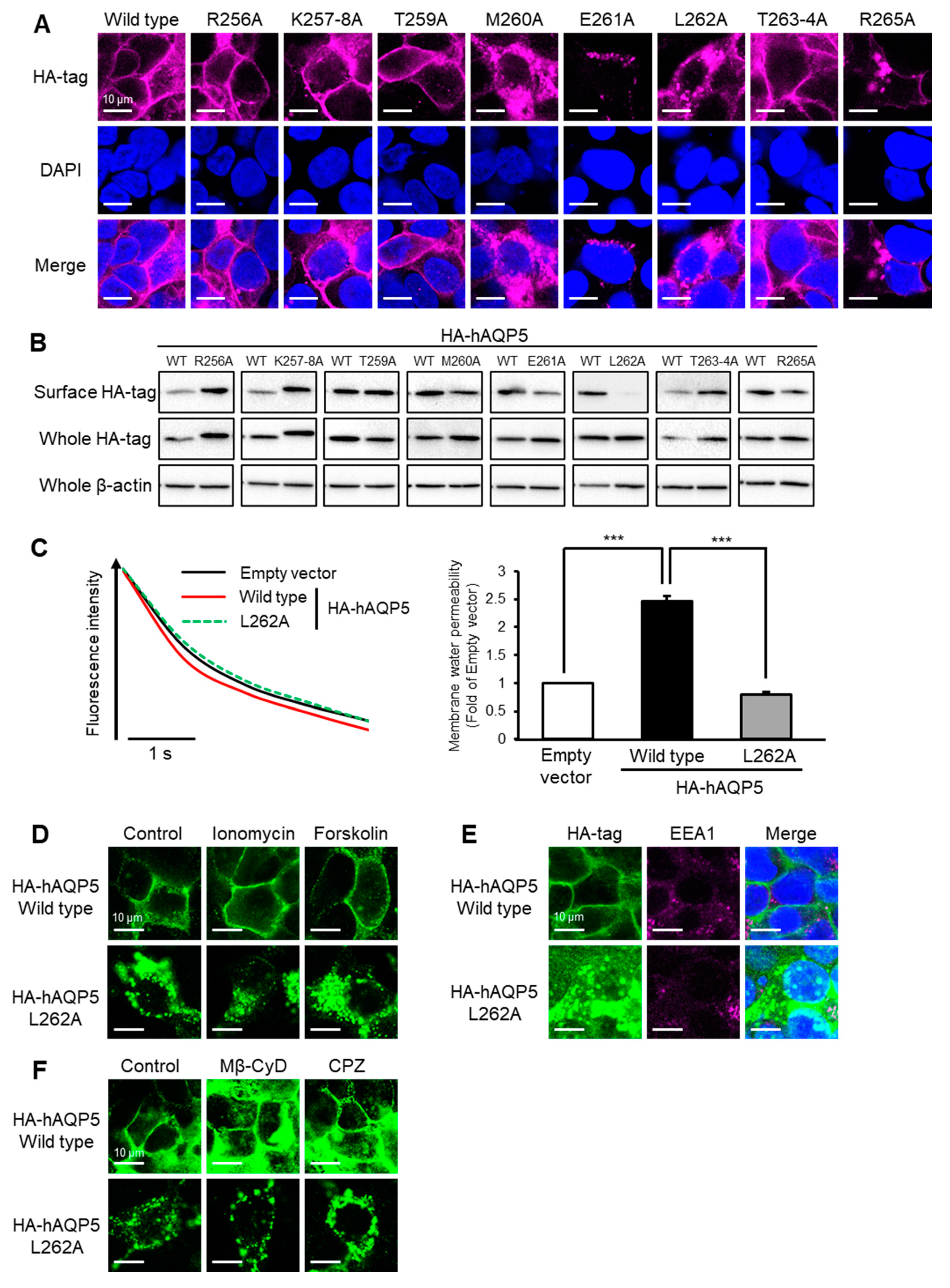
2.3. Leu262 Is Required for Plasma Membrane Localization of AQP5
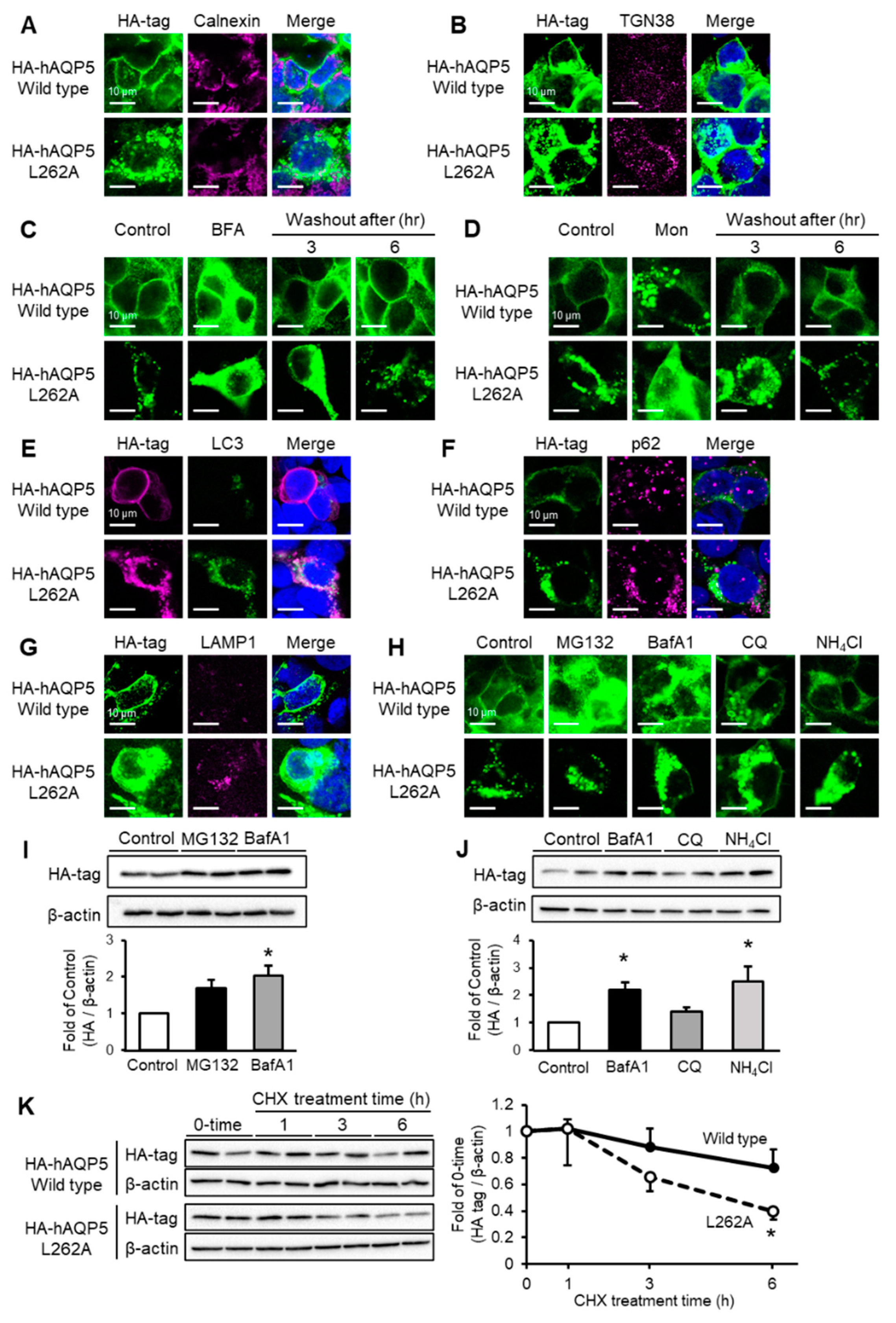
2.4. C-Terminal Domain Mutant of AQP5 Localized to Autophagosome or Lysosome and Was Degraded by Autophagy
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies
4.3. Immunofluorescence
4.4. Cell Surface Biotinylation Assay
4.5. Immunoblotting
4.6. Assay for Plasma Membrane Osmotic Water Permeability
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Verkman, A.S.; Mitra, A.K. Structure and function of aquaporin water channels. Am. J. Physiol. Renal. Physiol. 2000, 278, F13–F28. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. More than just water channels: Unexpected cellular roles of aquaporins. J. Cell Sci. 2005, 118, 3225–3232. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Song, Y.; Gillespie, A.; Carlson, E.J.; Epstein, C.J.; Verkman, A.S. Defective secretion of saliva in transgenic mice lacking aquaporin-5 water channels. J. Biol. Chem. 1999, 274, 20071–20074. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Fukuda, N.; Song, Y.; Matthay, M.A.; Verkman, A.S. Lung fluid transport in aquaporin-5 knockout mice. J. Clin. Investig. 2000, 105, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Verkman, A.S. Aquaporin-5 dependent fluid secretion in airway submucosal glands. J. Biol. Chem. 2001, 276, 41288–41292. [Google Scholar] [CrossRef] [Green Version]
- Groneberg, D.A.; Gerber, A.; Fischer, A. Trafficking of lacrimal aquaporin-5 in Sjögren’s syndrome. Lancet 2001, 357, 2054–2055. [Google Scholar] [CrossRef]
- Steinfeld, S.; Cogan, E.; King, L.S.; Agre, P.; Kiss, R.; Delporte, C. Abnormal distribution of aquaporin-5 water channel protein in salivary glands from Sjögren’s syndrome patients. Lab Investig. 2001, 81, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Tsubota, K.; Hirai, S.; King, L.S.; Agre, P.; Ishida, N. Defective cellular trafficking of lacrimal gland aquaporin-5 in Sjögren’s syndrome. Lancet 2001, 357, 688–689. [Google Scholar] [CrossRef]
- Ichiyama, T.; Nakatani, E.; Tatsumi, K.; Hideshima, K.; Urano, T.; Nariai, Y.; Sekine, J. Expression of aquaporin 3 and 5 as a potential marker for distinguishing dry mouth from Sjögren’s syndrome. J. Oral. Sci. 2018, 60, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Ohinata, A.; Nagai, K.; Nomura, J.; Hashimoto, K.; Hisatsune, A.; Miyata, T.; Isohama, Y. Lipopolysaccharide changes the subcellular distribution of aquaporin 5 and increases plasma membrane water permeability in mouse lung epithelial cells. Biochem. Biophys. Res. Commun. 2005, 326, 521–526. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Yuan, Z.; Inoue, N.; Skowronski, M.T.; Nakae, Y.; Shono, M.; Cho, G.; Yasui, M.; Agre, P.; Nielsen, S. Identification of AQP5 in lipid rafts and its translocation to apical membranes by activation of M3 mAChRs in interlobular ducts of rat parotid gland. Am. J. Physiol. Cell Physiol. 2005, 289, C1303–C1311. [Google Scholar] [CrossRef]
- Nagai, K.; Watanabe, M.; Seto, M.; Hisatsune, A.; Miyata, T.; Isohama, Y. Nitric oxide decreases cell surface expression of aquaporin-5 and membrane water permeability in lung epithelial cells. Biochem. Biophys. Res. Commun. 2007, 354, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Luna, E.J.; Hitt, A.L. Cytoskeleton--plasma membrane interactions. Science 1992, 258, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Anitei, M.; Hoflack, B. Bridging membrane and cytoskeleton dynamics in the secretory and endocytic pathways. Nat. Cell Biol. 2011, 14, 11–19. [Google Scholar] [CrossRef]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell. Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef]
- Seabra, M.C.; Mules, E.H.; Hume, A.N. Rab GTPases, intracellular traffic and disease. Trends Mol. Med. 2002, 8, 23–30. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell. Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Langemeyer, L.; Fröhlich, F.; Ungermann, C. Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol. 2018, 28, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Öberg, F.; Sjöhamn, J.; Hedfalk, K.; Bill, R.M.; Conner, A.C.; Conner, M.T.; Törnroth-Horsefield, S. Plasma Membrane Abundance of Human Aquaporin 5 Is Dynamically Regulated by Multiple Pathways. PLoS ONE 2015, 10, e0143027. [Google Scholar] [CrossRef]
- Wellner, R.B.; Hong, S.; Cotrim, A.P.; Swaim, W.D.; Baum, B.J. Modifying the NH2 and COOH termini of aquaporin-5: Effects on localization in polarized epithelial cells. Tissue Eng. 2005, 11, 1449–1458. [Google Scholar] [CrossRef]
- Wellner, R.B.; Cotrim, A.P.; Hong, S.; Swaim, W.D.; Baum, B.J. Localization of AQP5/AQP8 chimeras in MDCK-II cells: Exchange of the N- and C-termini. Biochem. Biophys. Res. Commun. 2005, 330, 172–177. [Google Scholar] [CrossRef]
- Kuwahara, M.; Asai, T.; Terada, Y.; Sasaki, S. The C-terminal tail of aquaporin-2 determines apical trafficking. Kidney Int. 2005, 68, 1999–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131, jcs217364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chino, H.; Mizushima, N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef]
- Conner, A.C.; Bill, R.M.; Conner, M.T. An emerging consensus on aquaporin translocation as a regulatory mechanism. Mol. Membr. Biol. 2013, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Moeller, H.B.; Aroankins, T.S.; Slengerik-Hansen, J.; Pisitkun, T.; Fenton, R.A. Phosphorylation and ubiquitylation are opposing processes that regulate endocytosis of the water channel aquaporin-2. J. Cell Sci. 2014, 127, 3174–3183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamsteeg, E.J.; Hendriks, G.; Boone, M.; Konings, I.B.; Oorschot, V.; van der Sluijs, P.; Klumperman, J.; Deen, P.M. Short-chain ubiquitination mediates the regulated endocytosis of the aquaporin-2 water channel. Proc. Natl. Acad. Sci. USA 2006, 103, 18344–18349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, T.; Azlina, A.; Javkhlan, P.; Yao, C.; Akamatsu, T.; Hosoi, K. Novel phosphorylation of aquaporin-5 at its threonine 259 through cAMP signaling in salivary gland cells. Am. J. Physiol. Cell Physiol. 2011, 301, C667–C678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Babst, M. Quality control: Quality control at the plasma membrane: One mechanism does not fit all. J. Cell Biol. 2014, 205, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Progida, C.; Bakke, O. Bidirectional traffic between the Golgi and the endosomes-machineries and regulation. J. Cell Sci. 2016, 129, 3971–3982. [Google Scholar] [CrossRef] [Green Version]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Apaja, P.M.; Lukacs, G.L. Protein quality control at the plasma membrane. Curr. Opin. Cell Biol. 2011, 23, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Zeitlin, P.L. Pharmacologic restoration of delta F508 CFTR-mediated chloride current. Kidney Int. 2000, 57, 832–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spanò, V.; Montalbano, A.; Carbone, A.; Scudieri, P.; Galietta, L.J.V.; Barraja, P. An overview on chemical structures as ΔF508-CFTR correctors. Eur. J. Med. Chem. 2019, 180, 430–448. [Google Scholar] [CrossRef]
- Stoops, E.H.; Caplan, M.J. Trafficking to the apical and basolateral membranes in polarized epithelial cells. J. Am. Soc. Nephrol. 2014, 25, 1375–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, T.; Sasaki, S.; Uchida, S. Polarized trafficking of the aquaporin-3 water channel is mediated by an NH2-terminal sorting signal. Am. J. Physiol. Cell Physiol. 2006, 290, C298–C304. [Google Scholar] [CrossRef] [PubMed]
- Madrid, R.; Le Maout, S.; Barrault, M.B.; Janvier, K.; Benichou, S.; Mérot, J. Polarized trafficking and surface expression of the AQP4 water channel are coordinated by serial and regulated interactions with different clathrin-adaptor complexes. EMBO J. 2001, 20, 7008–7021. [Google Scholar] [CrossRef] [Green Version]
- Horsefield, R.; Nordén, K.; Fellert, M.; Backmark, A.; Törnroth-Horsefield, S.; Terwisscha van Scheltinga, A.C.; Kvassman, J.; Kjellbom, P.; Johanson, U.; Neutze, R. High-resolution x-ray structure of human aquaporin 5. Proc. Natl. Acad. Sci. USA 2008, 105, 13327–13332. [Google Scholar] [CrossRef] [Green Version]
- Fenton, R.A.; Moeller, H.B.; Nielsen, S.; de Groot, B.L.; Rützler, M. A plate reader-based method for cell water permeability measurement. Am. J. Physiol. Renal. Physiol. 2010, 298, F224–F230. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | 221 224 235 245 255 265 |
|---|---|
| HA-hAQP5-WT | -YFYLLFPNSLSLSERVAIIKGTYEPNENWEEQREERKKTMELTTR |
| HA-hAQP5 1-255 | -YFYLLFPNSLSLSERVAIIKGTYEPNENWEEQREE |
| HA-hAQP5 1-245 | -YFYLLFPNSLSLSERVAIIKGTYEP |
| HA-hAQP5 1-235 | -YFYLLFPNSLSLSER |
| HA-hAQP5 1-224 | -YFYL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muroi, S.-i.; Isohama, Y. C-Terminal Domain of Aquaporin-5 Is Required to Pass Its Protein Quality Control and Ensure Its Trafficking to Plasma Membrane. Int. J. Mol. Sci. 2021, 22, 13461. https://doi.org/10.3390/ijms222413461
Muroi S-i, Isohama Y. C-Terminal Domain of Aquaporin-5 Is Required to Pass Its Protein Quality Control and Ensure Its Trafficking to Plasma Membrane. International Journal of Molecular Sciences. 2021; 22(24):13461. https://doi.org/10.3390/ijms222413461
Chicago/Turabian StyleMuroi, Shin-ichi, and Yoichiro Isohama. 2021. "C-Terminal Domain of Aquaporin-5 Is Required to Pass Its Protein Quality Control and Ensure Its Trafficking to Plasma Membrane" International Journal of Molecular Sciences 22, no. 24: 13461. https://doi.org/10.3390/ijms222413461
APA StyleMuroi, S.-i., & Isohama, Y. (2021). C-Terminal Domain of Aquaporin-5 Is Required to Pass Its Protein Quality Control and Ensure Its Trafficking to Plasma Membrane. International Journal of Molecular Sciences, 22(24), 13461. https://doi.org/10.3390/ijms222413461