
Dynamic Coupling of Tyrosine 185 with the Bacteriorhodopsin Photocycle, as Revealed by Chemical Shifts, Assisted AF-QM/MM Calculations and Molecular Dynamic Simulations

 and
and
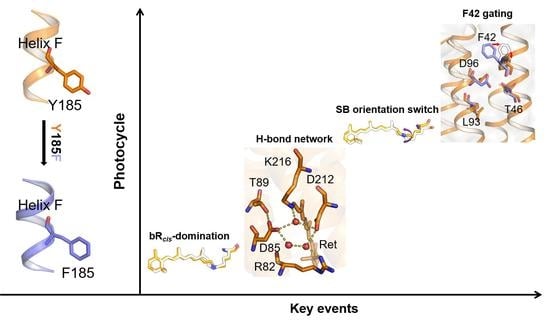
Abstract
:
1. Introduction
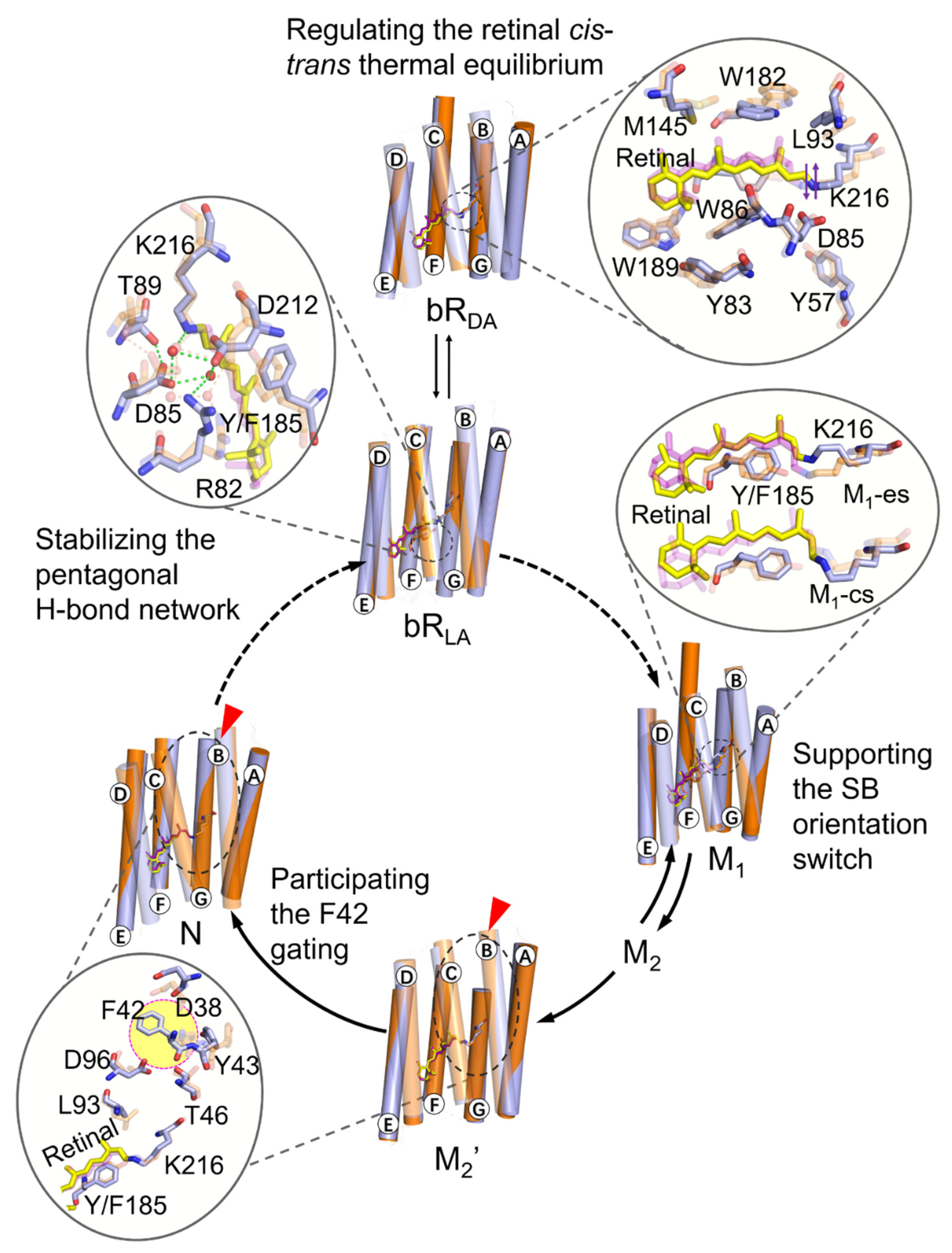
2. Results and Discussion
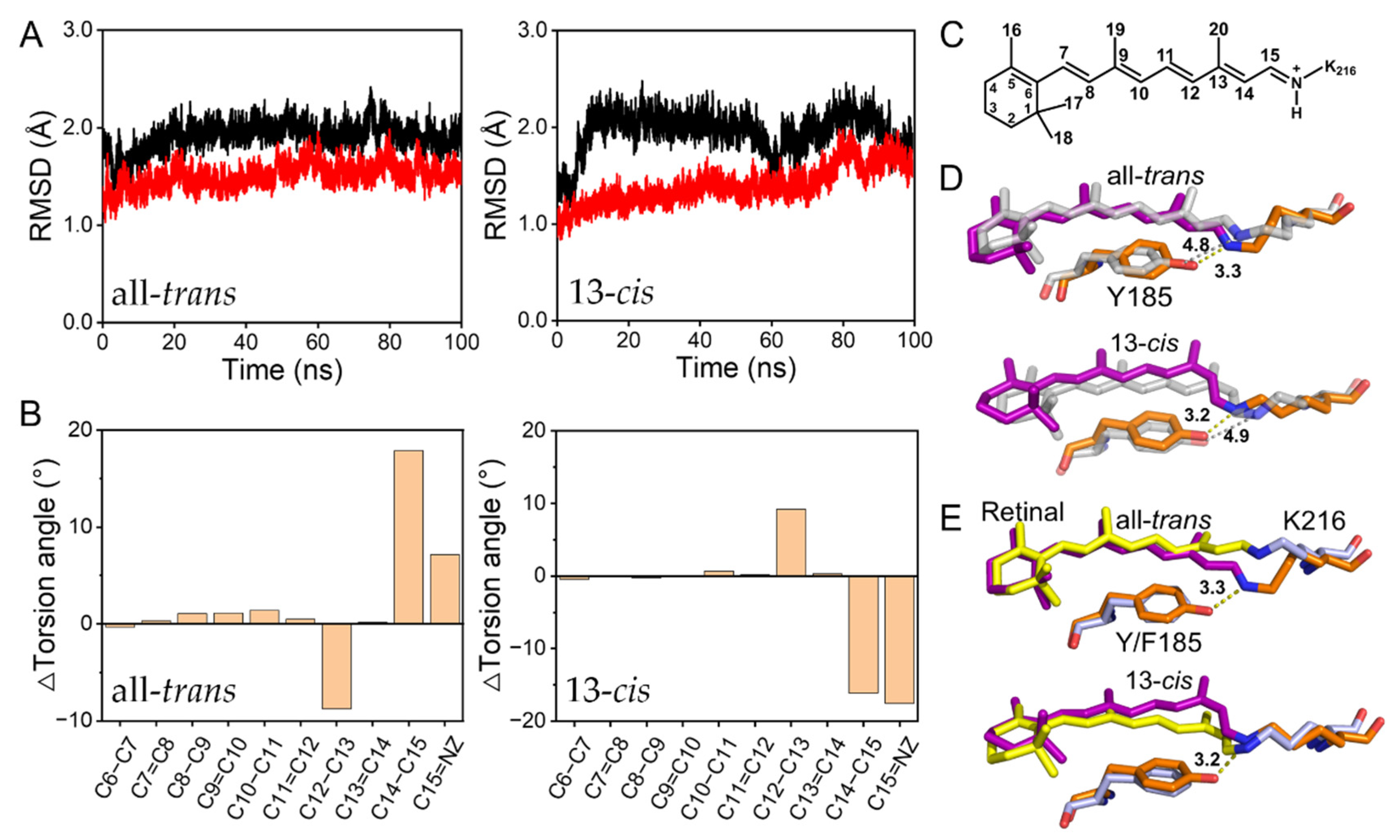
2.1. Coupling of Y185 with the Retinal Chromophore in the Dark-Adapted bR
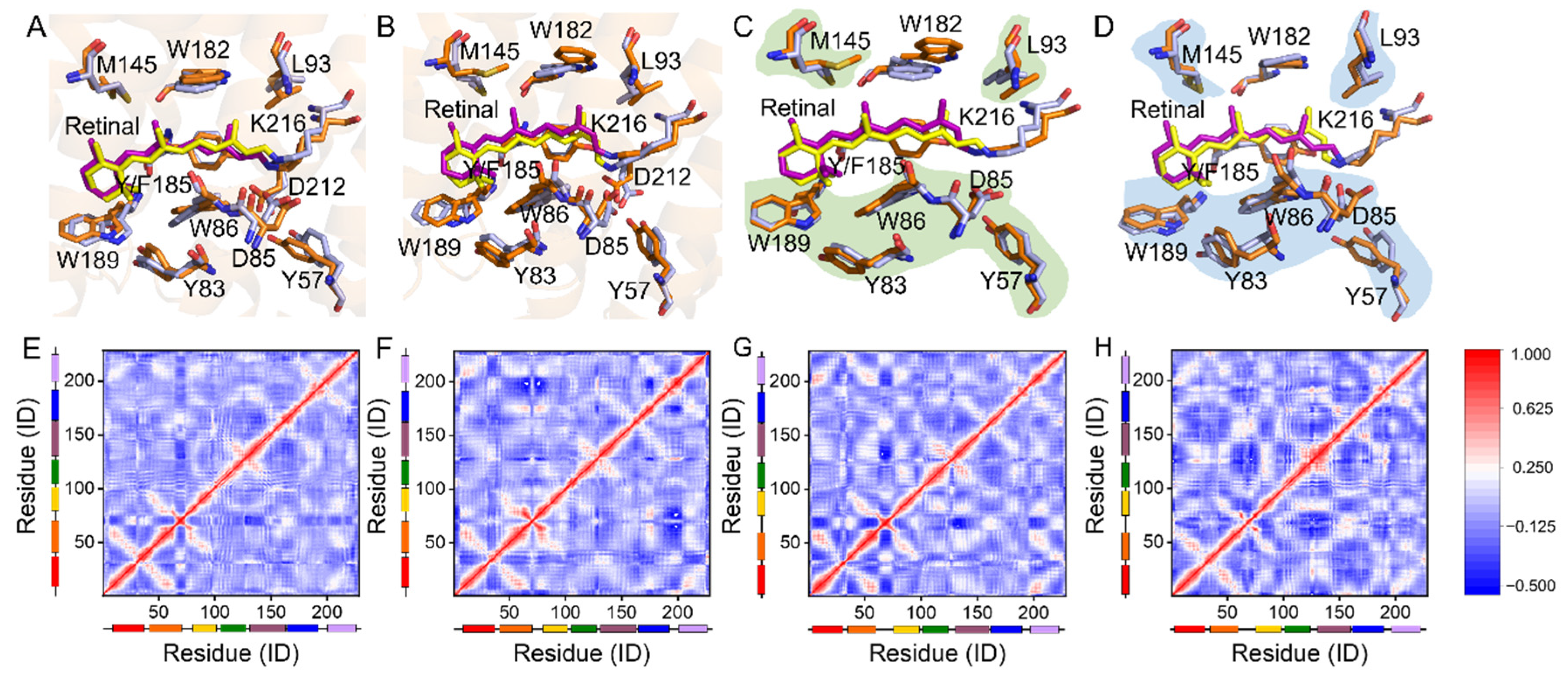
2.2. Regulation of the Extracellular Side Pentagonal H-Bond Network of the Retinal Binding Pocket
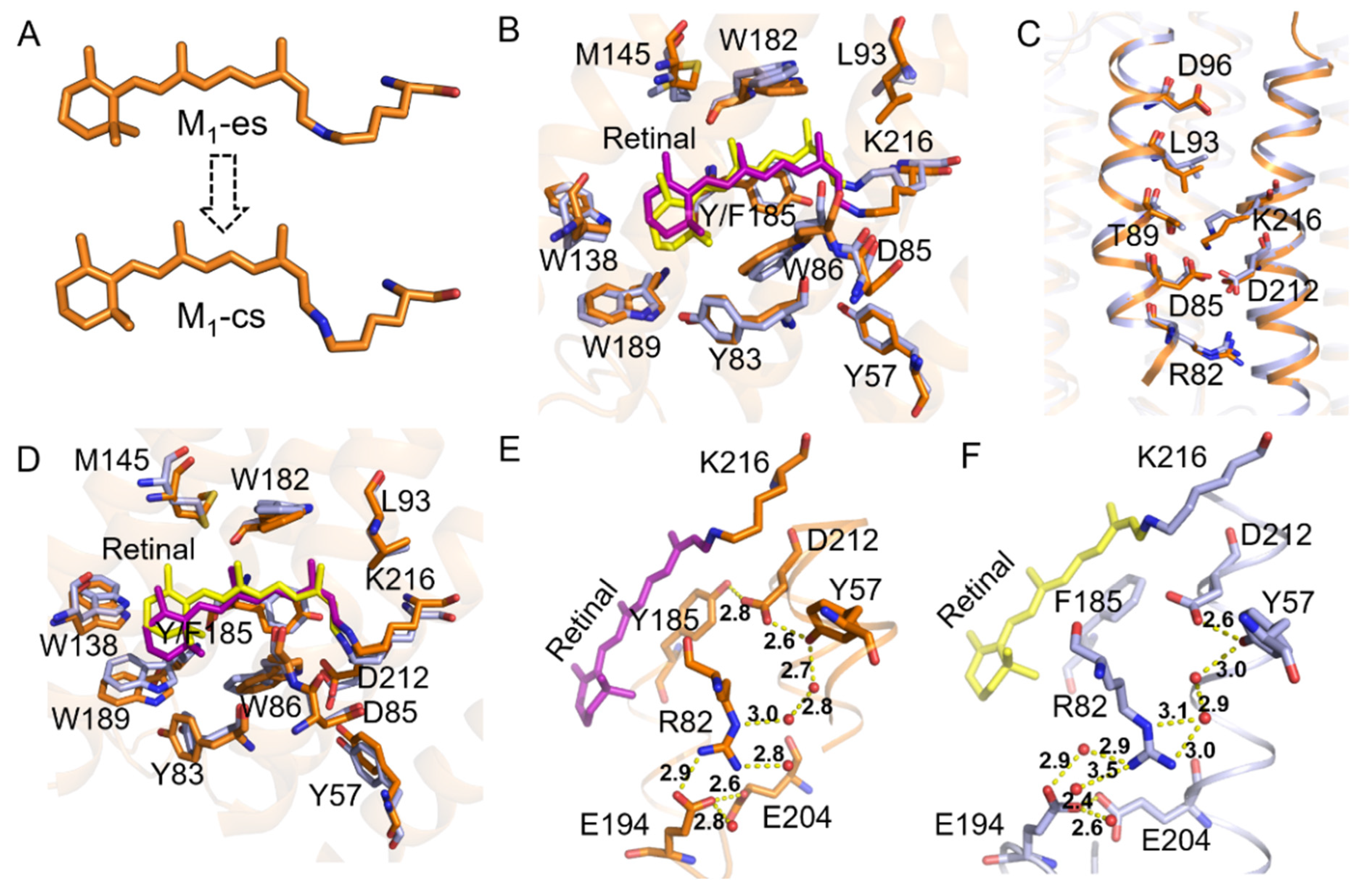
2.3. Coupling of Y185 with the M1 and M2 States
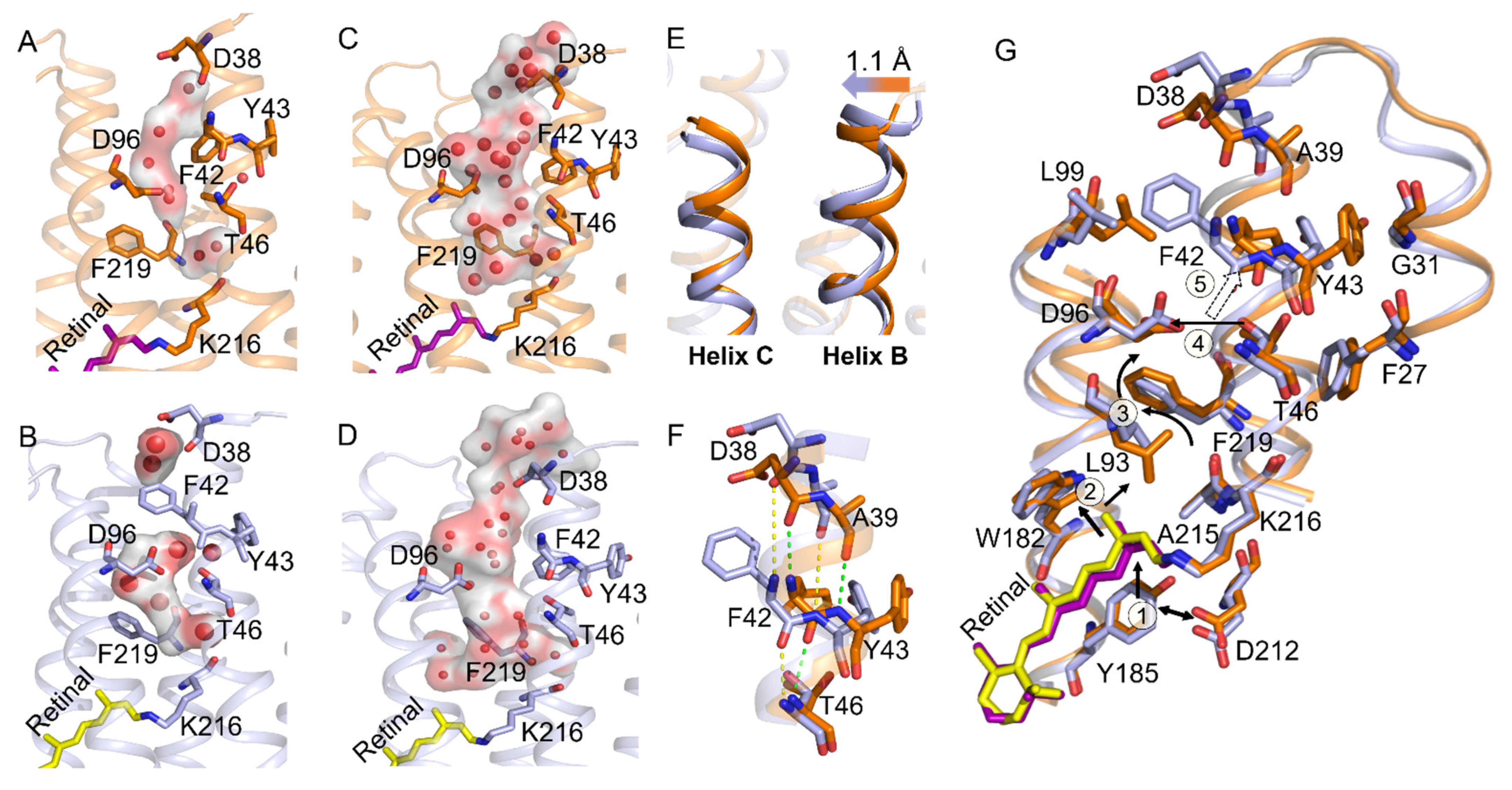
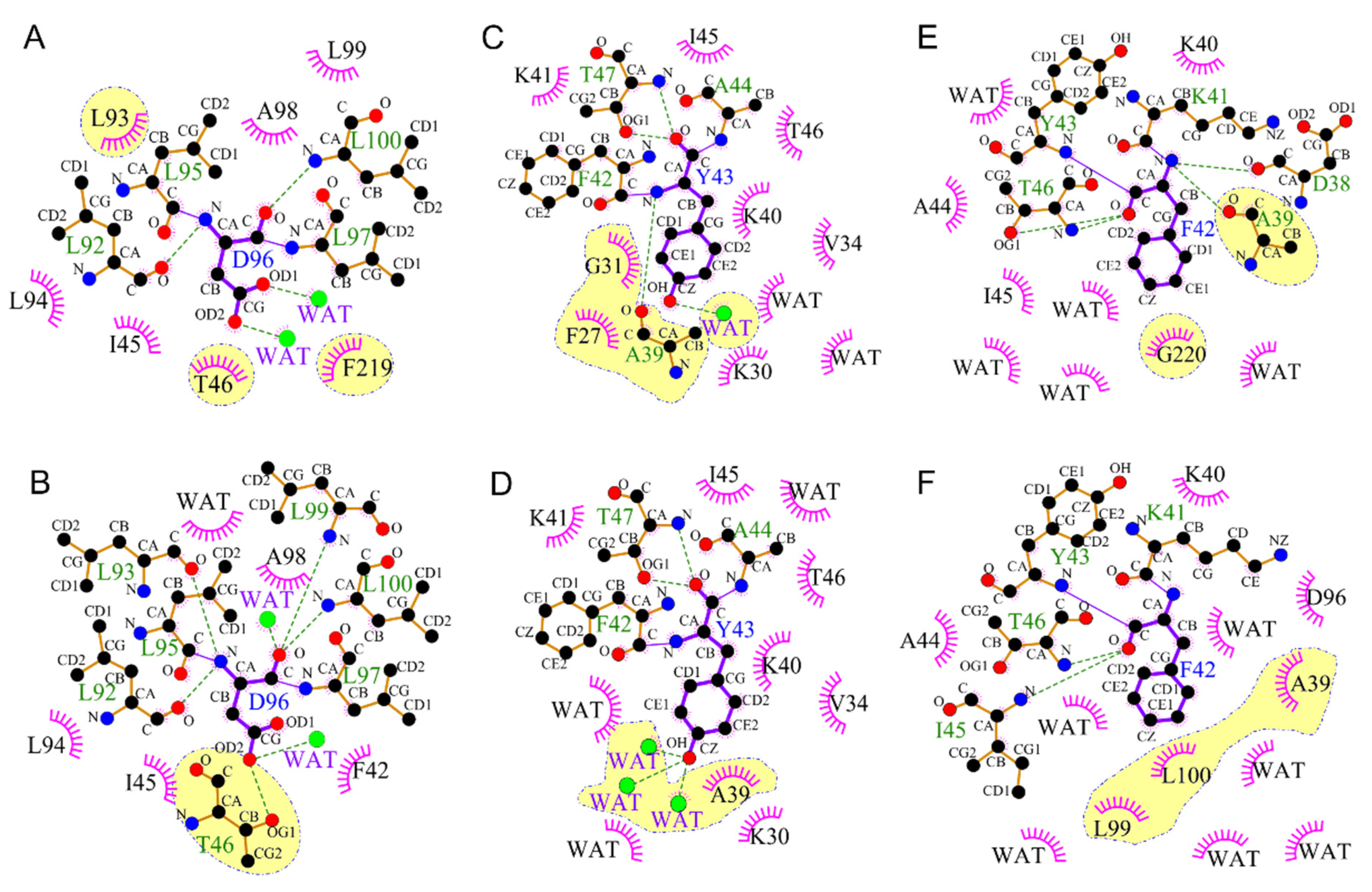
2.4. Participation in Opening the F42 Gate in the M2’ to N States
3. Conclusions
4. Materials and Methods
4.1. Molecular Models and Parameters
4.2. Automatic Fragmentation Quantum Mechanic/Molecular Mechanic Calculations
4.3. Molecular Dynamic Simulation
4.4. Analysis of the Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oesterhelt, D.; Stoeckenius, W. Functions of a New Photoreceptor Membrane. Proc. Natl. Acad. Sci. USA 1973, 70, 2853–2857. [Google Scholar] [CrossRef] [Green Version]
- Sperling, W.; Carl, P.; Rafferty, C.N.; Dencher, N.A. Photochemistry and dark equilibrium of retinal isomers and bacteriorhodopsin isomers. Eur. Biophys. J. 1977, 3, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Stoeckenius, W.; Lozier, R.H.; Bogomolni, R.A. Bacteriorhodopsin and the purple membrane of halobacteria. Biochim. Biophys. Acta (BBA) Rev. Bioenerg. 1979, 505, 215–278. [Google Scholar] [CrossRef]
- Lanyi, J.K. Bacteriorhodopsin. Annu. Rev. Physiol. 2004, 66, 665–688. [Google Scholar] [CrossRef] [PubMed]
- Lanyi, J.K. Mechanism of proton transport from crystallographic structures of the nine states of the bacteriorhodopsin photocycle. Biochim. Biophys. Acta 2004, 1658, 78. [Google Scholar]
- Lanyi, J.K. Proton transfers in the bacteriorhodopsin photocycle. Biochim. Biophys. Acta (BBA) Bioenerg. 2006, 1757, 1012–1018. [Google Scholar] [CrossRef] [Green Version]
- Luecke, H.; Schobert, B.; Richter, H.-T.; Cartailler, J.-P.; Lanyi, J.K. Structural Changes in Bacteriorhodopsin during Ion Transport at 2 Angstrom Resolution. Science 1999, 286, 255–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luecke, H.; Schobert, B.; Cartailler, J.-P.; Richter, H.-T.; Rosengarth, A.; Needleman, R.; Lanyi, J.K. Coupling photoisomerization of retinal to directional transport in bacteriorhodopsin. J. Mol. Biol. 2000, 300, 1237–1255. [Google Scholar] [CrossRef] [Green Version]
- Lanyi, J.K.; Schobert, B. Crystallographic Structure of the Retinal and the Protein after Deprotonation of the Schiff Base: The Switch in the Bacteriorhodopsin Photocycle. J. Mol. Biol. 2002, 321, 727–737. [Google Scholar] [CrossRef]
- Wang, T.; Sessions, A.O.; Lunde, C.S.; Rouhani, S.; Glaeser, R.M.; Duan, Y.; Facciotti, M.T. Deprotonation of D96 in Bacteriorhodopsin Opens the Proton Uptake Pathway. Structure 2013, 21, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Wickstrand, C.; Nogly, P.; Nango, E.; Iwata, S.; Standfuss, J.; Neutze, R. Bacteriorhodopsin: Structural Insights Revealed Using X-Ray Lasers and Synchrotron Radiation. Annu. Rev. Biochem. 2019, 88, 59–83. [Google Scholar] [CrossRef] [Green Version]
- Schobert, B.; Brown, L.; Lanyi, J.K. Crystallographic Structures of the M and N Intermediates of Bacteriorhodopsin: Assembly of a Hydrogen-bonded Chain of Water Molecules Between Asp-96 and the Retinal Schiff Base. J. Mol. Biol. 2003, 330, 553–570. [Google Scholar] [CrossRef]
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.; Tanaka, R.; et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 2016, 354, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Neutze, R.; Pebay-Peyroula, E.; Edman, K.; Royant, A.; Navarro, J.; Landau, E.M. Bacteriorhodopsin: A high-resolution structural view of vectorial proton transport. Biochim. Biophys. Acta (BBA) Biomembr. 2002, 1565, 144–167. [Google Scholar] [CrossRef] [Green Version]
- Wickstrand, C.; Dods, R.; Royant, A.; Neutze, R. Bacteriorhodopsin: Would the real structural intermediates please stand up? Biochim. Biophys. Acta (BBA) Gen. Subj. 2015, 1850, 536–553. [Google Scholar] [CrossRef] [Green Version]
- Lanyi, J.K.; Schobert, B. Mechanism of Proton Transport in Bacteriorhodopsin from Crystallographic Structures of the K, L, M1, M2, and M2′ Intermediates of the Photocycle. J. Mol. Biol. 2003, 328, 439–450. [Google Scholar] [CrossRef]
- Jang, D.J.; El-Sayed, M.A.; Stern, L.J.; Mogi, T.; Khorana, H.G. Effect of genetic modification of tyrosine-185 on the proton pump and the blue-to-purple transition in bacteriorhodopsin. Proc. Natl. Acad. Sci. USA 1990, 87, 4103–4107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothschild, K.; Roepe, P.; Ahl, P.L.; Earnest, T.N.; Bogomolni, R.A.; Das Gupta, S.K.; Mulliken, C.M.; Herzfeld, J. Evidence for a tyrosine protonation change during the primary phototransition of bacteriorhodopsin at low temperature. Proc. Natl. Acad. Sci. USA 1986, 83, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Ferrand, M.; Dianoux, A.J.; Petry, W.; Zaccai, G. Thermal motions and function of bacteriorhodopsin in purple membranes: Effects of temperature and hydration studied by neutron scattering. Proc. Natl. Acad. Sci. USA 1993, 90, 9668–9672. [Google Scholar] [CrossRef] [Green Version]
- Ferrand, M.; Zaccai, G.; Nina, M.; Smith, J.; Etchebest, C.; Roux, B. Structure and dynamics of bacteriorhodopsin. Comparison of simulation and experiment. FEBS Lett. 1993, 327, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Fitter, J.; Lechner, R.E.; Buldt, G.; Dencher, N.A. Internal molecular motions of bacteriorhodopsin: Hydration-induced flexibility studied by quasielastic incoherent neutron scattering using oriented purple membranes. Proc. Natl. Acad. Sci. USA 1996, 93, 7600–7605. [Google Scholar] [CrossRef] [Green Version]
- Fitter, J.; Lechner, R.; Dencher, N. Picosecond molecular motions in bacteriorhodopsin from neutron scattering. Biophys. J. 1997, 73, 2126–2137. [Google Scholar] [CrossRef] [Green Version]
- Reat, V.; Patzelt, H.; Ferrand, M.; Pfister, C.; Oesterhelt, D.; Zaccai, G. Dynamics of different functional parts of bacteriorhodopsin: H-2H labeling and neutron scattering. Proc. Natl. Acad. Sci. USA 1998, 95, 4970–4975. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Wang, H.; Peng, B.; Cui, H.; Gao, Y.; Iuga, D.; Judge, P.J.; Li, G.; Watts, A.; Zhao, X. Mediation mechanism of tyrosine 185 on the retinal isomerization equilibrium and the proton release channel in the seven-transmembrane receptor bacteriorhodopsin. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 1786–1795. [Google Scholar] [CrossRef]
- Ding, X.; Sun, C.; Cui, H.; Chen, S.; Gao, Y.; Yang, Y.; Wang, J.; He, X.; Iuga, D.; Tian, F.; et al. Functional roles of tyrosine 185 during the bacteriorhodopsin photocycle as revealed by in situ spectroscopic studies. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1859, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Wang, W. Molecular Dynamics Simulation of Membrane Proteins. Results Probl. Cell Differ. 2013, 805, 305–329. [Google Scholar] [CrossRef]
- Goossens, K.; De Winter, H. Molecular Dynamics Simulations of Membrane Proteins: An Overview. J. Chem. Inf. Model. 2018, 58, 2193–2202. [Google Scholar] [CrossRef] [PubMed]
- Chavent, M.; Duncan, A.; Sansom, M.S. Molecular dynamics simulations of membrane proteins and their interactions: From nanoscale to mesoscale. Curr. Opin. Struct. Biol. 2016, 40, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Ploeg, P.; Berendsen, H.J.C. Molecular dynamics simulation of a bilayer membrane. J. Chem. Phys. 1982, 76, 3271–3276. [Google Scholar] [CrossRef] [Green Version]
- Woolf, T.B.; Roux, B. Molecular dynamics simulation of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. USA 1994, 91, 11631–11635. [Google Scholar] [CrossRef] [Green Version]
- Woolf, T. Molecular dynamics of individual alpha-helices of bacteriorhodopsin in dimyristol phosphatidylocholine. I. Structure and dynamics. Biophys. J. 1997, 73, 2376–2392. [Google Scholar] [CrossRef] [Green Version]
- Dror, R.O.; Arlow, D.H.; Borhani, D.W.; Jensen, M.Ø.; Piana, S.; Shaw, D.E. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA 2009, 106, 4689–4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.; Valcourt, J.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R.; et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nat. Cell Biol. 2013, 503, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Freier, E.; Potschies, M.; Hofmann, E.; Gerwert, K. Directional Proton Transfer in Membrane Proteins Achieved through Protonated Protein-Bound Water Molecules: A Proton Diode. Angew. Chem. Int. Ed. 2010, 49, 6889–6893. [Google Scholar] [CrossRef] [PubMed]
- Del Val, C.; Bondar, L.; Bondar, A.-N. Coupling between inter-helical hydrogen bonding and water dynamics in a proton transporter. J. Struct. Biol. 2014, 186, 95–111. [Google Scholar] [CrossRef]
- Cui, Q.; Karplus, M. Molecular Properties from Combined QM/MM Methods. 2. Chemical Shifts in Large Molecules. J. Phys. Chem. B 2000, 104, 3721–3743. [Google Scholar] [CrossRef]
- Frank, A.; Möller, H.M.; Exner, T.E. Toward the Quantum Chemical Calculation of NMR Chemical Shifts of Proteins. 2. Level of Theory, Basis Set, and Solvents Model Dependence. J. Chem. Theory Comput. 2012, 8, 1480–1492. [Google Scholar] [CrossRef]
- Gao, Q.; Yokojima, S.; Kohno, T.; Ishida, T.; Fedorov, D.G.; Kitaura, K.; Fujihira, M.; Nakamura, S. Ab initio NMR chemical shift calculations on proteins using fragment molecular orbitals with electrostatic environment. Chem. Phys. Lett. 2007, 445, 331–339. [Google Scholar] [CrossRef]
- Gao, Q.; Yokojima, S.; Fedorov, D.G.; Kitaura, K.; Sakurai, M.; Nakamura, S. Fragment-Molecular-Orbital-Method-Based ab Initio NMR Chemical-Shift Calculations for Large Molecular Systems. J. Chem. Theory Comput. 2010, 6, 1428–1444. [Google Scholar] [CrossRef]
- He, X.; Wang, B.; Merz, K.M. Protein NMR Chemical Shift Calculations Based on the Automated Fragmentation QM/MM Approach. J. Phys. Chem. B 2009, 113, 10380–10388. [Google Scholar] [CrossRef]
- Jin, X.; Zhu, T.; Zhang, J.Z.H.; He, X. Automated Fragmentation QM/MM Calculation of NMR Chemical Shifts for Protein-Ligand Complexes. Front. Chem. 2018, 6, 150. [Google Scholar] [CrossRef]
- Zhu, T.; He, X.; Zhang, J.Z.H. Fragment density functional theory calculation of NMR chemical shifts for proteins with implicit solvation. Phys. Chem. Chem. Phys. 2012, 14, 7837–7845. [Google Scholar] [CrossRef]
- Swails, J.M.; Zhu, T.; He, X.; Case, D.A. AFNMR: Automated fragmentation quantum mechanical calculation of NMR chemical shifts for biomolecules. J. Biomol. NMR 2015, 63, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Luecke, H.; Schobert, B.; Richter, H.-T.; Cartailler, J.-P.; Lanyi, J.K. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 1999, 291, 899–911. [Google Scholar] [CrossRef] [Green Version]
- Essen, L.-O.; Siegert, R.; Lehmann, W.D.; Oesterhelt, D. Lipid patches in membrane protein oligomers: Crystal structure of the bacteriorhodopsin-lipid complex. Proc. Natl. Acad. Sci. USA 1998, 95, 11673–11678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, T.; Murakami, M.; Kouyama, T. Crystal Structure of the 13-cis Isomer of Bacteriorhodopsin in the Dark-adapted State. J. Mol. Biol. 2005, 352, 319–328. [Google Scholar] [CrossRef]
- Bryl, K.; Yoshihara, K. Two processes lead to a stable all- trans and 13- cis isomer equilibrium in dark-adapted bacteriorhodopsin; effect of high pressure on bacteriorhodopsin, bacteriorhodopsin mutant D96N and fluoro-bacteriorhodopsin analogues. Eur. Biophys. J. 2002, 31, 539–548. [Google Scholar] [CrossRef]
- Tsuda, M.; Ebrey, T. Effect of high pressure on the absorption spectrum and isomeric composition of bacteriorhodopsin. Biophys. J. 1980, 30, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Schulte, A.; Bradley, L., II. High-pressure near-infrared Raman spectroscopy of bacteriorhodopsin light to dark adaptation. Biophys. J. 1995, 69, 1554–1562. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Edelsbrunner, H.; Koehl, P. The weighted-volume derivative of a space-filling diagram. Proc. Natl. Acad. Sci. USA 2003, 100, 2203–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandori, H. Hydration switch model for the proton transfer in the Schiff base region of bacteriorhodopsin. Biochim. Biophys. Acta (BBA) Bioenerg. 2004, 1658, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Pebay-Peyroula, E.; Rummel, G.; Rosenbusch, J.P.; Landau, E.M. X-ray Structure of Bacteriorhodopsin at 2.5 Angstroms from Microcrystals Grown in Lipidic Cubic Phases. Science 1997, 277, 1676–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royant, A.; Edman, K.; Ursby, T.; Pebay-Peyroula, E.; Landau, E.M.; Neutze, R. Helix deformation is coupled to vectorial proton transport in the photocycle of bacteriorhodopsin. Nat. Cell Biol. 2000, 406, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Tanimoto, T.; Kandori, H. Water Molecules in the Schiff Base Region of Bacteriorhodopsin. J. Am. Chem. Soc. 2003, 125, 13312–13313. [Google Scholar] [CrossRef]
- Subramaniam, S.; Lindahl, M.; Bullough, P.; Faruqi, A.; Tittor, J.; Oesterhelt, D.; Brown, L.; Lanyi, J.; Henderson, R. Protein conformational changes in the bacteriorhodopsin photocycle. J. Mol. Biol. 1999, 287, 145–161. [Google Scholar] [CrossRef] [Green Version]
- Lanyi, J.K. Understanding Structure and Function in the Light-Driven Proton Pump Bacteriorhodopsin. J. Struct. Biol. 1998, 124, 164–178. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Bacteriorhodopsin—The movie. Nature 2000, 406, 569–570. [Google Scholar] [CrossRef]
- Bondar, A.-N.; del Val, C.; Freites, J.A.; Tobias, D.J.; White, S.H. Dynamics of SecY Translocons with Translocation-Defective Mutations. Structure 2010, 18, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Tanio, M.; Inoue, S.; Yokota, K.; Seki, T.; Tuzi, S.; Needleman, R.; Lanyi, J.K.; Naito, A.; Saitô, H. Long-Distance Effects of Site-Directed Mutations on Backbone Conformation in Bacteriorhodopsin from Solid State NMR of [1-13C]Val-Labeled Proteins. Biophys. J. 1999, 77, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Schätzler, B.; Dencher, N.A.; Tittor, J.; Oesterhelt, D.; Yaniv-Checover, S.; Nachliel, E.; Gutman, M. Subsecond Proton-Hole Propagation in Bacteriorhodopsin. Biophys. J. 2003, 84, 671–686. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.; Nachliel, E.; Gutman, M. The Role of Small Intraprotein Cavities in the Catalytic Cycle of Bacteriorhodopsin. Biophys. J. 2003, 85, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.; Henderson, R. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nat. Cell Biol. 2000, 406, 653–657. [Google Scholar] [CrossRef]
- Freier, E.; Wolf, S.; Gerwert, K. Proton transfer via a transient linear water-molecule chain in a membrane protein. Proc. Natl. Acad. Sci. USA 2011, 108, 11435–11439. [Google Scholar] [CrossRef] [Green Version]
- Maestro, Version 11.5; Schrödinger, LLC.: New York, NY, USA, 2018.
- DeLano, W.L. The PyMOL Molecular Graphics System; Delano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Feller, S.E.; MacKerell, A.D. An Improved Empirical Potential Energy Function for Molecular Simulations of Phospholipids. J. Phys. Chem. B 2000, 104, 7510–7515. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Im, W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.; Cerutti, D.; Cheatham, T.E.; Darden, T.; Duke, R.; Giese, T.J.; Gohlke, H.; Götz, A.; Homeyer, N. Amber 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, D.S.; Duke, R.; Freddolino, P.L.; Fan, H.; Lybrand, T.P. A Vulnerability in Popular Molecular Dynamics Packages Concerning Langevin and Andersen Dynamics. J. Chem. Theory Comput. 2008, 4, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Tang, S.; Case, D.A. Calculation of chemical shift anisotropy in proteins. J. Biomol. NMR 2011, 51, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- McDonald, I.K.; Thornton, J. Satisfying Hydrogen Bonding Potential in Proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ssNMR [25] | AF-QM/MM | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C10 | C11 | C14 | C15 | C10 | C11 | C14 | C15 | RMSE | |
| WT-bR | |||||||||
| bRcis | 130.0 | 139.0 | 110.0 | 163.2 | 129.5 | 138.6 | 109.5 | 162.7 | 0.522 |
| bRtrans | 133.2 | 135.4 | 122.7 | 160.0 | 132.8 | 135.9 | 123.2 | 160.5 | 0.489 |
| M1 | 129.4 | 129.4 | 125.7 | 164.0 | 128.9 | 128.9 | 125.3 | 163.5 | 0.491 |
| M2 | 129.4 | 129.4 | 124.0 | 164.8 | 128.9 | 128.9 | 123.5 | 164.3 | 0.504 |
| Y185F-bR | |||||||||
| bRcis | 130.0 | 138.0 | 111.8 | 165.4 | 129.5 | 137.6 | 111.3 | 165.0 | 0.428 |
| bRtrans | 132.6 | 135.1 | 123.0 | 160.2 | 132.1 | 134.7 | 122.6 | 159.8 | 0.438 |
| M1 | 129.5 | 129.5 | 125.7 | 164.0 | 129.0 | 129.0 | 125.2 | 163.5 | 0.443 |
| M2 | 129.5 | 129.5 | 124.7 | 165.0 | 129.0 | 129.0 | 124.2 | 164.5 | 0.505 |
| Pocket Volume/Å3 | Wild-Type | Y185F |
|---|---|---|
| bRtrans | 947.67 | 883.40 |
| bRcis | 968.09 | 989.80 |
| M1-es | 944.61 | 985.84 |
| M1-cs | 977.91 | 985.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Ding, X.; Sun, C.; Watts, A.; He, X.; Zhao, X. Dynamic Coupling of Tyrosine 185 with the Bacteriorhodopsin Photocycle, as Revealed by Chemical Shifts, Assisted AF-QM/MM Calculations and Molecular Dynamic Simulations. Int. J. Mol. Sci. 2021, 22, 13587. https://doi.org/10.3390/ijms222413587
Chen S, Ding X, Sun C, Watts A, He X, Zhao X. Dynamic Coupling of Tyrosine 185 with the Bacteriorhodopsin Photocycle, as Revealed by Chemical Shifts, Assisted AF-QM/MM Calculations and Molecular Dynamic Simulations. International Journal of Molecular Sciences. 2021; 22(24):13587. https://doi.org/10.3390/ijms222413587
Chicago/Turabian StyleChen, Sijin, Xiaoyan Ding, Chao Sun, Anthony Watts, Xiao He, and Xin Zhao. 2021. "Dynamic Coupling of Tyrosine 185 with the Bacteriorhodopsin Photocycle, as Revealed by Chemical Shifts, Assisted AF-QM/MM Calculations and Molecular Dynamic Simulations" International Journal of Molecular Sciences 22, no. 24: 13587. https://doi.org/10.3390/ijms222413587