
KRAS Affects Adipogenic Differentiation by Regulating Autophagy and MAPK Activation in 3T3-L1 and C2C12 Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
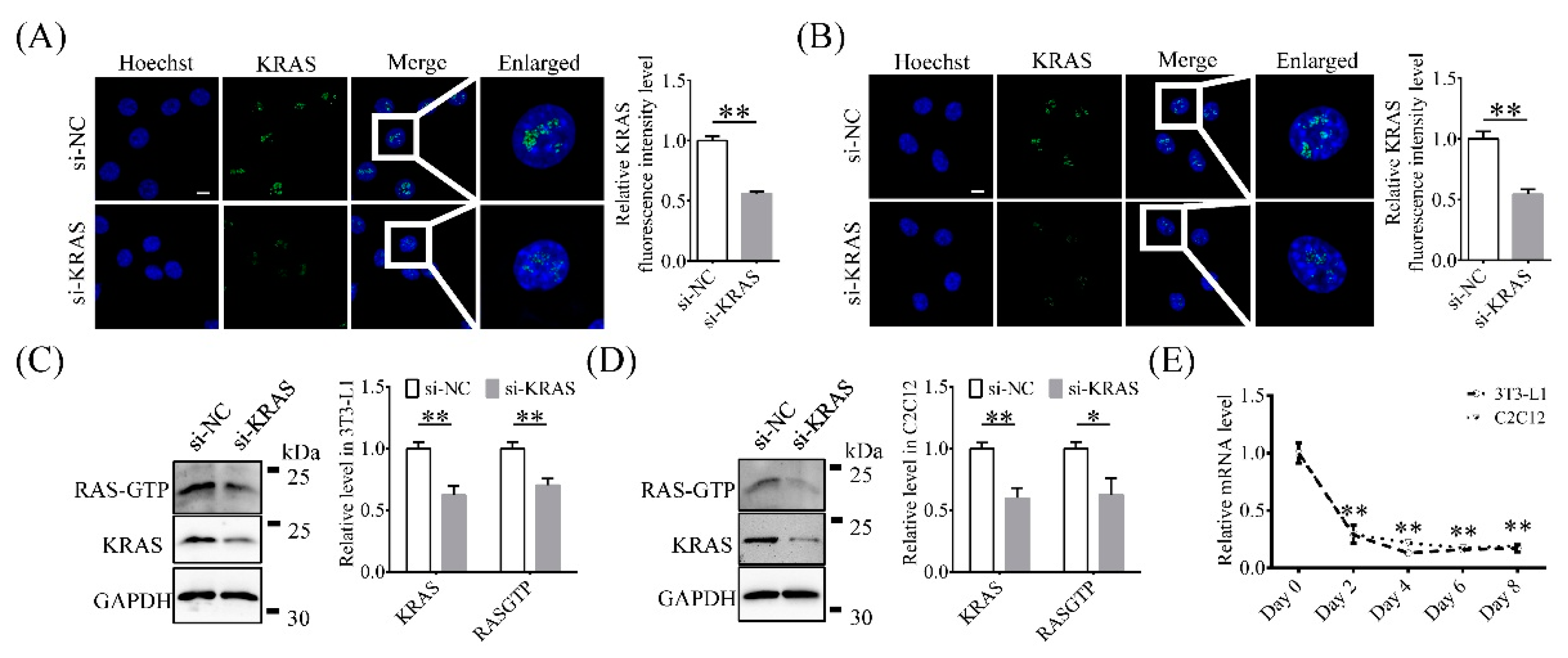
2.1. KRAS Is Expressed Mainly in 3T3-L1 and C2C12 Cell Nuclei
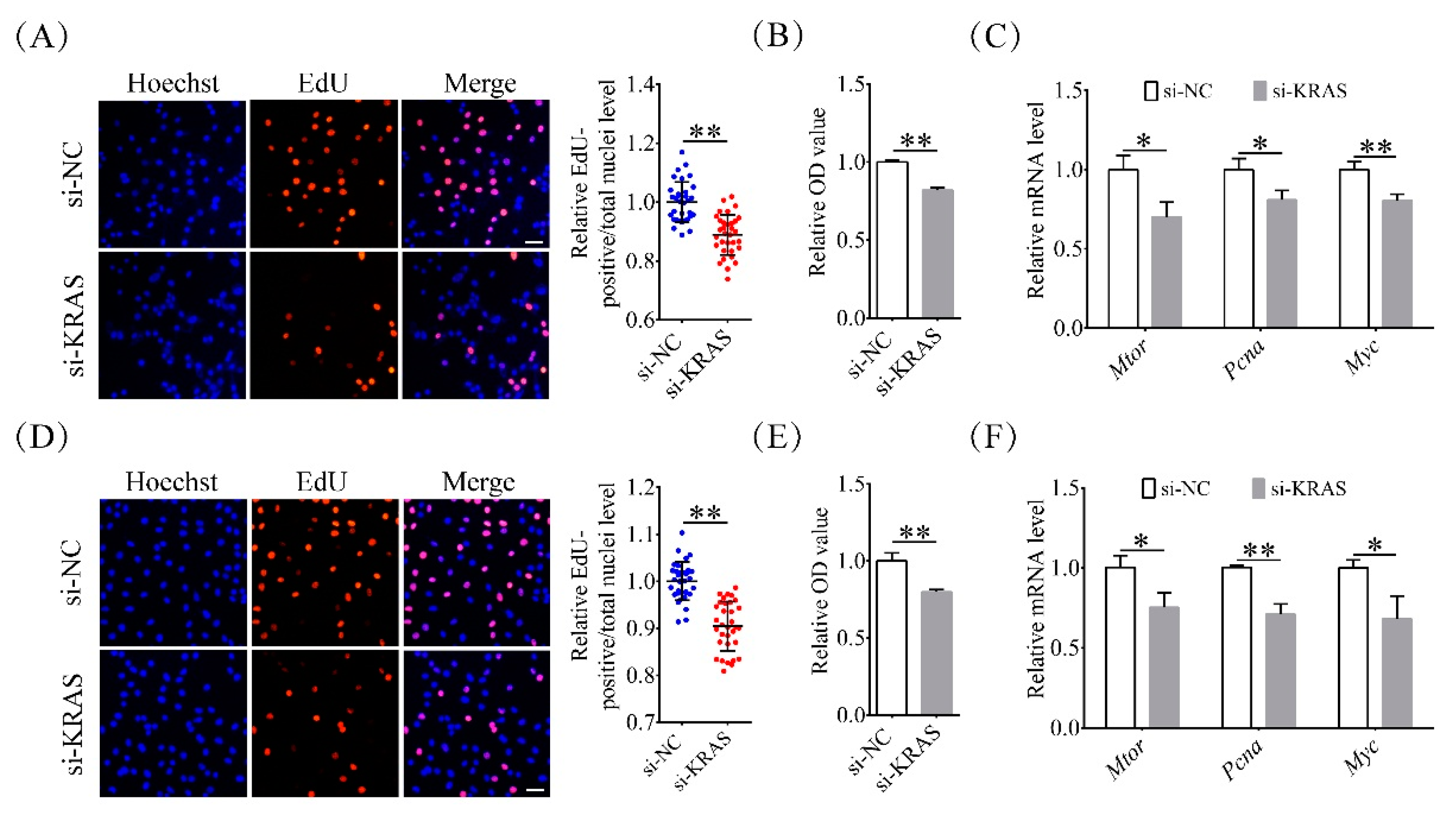
2.2. Inhibition of KRAS Enhanced Adipogenic Differentiation and Adipogenesis
2.3. Inhibition of KRAS Reduced Preadipocyte Proliferation
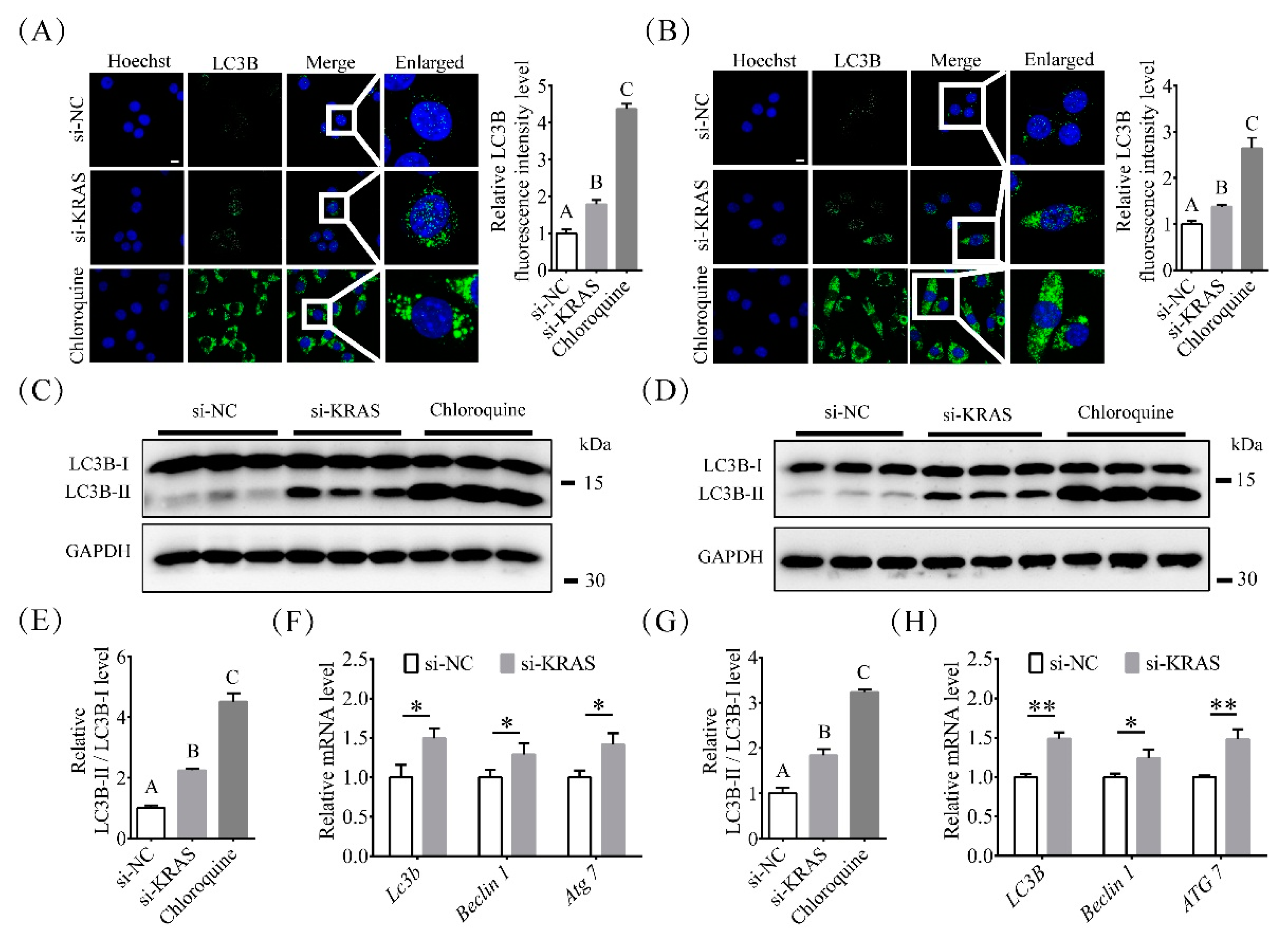
2.4. Inhibition of KRAS Enhanced Preadipocyte Autophagy
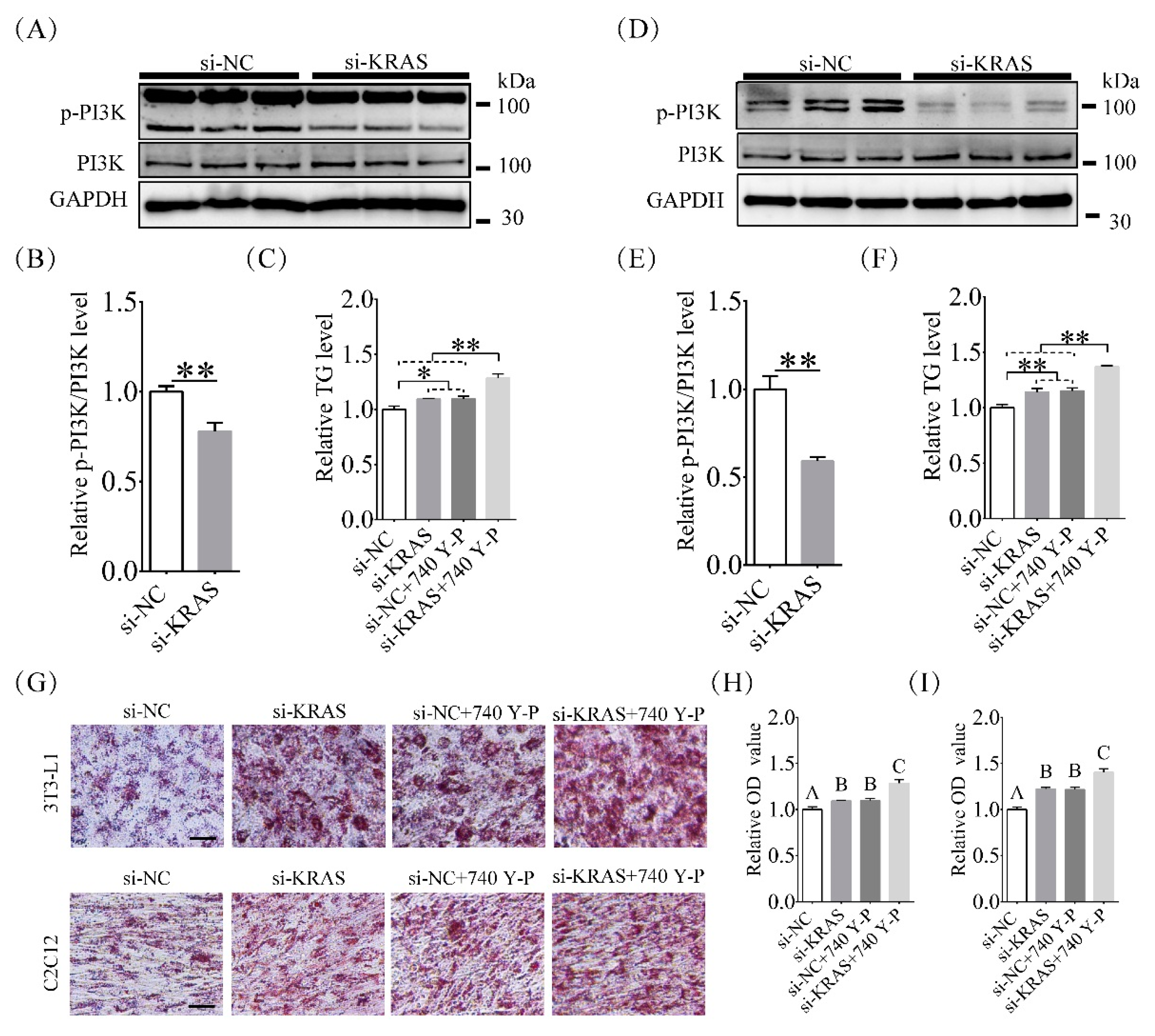
2.5. Inhibition of KRAS Decreased PI3K Phosphorylation Levels
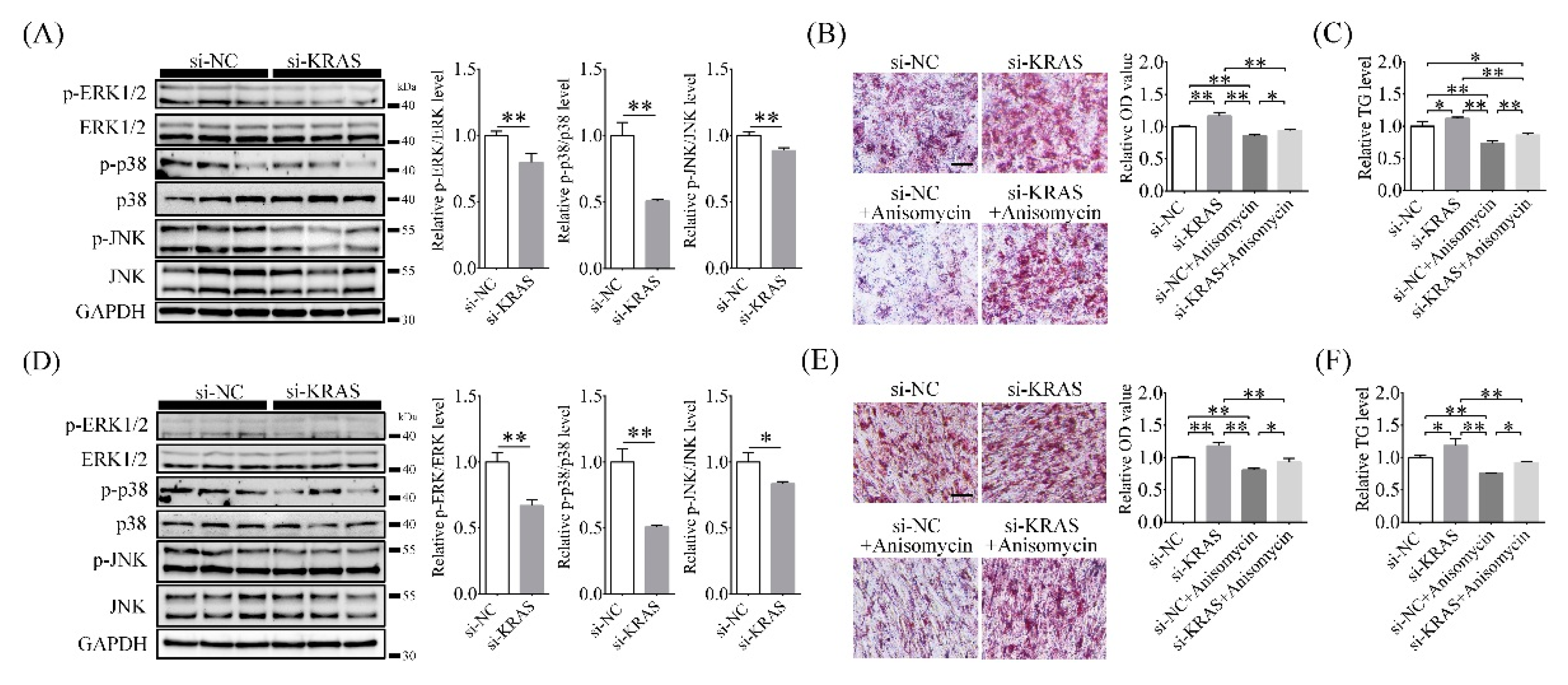
2.6. Inhibition of KRAS Decreased MAPK Phosphorylation Levels
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Adipogenic Differentiation Induction, and Chemical Treatments
4.2. Cell Transfection
4.3. RAS-GTP Level Assay
4.4. Autophagy Inhibition by Chloroquine
4.5. RNA Extraction and Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.6. Protein Separation and Western Blot Analysis
4.7. CCK-8 Assay
4.8. 5-Ethynyl-2′-Deoxyuridine (EdU) Assay
4.9. Triglyceride (TG) Assay
4.10. (Oil red O) ORO Staining and Quantification
4.11. Immunofluorescence
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foreyt, J.P.; Goodrick, G.K.; Gotto, A.M. Limitations of behavioral treatment of obesity: Review and analysis. J. Behav. Med. 1981, 4, 159–174. [Google Scholar] [CrossRef]
- Withrow, D.; Alter, D.A. The economic burden of obesity worldwide: A systematic review of the direct costs of obesity. Obes. Rev. 2011, 12, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Jaacks, L.M.; Vandevijvere, S.; Pan, A.; McGowan, C.J.; Wallace, C.; Imamura, F.; Mozaffarian, D.; Swinburn, B.; Ezzati, M. The obesity transition: Stages of the global epidemic. Lancet Diabetes Endocrinol. 2019, 7, 231–240. [Google Scholar] [CrossRef]
- Berghöfer, A.; Pischon, T.; Reinhold, T.; Apovian, C.M.; Sharma, A.M.; Willich, S.N. Obesity prevalence from a European perspective: A systematic review. BMC Public Health 2008, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llewellyn, A.; Simmonds, M.; Owen, C.G.; Woolacott, N. Childhood obesity as a predictor of morbidity in adulthood: A systematic review and meta-analysis. Obes. Rev. 2016, 17, 56–67. [Google Scholar] [CrossRef]
- de Ferranti, S.; Mozaffarian, D. The perfect storm: Obesity, adipocyte dysfunction, and metabolic consequences. Clin. Chem. 2008, 54, 945–955. [Google Scholar] [CrossRef] [Green Version]
- Sarantopoulos, C.N.; Banyard, D.A.; Ziegler, M.E.; Sun, B.; Shaterian, A.; Widgerow, A.D. Elucidating the Preadipocyte and Its Role in Adipocyte Formation: A Comprehensive Review. Stem Cell Rev. Rep. 2018, 14, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Valli, V.; Heilmann, K.; Danesi, F.; Bordoni, A.; Gerhäuser, C. Modulation of Adipocyte Differentiation and Proadipogenic Gene Expression by Sulforaphane, Genistein, and Docosahexaenoic Acid as a First Step to Counteract Obesity. Oxid. Med. Cell. Longev. 2018, 2018, 1617202. [Google Scholar] [CrossRef]
- Tchoukalova, Y.D.; Koutsari, C.; Karpyak, M.V.; Votruba, S.B.; Wendland, E.; Jensen, M.D. Subcutaneous adipocyte size and body fat distribution. Am. J. Clin. Nutr. 2008, 87, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Ren, X.; Han, P.; Hu, S.; Wang, J.; Yin, J. SiRNA against Fabp5 induces 3T3-L1 cells apoptosis during adipocytic induction. Mol. Biol. Rep. 2010, 37, 4003–4011. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, W.Q.; Guo, Y.Y.; Liu, Y.; Tang, Q.Q.; Guo, L. Krüppel-like factor 10 (KLF10) is transactivated by the transcription factor C/EBPβ and involved in early 3T3-L1 preadipocyte differentiation. J. Biol. Chem. 2018, 293, 14012–14021. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shi, X.E.; Song, C.; Sun, S.; Yang, G.; Li, X. BAMBI Promotes C2C12 Myogenic Differentiation by Enhancing Wnt/β-Catenin Signaling. Int. J. Mol. Sci. 2015, 16, 17734–17745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Park, Y.; Yoon, K.S.; Clark, J.M.; Park, Y. Permethrin alters adipogenesis in 3T3-L1 adipocytes and causes insulin resistance in C2C12 myotubes. J. Biochem. Mol. Toxicol. 2014, 28, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rong, Y.; Bao, L.; Nie, B.; Ren, G.; Zheng, C.; Amin, R.; Arnold, R.D.; Jeganathan, R.B.; Huggins, K.W. Suppression of adipocyte differentiation and lipid accumulation by stearidonic acid (SDA) in 3T3-L1 cells. Lipids Health Dis. 2017, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Song, Z.; Shao, W.; Du, W.W.; Zhao, L.R.; Zeng, K.; Yang, B.B.; Jin, T. Curcumin represses mouse 3T3-L1 cell adipogenic differentiation via inhibiting miR-17-5p and stimulating the Wnt signalling pathway effector Tcf7l2. Cell Death Dis. 2017, 8, e2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ojeda, F.J.; Rupérez, A.I.; Gomez-Llorente, C.; Gil, A.; Aguilera, C.M. Cell Models and Their Application for Studying Adipogenic Differentiation in Relation to Obesity: A Review. Int. J. Mol. Sci. 2016, 17, 1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhai, Y.; Wang, J. The Role of PPAR and Its Cross-Talk with CAR and LXR in Obesity and Atherosclerosis. Int. J. Mol. Sci. 2018, 19, 1260. [Google Scholar] [CrossRef] [Green Version]
- Harmon, A.W.; Patel, Y.M.; Harp, J.B. Genistein inhibits CCAAT/enhancer-binding protein beta (C/EBPbeta) activity and 3T3-L1 adipogenesis by increasing C/EBP homologous protein expression. Biochem. J. 2002, 367, 203–208. [Google Scholar] [CrossRef]
- Guo, L.; Li, X.; Tang, Q.Q. Transcriptional regulation of adipocyte differentiation: A central role for CCAAT/enhancer-binding protein (C/EBP) β. J. Biol. Chem. 2015, 290, 755–761. [Google Scholar] [CrossRef] [Green Version]
- Krens, S.F.; Corredor-Adámez, M.; He, S.; Snaar-Jagalska, B.E.; Spaink, H.P. ERK1 and ERK2 MAPK are key regulators of distinct gene sets in zebrafish embryogenesis. BMC Genom. 2008, 9, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal. Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Bost, F.; Aouadi, M.; Caron, L.; Binétruy, B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie 2005, 87, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189–198. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, J.; Wang, J. KRAS gene silencing inhibits the activation of PI3K-Akt-mTOR signaling pathway to regulate breast cancer cell epithelial-mesenchymal transition, proliferation and apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3085–3096. [Google Scholar]
- Al-Ghamdi, S.; Albasri, A.; Cachat, J.; Ibrahem, S.; Muhammad, B.A.; Jackson, D.; Nateri, A.S.; Kindle, K.B.; Ilyas, M. Cten is targeted by Kras signalling to regulate cell motility in the colon and pancreas. PLoS ONE 2011, 6, e20919. [Google Scholar] [CrossRef]
- Ferino, A.; Rapozzi, V.; Xodo, L.E. The ROS-KRAS-Nrf2 axis in the control of the redox homeostasis and the intersection with survival-apoptosis pathways: Implications for photodynamic therapy. J. Photochem. Photobiol. B Biol. 2020, 202, 111672. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef]
- Wu, H.Z.; Xiao, J.Q.; Xiao, S.S.; Cheng, Y. KRAS: A Promising Therapeutic Target for Cancer Treatment. Curr. Top. Med. Chem. 2019, 19, 2081–2097. [Google Scholar] [CrossRef]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.H.; Chen, C.Z.; Wang, Y.; Peng, Y.X.; Wang, W.H.; Yuan, B.; Gao, Y.; Jiang, H.; Zhang, J.B. COL1A1 affects apoptosis by regulating oxidative stress and autophagy in bovine cumulus cells. Theriogenology 2019, 139, 81–89. [Google Scholar] [CrossRef]
- Choe, W.K.; Kang, B.T.; Kim, S.O. Water-extracted plum (Prunus salicina L. cv. Soldam) attenuates adipogenesis in murine 3T3-L1 adipocyte cells through the PI3K/Akt signaling pathway. Exp. Ther. Med. 2018, 15, 1608–1615. [Google Scholar] [CrossRef] [Green Version]
- Mi, L.; Chen, Y.; Zheng, X.; Li, Y.; Zhang, Q.; Mo, D.; Yang, G. MicroRNA-139-5p Suppresses 3T3-L1 Preadipocyte Differentiation Through Notch and IRS1/PI3K/Akt Insulin Signaling Pathways. J. Cell. Biochem. 2015, 116, 1195–1204. [Google Scholar] [CrossRef]
- Wu, M.; Liu, D.; Zeng, R.; Xian, T.; Lu, Y.; Zeng, G.; Sun, Z.; Huang, B.; Huang, Q. Epigallocatechin-3-gallate inhibits adipogenesis through down-regulation of PPARγ and FAS expression mediated by PI3K-AKT signaling in 3T3-L1 cells. Eur. J. Pharmacol. 2017, 795, 134–142. [Google Scholar] [CrossRef]
- Kim, S.W.; Muise, A.M.; Lyons, P.J.; Ro, H.S. Regulation of adipogenesis by a transcriptional repressor that modulates MAPK activation. J. Biol. Chem. 2001, 276, 10199–10206. [Google Scholar] [CrossRef] [Green Version]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J., Jr.; Liu, X.S.; et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [Green Version]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Charó, N.L.; Rodríguez Ceschan, M.I.; Galigniana, N.M.; Toneatto, J.; Piwien-Pilipuk, G. Organization of nuclear architecture during adipocyte differentiation. Nucleus 2016, 7, 249–269. [Google Scholar] [CrossRef] [Green Version]
- Marion-Letellier, R.; Savoye, G.; Ghosh, S. Fatty acids, eicosanoids and PPAR gamma. Eur. J. Pharmacol. 2016, 785, 44–49. [Google Scholar] [CrossRef]
- Bouaboula, M.; Hilairet, S.; Marchand, J.; Fajas, L.; Le Fur, G.; Casellas, P. Anandamide induced PPARgamma transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur. J. Pharmacol. 2005, 517, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.M.; He, A.; Tan, M.; Wang, J.; Lu, D.; Razani, B.; Lodhi, I.J. MED19 Regulates Adipogenesis and Maintenance of White Adipose Tissue Mass by Mediating PPARγ-Dependent Gene Expression. Cell Rep. 2020, 33, 108228. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding adipocyte differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar] [CrossRef] [Green Version]
- Pairault, J.; Green, H. A study of the adipose conversion of suspended 3T3 cells by using glycerophosphate dehydrogenase as differentiation marker. Proc. Natl. Acad. Sci. USA 1979, 76, 5138–5142. [Google Scholar] [CrossRef] [Green Version]
- Altiok, S.; Xu, M.; Spiegelman, B.M. PPARgamma induces cell cycle withdrawal: Inhibition of E2F/DP DNA-binding activity via down-regulation of PP2A. Genes Dev. 1997, 11, 1987–1998. [Google Scholar] [CrossRef] [Green Version]
- Freytag, S.O.; Dang, C.V.; Lee, W.M. Definition of the activities and properties of c-myc required to inhibit cell differentiation. Cell Growth Differ. 1990, 1, 339–343. [Google Scholar]
- Freytag, S.O.; Geddes, T.J. Reciprocal regulation of adipogenesis by Myc and C/EBP alpha. Science 1992, 256, 379–382. [Google Scholar] [CrossRef]
- Jasaszwili, M.; Wojciechowicz, T.; Billert, M.; Strowski, M.Z.; Nowak, K.W.; Skrzypski, M. Effects of adropin on proliferation and differentiation of 3T3-L1 cells and rat primary preadipocytes. Mol. Cell. Endocrinol. 2019, 496, 110532. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.J.; Pan, H.; Zhang, X.Z.; Li, N.S.; Wang, L.J.; Yang, H.B.; Gong, F.Y. The effect of myostatin on proliferation and lipid accumulation in 3T3-L1 preadipocytes. J. Mol. Endocrinol. 2015, 54, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Kang, S.E.; Ahn, K.S.; Um, J.Y.; Yang, W.M.; Yun, M.; Lee, S.G. Inhibition of the PI3K-AKT-mTOR pathway suppresses the adipocyte-mediated proliferation and migration of breast cancer cells. J. Cancer 2020, 11, 2552–2559. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Tang, G.; Zhang, Q.; Yong, W.; Zhang, W.; Xiao, J.; Wei, C.; He, C.; Yang, G.; Pang, W. A Novel lnc-RNA, Named lnc-ORA, Is Identified by RNA-Seq Analysis, and Its Knockdown Inhibits Adipogenesis by Regulating the PI3K/AKT/mTOR Signaling Pathway. Cells 2019, 8, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janani, C.; Ranjitha Kumari, B.D. PPAR gamma gene—A review. Diabetes Metab. Syndr. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Alonso-Vale, M.I.; Peres, S.B.; Vernochet, C.; Farmer, S.R.; Lima, F.B. Adipocyte differentiation is inhibited by melatonin through the regulation of C/EBPbeta transcriptional activity. J. Pineal Res. 2009, 47, 221–227. [Google Scholar] [CrossRef]
- Bezy, O.; Vernochet, C.; Gesta, S.; Farmer, S.R.; Kahn, C.R. TRB3 blocks adipocyte differentiation through the inhibition of C/EBPbeta transcriptional activity. Mol. Cell. Biol. 2007, 27, 6818–6831. [Google Scholar] [CrossRef] [Green Version]
- Deng, B.; Wen, J.; Ding, Y.; Gao, Q.; Huang, H.; Ran, Z.; Qian, Y.; Peng, J.; Jiang, S. Functional analysis of pig myostatin gene promoter with some adipogenesis- and myogenesis-related factors. Mol. Cell. Biochem. 2012, 363, 291–299. [Google Scholar] [CrossRef]
- Chen, H.C.; Farese, R.V., Jr. Inhibition of triglyceride synthesis as a treatment strategy for obesity: Lessons from DGAT1-deficient mice. Arter. Thromb. Vasc. Biol. 2005, 25, 482–486. [Google Scholar] [CrossRef] [Green Version]
- Flowers, M.T.; Ade, L.; Strable, M.S.; Ntambi, J.M. Combined deletion of SCD1 from adipose tissue and liver does not protect mice from obesity. J. Lipid Res. 2012, 53, 1646–1653. [Google Scholar] [CrossRef] [Green Version]
- Ralston, J.C.; Badoud, F.; Cattrysse, B.; McNicholas, P.D.; Mutch, D.M. Inhibition of stearoyl-CoA desaturase-1 in differentiating 3T3-L1 preadipocytes upregulates elongase 6 and downregulates genes affecting triacylglycerol synthesis. Int. J. Obes. 2014, 38, 1449–1456. [Google Scholar] [CrossRef]
- Pang, J.; Choi, Y.; Park, T. Ilex paraguariensis extract ameliorates obesity induced by high-fat diet: Potential role of AMPK in the visceral adipose tissue. Arch. Biochem. Biophys. 2008, 476, 178–185. [Google Scholar] [CrossRef]
- Baerga, R.; Zhang, Y.; Chen, P.H.; Goldman, S.; Jin, S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 2009, 5, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Česen, M.H.; Pegan, K.; Spes, A.; Turk, B. Lysosomal pathways to cell death and their therapeutic applications. Exp. Cell Res. 2012, 318, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Athar, M.; Chaudry, I.H. When apoptosis meets autophagy: Deciding cell fate after trauma and sepsis. Trends Mol. Med. 2009, 15, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Kim, Y.J.; Kim, C.L.; Lee, G.M. Differential induction of autophagy in caspase-3/7 down-regulating and Bcl-2 overexpressing recombinant CHO cells subjected to sodium butyrate treatment. J. Biotechnol. 2012, 161, 34–41. [Google Scholar] [CrossRef]
- Przyklenk, K.; Dong, Y.; Undyala, V.V.; Whittaker, P. Autophagy as a therapeutic target for ischaemia /reperfusion injury? Concepts, controversies, and challenges. Cardiovasc. Res. 2012, 94, 197–205. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J. Biol. Chem. 2004, 279, 47704–47710. [Google Scholar] [CrossRef] [Green Version]
- McCurdy, C.E.; Klemm, D.J. Adipose tissue insulin sensitivity and macrophage recruitment: Does PI3K pick the pathway? Adipocyte 2013, 2, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Derossi, D.; Williams, E.J.; Green, P.J.; Dunican, D.J.; Doherty, P. Stimulation of mitogenesis by a cell-permeable PI 3-kinase binding peptide. Biochem. Biophys. Res. Commun. 1998, 251, 148–152. [Google Scholar] [CrossRef]
- Atkins, C.M.; Selcher, J.C.; Petraitis, J.J.; Trzaskos, J.M.; Sweatt, J.D. The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1998, 1, 602–609. [Google Scholar] [CrossRef]
- Furtado, L.M.; Somwar, R.; Sweeney, G.; Niu, W.; Klip, A. Activation of the glucose transporter GLUT4 by insulin. Biochem. Cell Biol. 2002, 80, 569–578. [Google Scholar] [CrossRef]
- Michelle Furtado, L.; Poon, V.; Klip, A. GLUT4 activation: Thoughts on possible mechanisms. Acta Physiol. Scand. 2003, 178, 287–296. [Google Scholar] [CrossRef]
- Chung, S.; Brown, J.M.; Sandberg, M.B.; McIntosh, M. Trans-10,cis-12 CLA increases adipocyte lipolysis and alters lipid droplet-associated proteins: Role of mTOR and ERK signaling. J. Lipid Res. 2005, 46, 885–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo, M.E.; Lee, M.J.; Sullivan, S.; Feng, J.; Schneider, S.H.; Greenberg, A.S.; Fried, S.K. Tumor necrosis factor alpha and glucocorticoid synergistically increase leptin production in human adipose tissue: Role for p38 mitogen-activated protein kinase. J. Clin. Endocrinol. Metab. 2006, 91, 1484–1490. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, A.; Czech, M.P.; Davis, R.J. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev. 2004, 18, 1976–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardwell, L. Mechanisms of MAPK signalling specificity. Biochem. Soc. Trans. 2006, 34, 837–841. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, J.J.; Li, J.; Park, K.; Qian, X.; Ma, J.X.; Zhang, S.X. Pigment epithelium-derived factor suppresses adipogenesis via inhibition of the MAPK/ERK pathway in 3T3-L1 preadipocytes. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1378–E1387. [Google Scholar] [CrossRef] [Green Version]
- Lai, P.Y.; Tsai, C.B.; Tseng, M.J. Active form Notch4 promotes the proliferation and differentiation of 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun. 2013, 430, 1132–1139. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.; Kim, Y.H.; Lee, N.H.; Song, K.M. Irisin promotes C2C12 myoblast proliferation via ERK-dependent CCL7 upregulation. PLoS ONE 2019, 14, e0222559. [Google Scholar] [CrossRef] [Green Version]
- Camp, H.S.; Tafuri, S.R. Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J. Biol. Chem. 1997, 272, 10811–10816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, E.; Kim, J.B.; Sarraf, P.; Spiegelman, B.M. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 1996, 274, 2100–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, Y.; Sato, F.; Fukunaga, H.; Iwasaki, Y.; Chiba, Y.; Tebakari, M.; Daigo, Y.; Kawashima, J.; Kamei, J. Placental extract suppresses differentiation of 3T3-L1 preadipocytes to mature adipocytes via accelerated activation of p38 MAPK during the early phase of adipogenesis. Nutr. Metab. 2019, 16, 1–13. [Google Scholar] [CrossRef]
- Wang, Z.; Ka, S.O.; Lee, Y.; Park, B.H.; Bae, E.J. Butein induction of HO-1 by p38 MAPK/Nrf2 pathway in adipocytes attenuates high-fat diet induced adipose hypertrophy in mice. Eur. J. Pharmacol. 2017, 799, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Kim, J.K.; Jeon, J.H.; Yoon, S.R.; Choi, I.; Yang, Y. c-Jun N-terminal kinase is involved in the suppression of adiponectin expression by TNF-alpha in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2005, 327, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Kang, C.; Goh, J.; Chae, S.I.; Kim, H.C.; Lee, T.J.; Abd El-Aty, A.M.; Jeong, J.H. WISP1 promotes non-alcoholic fatty liver disease and skeletal muscle insulin resistance via TLR4/JNK signaling. J. Cell. Physiol. 2018, 233, 6077–6087. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Chen, J.; Hai, T. The regulation of ATF3 gene expression by mitogen-activated protein kinases. Biochem. J. 2007, 401, 559–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.Y.; Lee, J.H.; Min, B.; Lee, Y.H. The translation inhibitor anisomycin induces Elk-1-mediated transcriptional activation of egr-1 through multiple mitogen-activated protein kinase pathways. Exp. Mol. Med. 2006, 38, 677–685. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.; Chen, C.-Z.; Peng, Y.; Li, Z.; Gao, Y.; Liang, S.; Yuan, B.; Kim, N.-H.; Jiang, H.; Zhang, J.-B. KRAS Affects Adipogenic Differentiation by Regulating Autophagy and MAPK Activation in 3T3-L1 and C2C12 Cells. Int. J. Mol. Sci. 2021, 22, 13630. https://doi.org/10.3390/ijms222413630
Yu W, Chen C-Z, Peng Y, Li Z, Gao Y, Liang S, Yuan B, Kim N-H, Jiang H, Zhang J-B. KRAS Affects Adipogenic Differentiation by Regulating Autophagy and MAPK Activation in 3T3-L1 and C2C12 Cells. International Journal of Molecular Sciences. 2021; 22(24):13630. https://doi.org/10.3390/ijms222413630
Chicago/Turabian StyleYu, Wenjie, Cheng-Zhen Chen, Yanxia Peng, Ze Li, Yan Gao, Shuang Liang, Bao Yuan, Nam-Hyung Kim, Hao Jiang, and Jia-Bao Zhang. 2021. "KRAS Affects Adipogenic Differentiation by Regulating Autophagy and MAPK Activation in 3T3-L1 and C2C12 Cells" International Journal of Molecular Sciences 22, no. 24: 13630. https://doi.org/10.3390/ijms222413630
APA StyleYu, W., Chen, C.-Z., Peng, Y., Li, Z., Gao, Y., Liang, S., Yuan, B., Kim, N.-H., Jiang, H., & Zhang, J.-B. (2021). KRAS Affects Adipogenic Differentiation by Regulating Autophagy and MAPK Activation in 3T3-L1 and C2C12 Cells. International Journal of Molecular Sciences, 22(24), 13630. https://doi.org/10.3390/ijms222413630