
Angiotensin Type 2 and Mas Receptor Activation Prevents Myocardial Fibrosis and Hypertrophy through the Reduction of Inflammatory Cell Infiltration and Local Sympathetic Activity in Angiotensin II-Dependent Hypertension

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Effects of C21 and Ang 1-7 Administration on Systolic Blood Pressure, Serological Parameters, Glomerular Filtration Rate, Heart Weight and Heart/Body Weight Ratio in Ang II Treated Rats for 1 and 4 Weeks
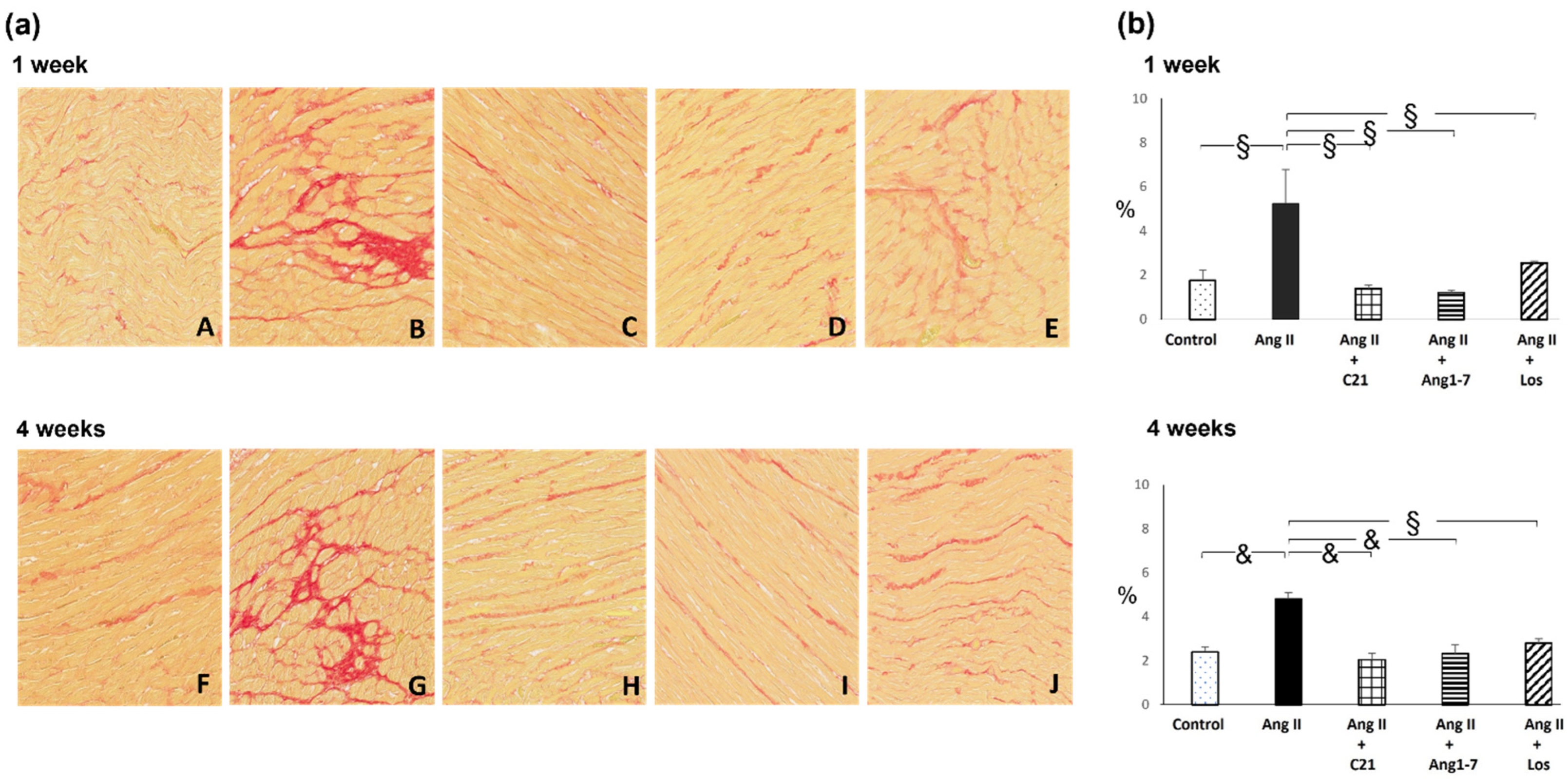
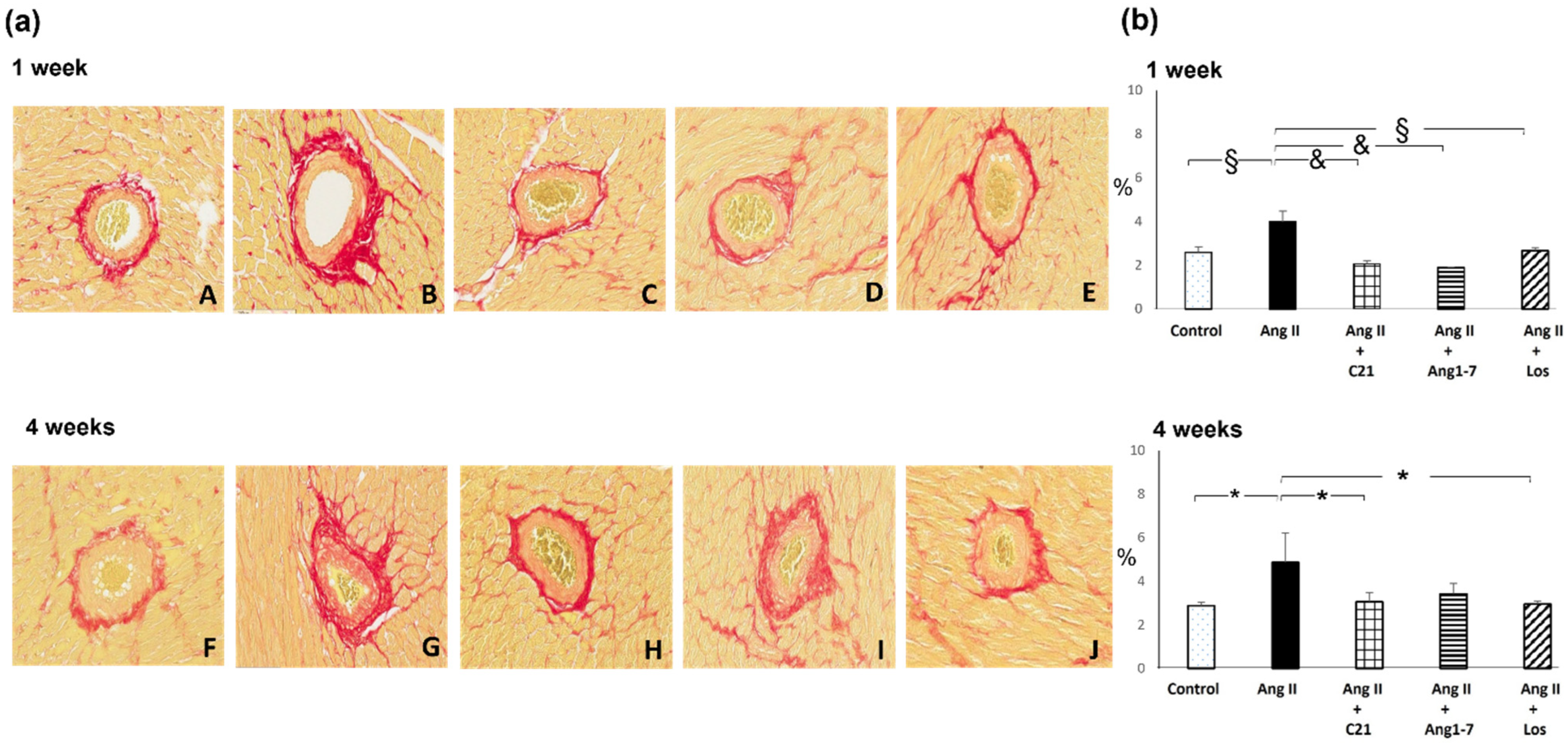
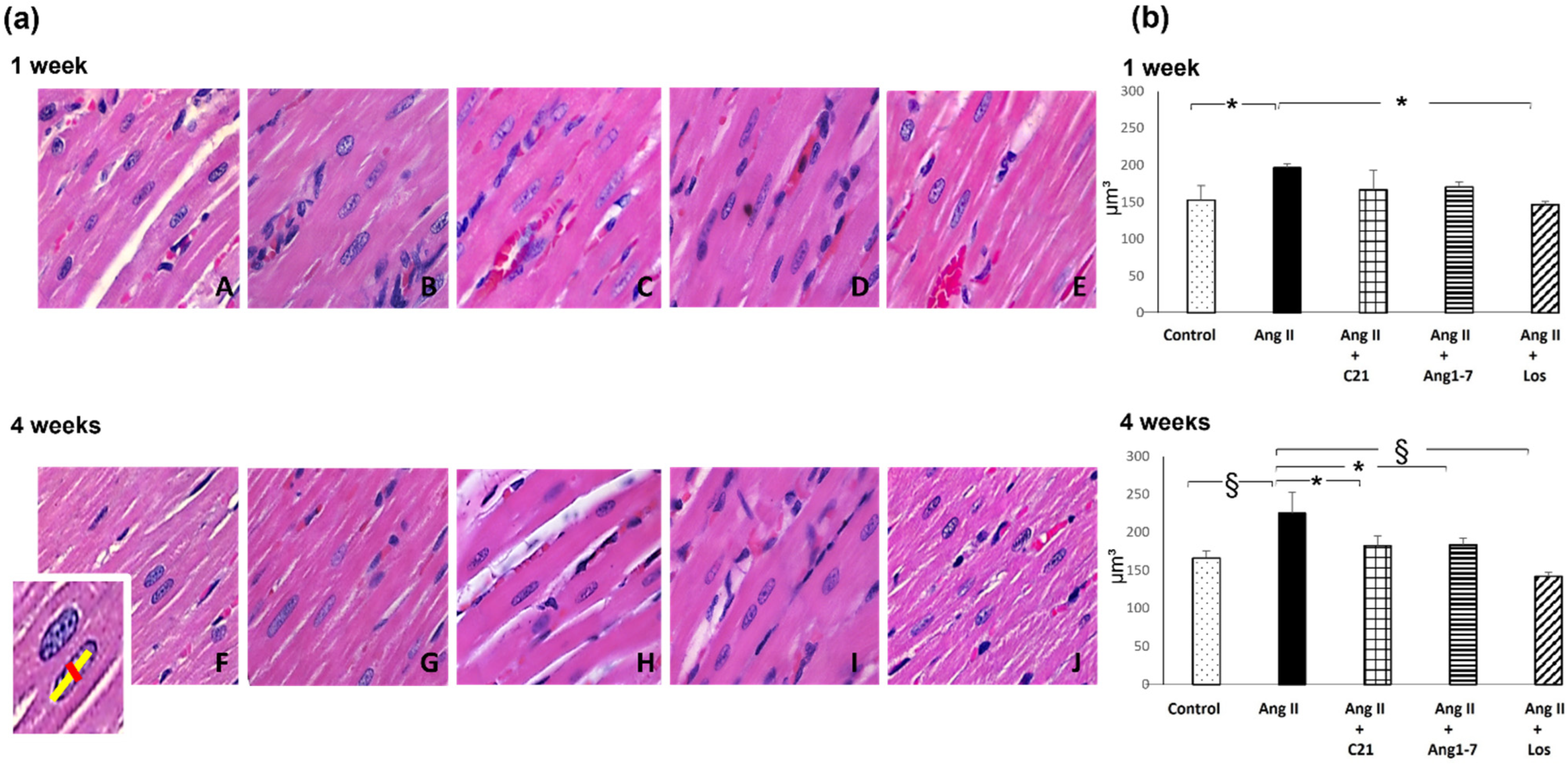
2.2. Effects of C21 and Ang 1-7 Administration on Myocardial Fibrosis and Hypertrophy in Ang II Treated Rats for 1 and 4 Weeks
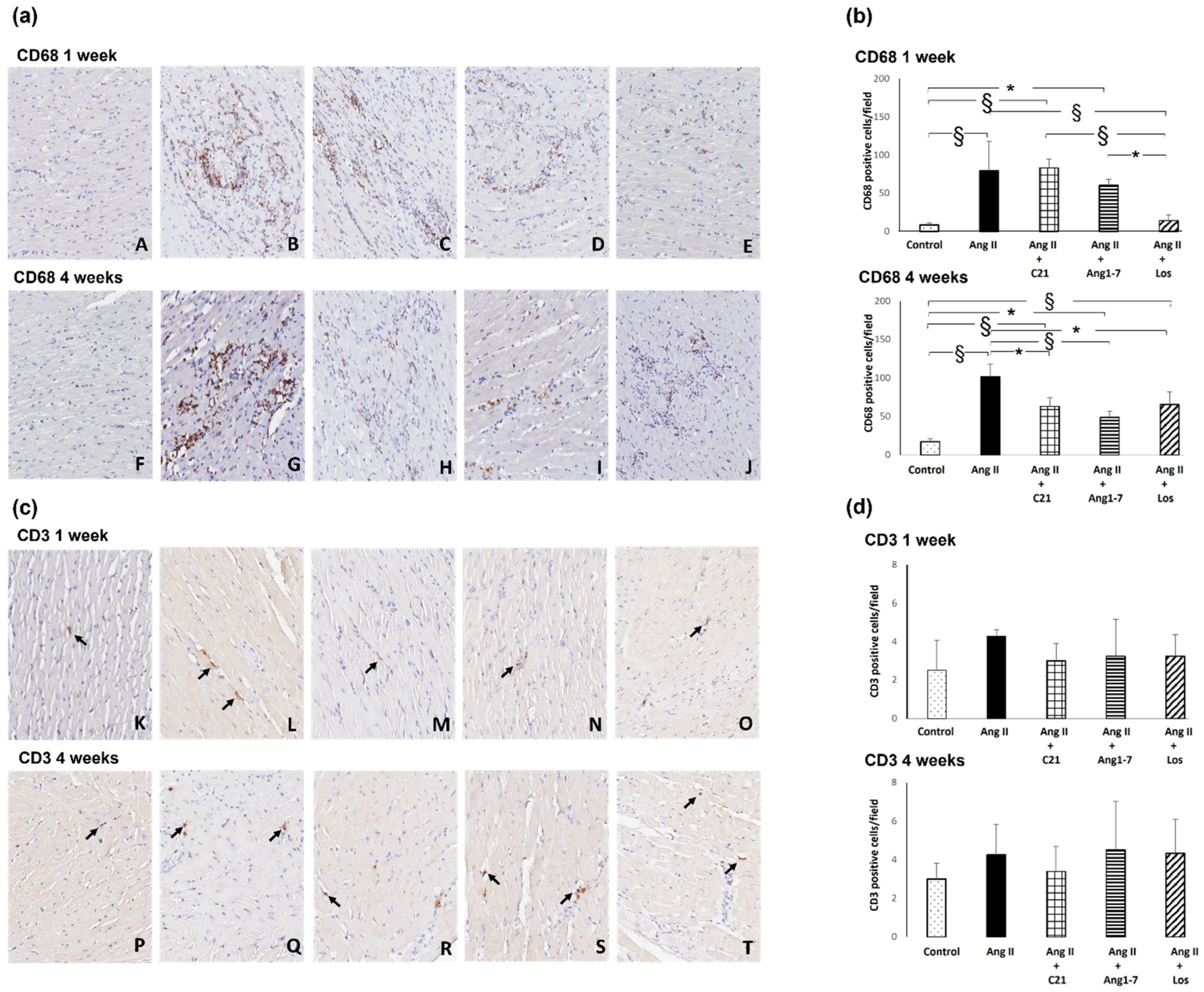
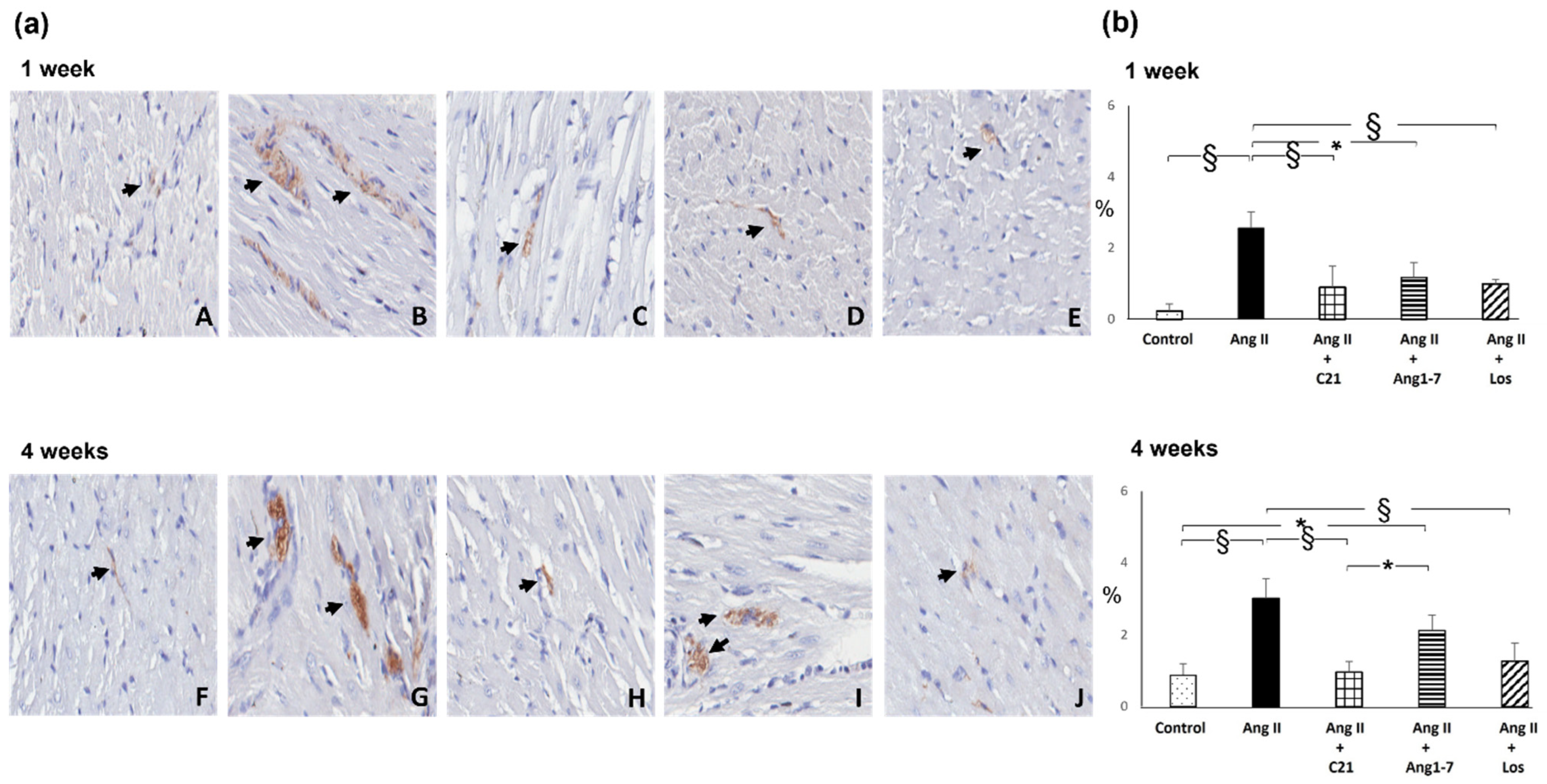
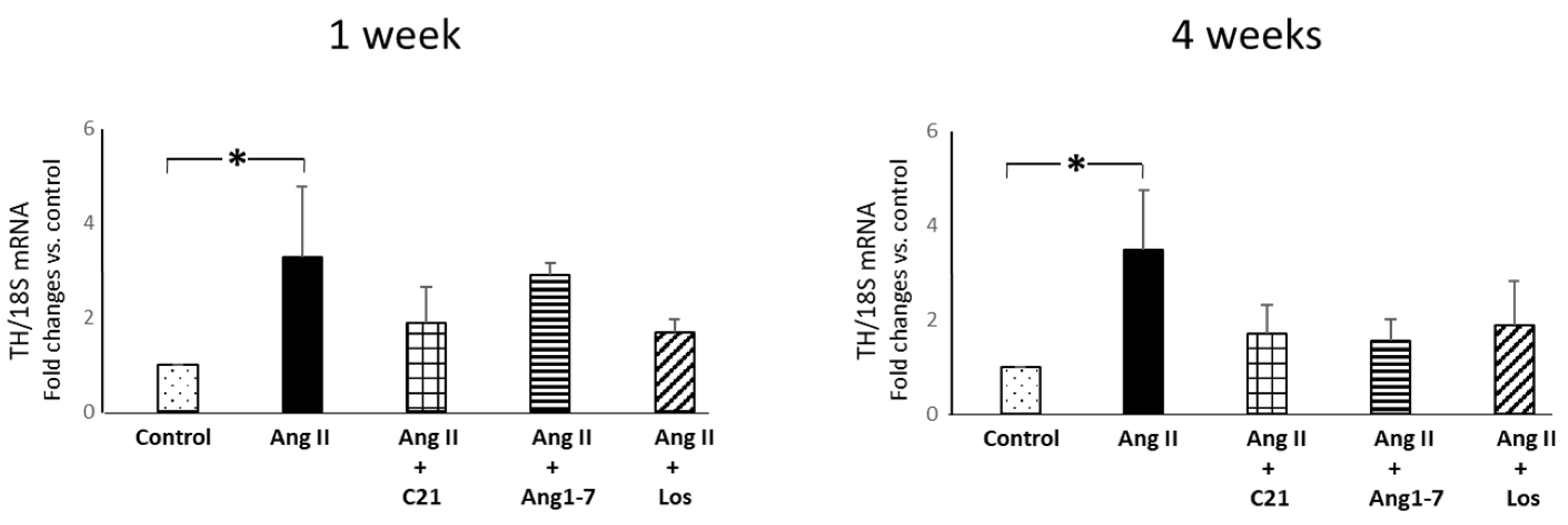
2.3. Effects of C21 and Ang 1-7 Administration on Myocardial Inflammatory Cell Infiltration and Tyrosine Hydroxylase Expression in Ang II Treated Rats for 1 and 4 Weeks
3. Discussion
4. Materials and Methods
4.1. Experimental Model of Ang II-Dependent Hypertension
4.2. Morphometric Analysis of Myocardial Interstitial and Perivascular Fibrosis
4.3. Histological Analysis and Morphometric Evaluation of Myocardial Hypertrophy
4.4. Immunohistochemical Evaluation of Myocardial Monocyte/Macrophage Infiltration, T Lymphocytes and Tyrosine Hydroxylase Expression
4.5. Myocardial mRNA Expression of Tyrosine Hydroxylase
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Te Riet, L.; van Esch, J.H.M.; Roks, A.J.M.; van den Meiracker, A.H.; Danser, A.H.J. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; De Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. The Task Force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension. J. Hypertens. 2018, 36, 1953–2041. [Google Scholar] [CrossRef] [Green Version]
- Lorell, B.H.; Carabello, B.A. Left ventricular hypertrophy: Pathogenesis, detection, and prognosis. Circulation 2000, 102, 470–479. [Google Scholar] [CrossRef]
- Martos, R.; Baugh, J.; Ledwidge, M.; O’Loughlin, C.; Conlon, C.; Patle, A.; Donnelly, S.C.; McDonald, K. Diastolic heart failure: Evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 2007, 115, 888–895. [Google Scholar] [CrossRef] [Green Version]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rupérez, M.; Esteban, V.; Rodríguez-Vita, J.; Sánchez-López, E.; Carvajal, G.; Egido, J. Angiotensin II: A key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol. Dial. Transplant. 2006, 21, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Durango, N.; Fuentes, C.A.; Castillo, A.E.; González-Gómez, L.M.; Vecchiola, A.; Fardella, C.E.; Kalergis, A.M. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: Molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int. J. Mol. Sci. 2016, 17, 797. [Google Scholar] [CrossRef] [Green Version]
- Unger, T.; Steckelings, U.M.; dos Santos, R.A.S. The Protective Arm of the Renin Angiotensin System (RAS): In Functional Aspects and Therapeutic Implication; Academic Press: New York, NY, USA, 2015. [Google Scholar]
- McKinney, C.A.; Fattah, C.; Loughrey, C.M.; Milligan, G.; Nicklin, S.A. Angiotensin-(1-7) and angiotensin-(1-9): Function in cardiac and vascular remodelling. Clin. Sci. 2014, 126, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Povlsen, A.L.; Grimm, D.; Wehland, M.; Infanger, M.; Krüger, M. The vasoactive Mas receptor in essential hypertension. J. Clin. Med. 2020, 9, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumners, C.; Peluso, A.A.; Haugaard, A.H.; Bertelsen, J.B.; Steckelings, U.M. Anti-fibrotic mechanisms of angiotensin AT2-receptor stimulation. Acta Physiol. 2019, 227, e13280. [Google Scholar] [CrossRef] [Green Version]
- Kaschina, E.; Grzesiak, A.; Li, J.; Foryst-Ludwig, A.; Timm, M.; Rompe, F.; Sommerfeld, M.; Kemnitz, U.R.; Curato, C.; Namsolleck, P.; et al. Angiotensin II type 2 receptor stimulation: A novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 2008, 118, 2523–2532. [Google Scholar] [CrossRef] [Green Version]
- Lauer, D.; Slavic, S.; Sommerfeld, M.; Thöne-Reineke, C.; Sharkovska, Y.; Hallberg, A.; Dahlöf, B.; Kintscher, U.; Unger, T.; Steckelings, U.M.; et al. Angiotensin type 2 receptor stimulation ameliorates left ventricular fibrosis and dysfunction via regulation of tissue inhibitor of matrix metalloproteinase 1/matrix metalloproteinase 9 axis and transforming growth factor β1 in the rat heart. Hypertension 2014, 63, e60–e67. [Google Scholar] [CrossRef] [Green Version]
- Castoldi, G.; di Gioia, C.R.T.; Roma, F.; Carletti, R.; Manzoni, G.; Stella, A.; Zerbini, G.; Perseghin, G. Activation of angiotensin type 2 (AT2) receptors prevents myocardial hypertrophy in Zucker diabetic fatty rats. Acta Diabetol. 2019, 56, 97–104. [Google Scholar] [CrossRef]
- Paulis, L.; Becker, S.T.R.; Lucht, K.; Schwengel, K.; Slavic, S.; Kaschina, E.; Thöne-Reineke, C.; Dahlöf, B.; Baulmann, J.; Unger, T.; et al. Direct angiotensin II type 2 receptor stimulation in Nω-nitro-L-arginine-methyl ester-induced hypertension. The effect on pulse wave velocity and aortic remodeling. Hypertension 2012, 59, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.; Leibowitz, A.; Yamamoto, N.; Rautureau, Y.; Paradis, P.; Schiffrin, E.L. Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension 2012, 59, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dopona, E.P.B.; Rocha, V.F.; Furukawa, L.N.S.; Oliveira, I.B.; Heimann, J.C. Myocardial hypertrophy induced by high salt consumption is prevented by angiotensin II AT2 receptor agonist. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, G.; di Gioia, C.R.T.; Carletti, R.; Roma, F.; Zerbini, G.; Stella, A. Angiotensin type-2 (AT-2)-receptor activation reduces renal fibrosis in cyclosporine nephropathy: Evidence for blood pressure independent effect. Biosci. Rep. 2016, 36, e00403. [Google Scholar] [CrossRef] [PubMed]
- Grobe, J.L.; Mecca, A.P.; Lingis, M.; Shenoy, V.; Bolton, T.A.; Machado, J.M.; Speth, R.C.; Raizada, M.K.; Katovich, M.J. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1-7). Am. J. Physiol. Heart. Circ. Physiol. 2007, 292, H736–H742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Yin, A.; Zhang, Q.; Zhong, T.; O’Rourke, S.T.; Sun, C. Angiotensin-(1-7) attenuates angiotensin II-induced cardiac hypertrophy via a Sirt3-dependent mechanism. Am. J. Physiol. Heart. Circ. Physiol. 2017, 312, H980–H991. [Google Scholar] [CrossRef]
- Shah, A.; Oh, Y.B.; Lee, S.H.; Lim, J.M.; Kim, S.H. Angiotensin-(1-7) attenuates hypertension in exercise-trained renal hypertensive rats. Am. J. Physiol. Heart. Circ. Physiol. 2012, 302, H2372–H2380. [Google Scholar] [CrossRef] [Green Version]
- Mori, J.; Patel, V.B.; Abo Alrob, O.; Basu, R.; Altamimi, T.; DesAulniers, J.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ. Heart. Fail. 2014, 7, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Grobe, J.L.; Mecca, A.P.; Mao, H.; Katovich, M.J. Chronic angiotensin-(1-7) prevents cardiac fibrosis in DOCA-salt model of hypertension. Am. J. Physiol. Heart. Circ. Physiol. 2006, 290, H2417–H2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelosa, P.; Pignieri, A.; Fändriks, L.; de Gasparo, M.; Hallberg, A.; Banfi, C.; Castiglioni, L.; Turolo, L.; Guerrini, U.; Tremoli, E.; et al. Stimulation of AT2 receptor exerts beneficial effects in stroke-prone rats: Focus on renal damage. J. Hypertens. 2009, 27, 2444–2451. [Google Scholar] [CrossRef]
- Hilliard, L.M.; Chow, C.L.E.; Mirabito, K.M.; Steckelings, U.M.; Unger, T.; Widdop, R.E.; Denton, K.M. Angiotensin type 2 receptor stimulation increases renal function in female, but not male, spontaneously hypertensive rats. Hypertension 2014, 64, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Matavelli, L.C.; Huang, J.; Siragy, H.M. Angiotensin AT2 receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension 2011, 57, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Assersen, K.B.; Sumners, C.; Steckelings, U.M. The renin-angiotensin system in hypertension, a constantly renewing classic: Focus on the angiotensin AT2-receptor. Can. J. Cardiol. 2020, 36, 683–693. [Google Scholar] [CrossRef]
- Sumners, C.; de Kloet, A.D.; Krause, E.G.; Unger, T.; Steckelings, U.M. Angiotensin type 2 receptors: Blood pressure regulation and end organ damage. Curr. Opin. Pharmacol. 2015, 21, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Simões e Silva, A.C.; Silveira, K.D.; Ferreira, A.J.; Teixeira, M.M. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2021, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Schlaich, M.P.; Kaye, D.M.; Lambert, E.; Sommerville, M.; Socratous, F.; Esler, M.D. Relation between cardiac sympathetic activity and hypertensive left ventricular hypertrophy. Circulation. 2003, 108, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sun, Y.; Dong, M.; Yang, X.; Meng, X.; Niu, R.; Guan, J.; Zhang, Y.; Zhang, C. Comparison of angiotensin-(1-7), losartan and their combination on atherosclerotic plaque formation in apolipoprotein E knockout mice. Atherosclerosis 2015, 240, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, G.; di Gioia, C.; Giollo, F.; Carletti, R.; Bombardi, C.; Antoniotti, M.; Roma, F.; Zerbini, G.; Stella, A. Different regulation of miR-29a-3p in glomeruli and tubules in an experimental model of angiotensin II-dependent hypertension: Potential role in renal fibrosis. Clin. Exp. Pharmacol. Physiol. 2016, 43, 335–342. [Google Scholar] [CrossRef]
- Benter, I.F.; Yousif, M.H.; Cojocel, C.; Al-Maghrebi, M.; Diz, D.I. Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. Am. J. Physiol. Heart. Circ. Physiol. 2007, 292, 666–672. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Singh, T.; Sharma, P.L. Angiotensin (1-7)/Mas receptor axis activation ameliorates the changes in fatty acid composition in diabetic rats with nephropathy. J. Exp. Pharmacol. 2010, 2, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Serra, A.J.; Higuchi, M.L.; Ihara, S.S.; Antônio, E.L.; Santos, M.H.; Bombig, M.T.; Tucci, P.J. Exercise training prevents β-adrenergic hyperactivity-induced myocardial hypertrophy and lesions. Eur. J. Heart Fail. 2008, 10, 534–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castoldi, G.; Carletti, R.; Ippolito, S.; Colzani, M.; Barzaghi, F.; Stella, A.; Zerbini, G.; Perseghin, G.; Zatti, G.; di Gioia, C.R.T. Sodium-glucose cotransporter 2 inhibition prevents renal fibrosis in cyclosporine nephropathy. Acta Diabetol. 2021, 58, 1059–1070. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Control | Ang II | Ang II+C21 | Ang II+Ang 1-7 | Ang II+Los |
|---|---|---|---|---|---|
| SBP (mmHg) | 146.2 ± 2.3 | 198.3 ± 4.4 ‡,ζ | 193.7 ± 2.3 ‡,ζ | 194.3 ± 2.3 ‡,ζ | 148.1 ± 1.2 |
| BW (g) | 353.5 ± 25.3 | 363.3 ± 2.4 δ | 317.8 ± 9.7 ^ | 344.0 ± 2.1 | 372.0 ± 8.2 |
| Heart Weight (g) | 1.11 ± 0.06 | 1.26 ± 0.01 *,^ | 1.22 ± 0.06 | 1.25 ± 0.03 * | 1.06 ± 0.02 |
| Heart/Body Weight (mg/g) | 3.16 ± 0.20 | 3.46 ± 0.05 ^ | 3.84 ± 0.15 †,ζ | 3.63 ± 0.09 *,^ | 2.85 ± 0.05 |
| GFR (mL/min) | 1.35 ± 0.19 | 1.23 ± 0.02 | 1.30 ± 0.08 | 1.45 ± 0.11 | 1.47± 0.10 |
| Plasma | |||||
| Glucose (mg/dL) | 139.4 ± 3.5 | 140.8 ± 6.2 | 127.2 ± 6.3 | 129.7 ± 5.2 | 132.5 ± 8.5 |
| Sodium (mEq/L) | 140.0 ± 0.43 | 141.7 ± 0.35 | 139.9 ± 0.70 | 141.1 ± 0.80 | 138.2 ± 0.81 |
| Potassium (mEq/L) | 4.40 ± 0.47 | 4.26 ± 0.35 | 4.94 ± 0.66 | 4.20 ± 0.36 | 4.18 ± 0.18 |
| Calcium (mg/dL) | 7.64 ± 0.80 | 6.88 ± 0.18 | 4.75 ± 0.79 | 6.00 ± 0.88 | 7.94 ± 0.48 |
| Phosphate (mg/dL) | 6.41 ± 0.56 | 6.70 ± 0.59 | 6.93 ± 1.22 | 5.46 ± 0.81 | 6.20 ± 0.44 |
| Cholesterol (mg/dL) | 58.4 ± 4.48 | 65.1 ± 9.00 | 47.3 ± 3.3 | 55.3 ± 3.9 | 52.3 ± 4.5 |
| Triglycerides (mg/dL) | 114.2 ± 36.2 | 91.9 ± 21.1 | 63.5 ± 2.3 | 75.9 ± 12.7 | 72.6 ± 9.6 |
| CRP (µg/mL) | 34.1 ± 0.67 | 36.0 ± 0.63 | 48.2 ± 12.6 | 36.1 ± 5.5 | 43.6 ± 14.9 |
| MCP-1 (pg/mL) | 253.3 ± 6.66 | 287.9 ± 9.44 | 269.5 ± 5.85 | 253.4 ± 5.11 | 276.9 ± 23.5 |
| Parameters | Control | Ang II | Ang II+C21 | Ang II+Ang 1-7 | Ang II+Los |
|---|---|---|---|---|---|
| SBP (mmHg) | 145.0 ± 2.8 | 201.2 ± 3.9 ‡,ζ | 195.4 ± 2.0 ‡,ζ | 196.8 ± 4.2 ‡,ζ | 138.1 ± 3.5 |
| BW (g) | 449.0 ± 11.3 | 369.6 ± 9.6 ‡ | 368.4 ± 10.9 ‡ | 354.0 ± 8.4 ‡ | 392.1 ± 13.2 † |
| Heart Weight (g) | 1.32 ± 0.05 | 1.40 ± 0.02 ^ | 1.45 ± 0.05 ^ | 1.35 ± 0.03 ^ | 1.14 ± 0.05 * |
| Heart/Body Weight (mg/g) | 2.94 ± 0.07 | 3.79 ± 0.08 †,ζ | 3.94 ± 0.13 ‡,ζ | 3.86 ± 0.16 ‡,ζ | 2.91± 0.06 |
| GFR (mL/min) | 1.91 ± 0.19 | 1.41 ± 0.12 * | 1.34 ± 0.14† | 1.45 ± 0.12 * | 1.35 ± 0.12 * |
| Plasma | |||||
| Glucose (mg/dL) | 122.7 ± 4.8 | 130.0 ± 12.0 | 115.7 ± 7.5 | 113.6 ± 8.9 | 133.1 ± 9.6 |
| Sodium (mEq/L) | 138.6 ± 0.94 | 137.1 ± 2.37 | 138.5 ± 1.72 | 137.7 ± 0.91 | 141.2 ± 1.06 |
| Potassium (mEq/L) | 4.24 ± 0.18 | 3.58 ± 0.51 | 3.90 ± 0.33 | 4.62 ± 0.43 | 4.50 ± 0.32 |
| Calcium (mg/dL) | 7.87 ± 0.24 | 7.57 ± 0.46 | 7.65 ± 0.29 | 7.44 ± 0.41 | 7.88 ± 0.27 |
| Phosphate (mg/dL) | 6.40 ± 0.36 | 6.21 ± 0.65 | 6.18 ± 0.85 | 6.09 ± 0.92 | 7.06 ± 0.43 |
| Cholesterol (mg/dL) | 44.1 ± 2.3 | 49.5 ± 5.4 | 48.8 ± 2.3 | 45.3 ± 2.6 | 48.0 ± 2.3 |
| Triglycerides (mg/dL) | 111.9 ± 13.0 | 90.8 ± 20.6 | 91.3 ± 14.7 | 82.0 ± 10.3 | 86.7 ± 11.2 |
| CRP (µg/mL) | 31.8 ± 2.43 | 27.4 ± 1.85 | 43.9 ± 4.13 | 42.7 ± 8.17 | 43.1 ± 6.69 |
| MCP-1 (pg/mL) | 261.2 ± 7.56 | 270.4 ± 9.83 | 280.0 ± 9.36 | 263.5 ± 5.29 | 285.2 ± 19.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castoldi, G.; Carletti, R.; Ippolito, S.; Stella, A.; Zerbini, G.; Pelucchi, S.; Zatti, G.; di Gioia, C.R.T. Angiotensin Type 2 and Mas Receptor Activation Prevents Myocardial Fibrosis and Hypertrophy through the Reduction of Inflammatory Cell Infiltration and Local Sympathetic Activity in Angiotensin II-Dependent Hypertension. Int. J. Mol. Sci. 2021, 22, 13678. https://doi.org/10.3390/ijms222413678
Castoldi G, Carletti R, Ippolito S, Stella A, Zerbini G, Pelucchi S, Zatti G, di Gioia CRT. Angiotensin Type 2 and Mas Receptor Activation Prevents Myocardial Fibrosis and Hypertrophy through the Reduction of Inflammatory Cell Infiltration and Local Sympathetic Activity in Angiotensin II-Dependent Hypertension. International Journal of Molecular Sciences. 2021; 22(24):13678. https://doi.org/10.3390/ijms222413678
Chicago/Turabian StyleCastoldi, Giovanna, Raffaella Carletti, Silvia Ippolito, Andrea Stella, Gianpaolo Zerbini, Sara Pelucchi, Giovanni Zatti, and Cira R. T. di Gioia. 2021. "Angiotensin Type 2 and Mas Receptor Activation Prevents Myocardial Fibrosis and Hypertrophy through the Reduction of Inflammatory Cell Infiltration and Local Sympathetic Activity in Angiotensin II-Dependent Hypertension" International Journal of Molecular Sciences 22, no. 24: 13678. https://doi.org/10.3390/ijms222413678
APA StyleCastoldi, G., Carletti, R., Ippolito, S., Stella, A., Zerbini, G., Pelucchi, S., Zatti, G., & di Gioia, C. R. T. (2021). Angiotensin Type 2 and Mas Receptor Activation Prevents Myocardial Fibrosis and Hypertrophy through the Reduction of Inflammatory Cell Infiltration and Local Sympathetic Activity in Angiotensin II-Dependent Hypertension. International Journal of Molecular Sciences, 22(24), 13678. https://doi.org/10.3390/ijms222413678