Importance of Both Imprinted Genes and Functional Heterogeneity in Pancreatic Beta Cells: Is There a Link?

Abstract
:1. Introduction
1.1. Genomic Imprinting
1.2. Human Imprinting Disorders
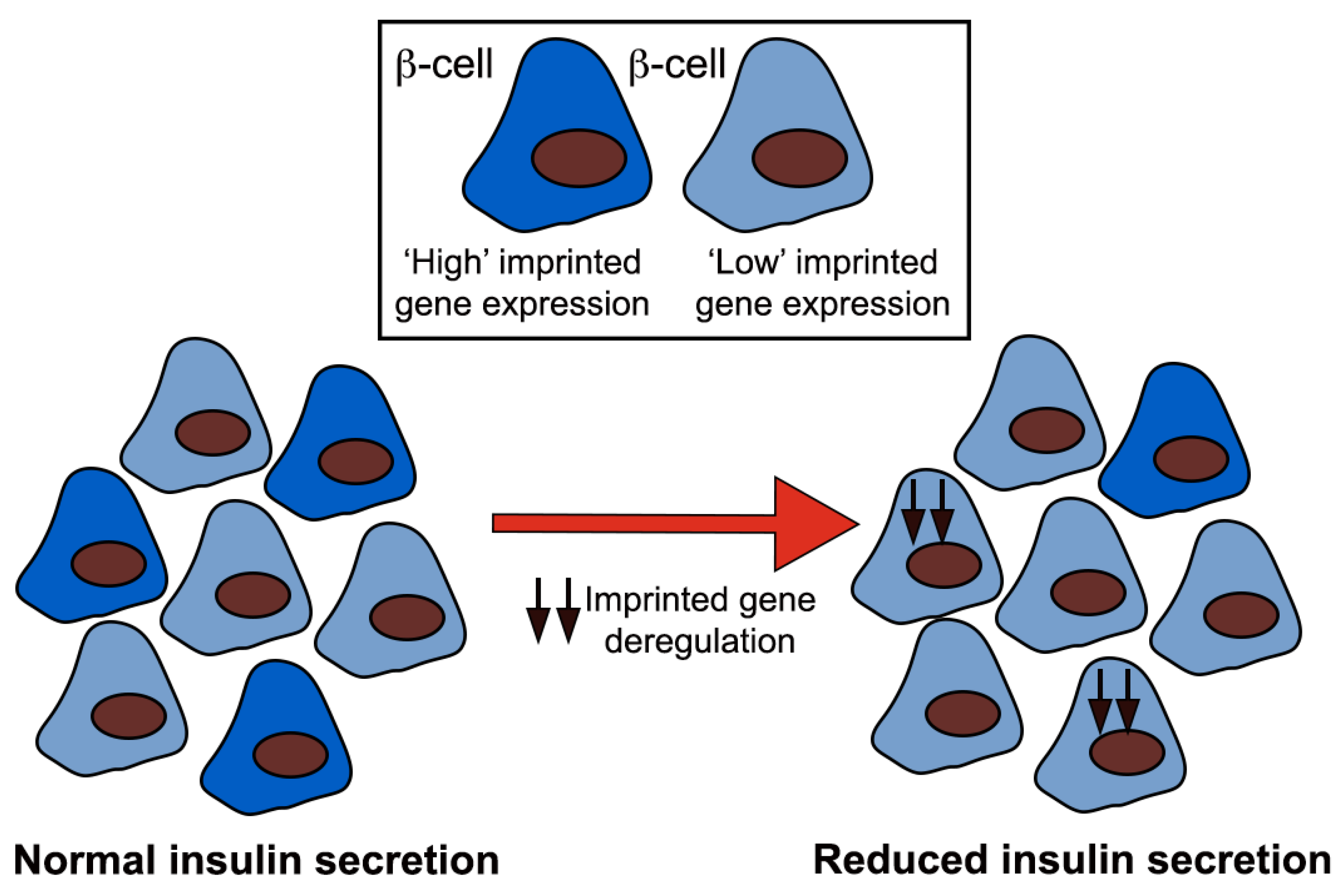
1.3. Imprinted Genes and Pancreatic Beta Cells
1.4. Beta Cell Heterogeneity
1.5. Transcriptomic Diversity between Beta Cell Subpopulations
Author Contributions
Funding
Conflicts of Interest
References
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leete, P.; Willcox, A.; Krogvold, L.; Dahl-Jorgensen, K.; Foulis, A.K.; Richardson, S.J.; Morgan, N.G. Differential Insulitic Profiles Determine the Extent of beta-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes 2016, 65, 1362–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10 (Suppl. 4), 32–42. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Pullen, T.J.; Hodson, D.J.; Martinez-Sanchez, A. Pancreatic beta-cell identity, glucose sensing and the control of insulin secretion. Biochem. J. 2015, 466, 203–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Janson, J.; Soeller, W.C.; Butler, P.C. Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: Evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 2003, 52, 2304–2314. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specialisations of the pancreatic beta cell: Gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Iynedjian, P.B. Molecular physiology of mammalian glucokinase. Cell Mol. Life Sci. 2009, 66, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Semplici, F.; Ravier, M.A.; Bellomo, E.A.; Pullen, T.J.; Gilon, P.; Sekler, I.; Rizzuto, R.; Rutter, G.A. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic beta-cells. PLoS ONE 2012, 7, e39722. [Google Scholar] [CrossRef] [Green Version]
- Rorsman, P.; Ashcroft, F.M. Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.C. The dual control of insulin secretion by glucose involves triggering and amplifying pathways in beta-cells. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. 1), S27–S31. [Google Scholar] [CrossRef]
- Ferdaoussi, M.; Dai, X.; Jensen, M.V.; Wang, R.; Peterson, B.S.; Huang, C.; Ilkayeva, O.; Smith, N.; Miller, N.; Hajmrle, C.; et al. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional beta cells. J. Clin. Investig. 2015, 125, 3847–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentki, M.; Corkey, B.E.; Madiraju, S.R.M. Lipid-associated metabolic signalling networks in pancreatic beta cell function. Diabetologia 2020, 63, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Guerra, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.S.; Stein, R.W. Evidence for Loss in Identity, De-Differentiation, and Trans-Differentiation of Islet beta-Cells in Type 2 Diabetes. Front. Genet. 2017, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullen, T.J.; Huising, M.O.; Rutter, G.A. Analysis of Purified Pancreatic Islet Beta and Alpha Cell Transcriptomes Reveals 11beta-Hydroxysteroid Dehydrogenase (Hsd11b1) as a Novel Disallowed Gene. Front. Genet. 2017, 8, 41. [Google Scholar] [CrossRef]
- Ishida, E.; Kim-Muller, J.Y.; Accili, D. Pair Feeding, but Not Insulin, Phloridzin, or Rosiglitazone Treatment, Curtails Markers of beta-Cell Dedifferentiation in db/db Mice. Diabetes 2017, 66, 2092–2101. [Google Scholar] [CrossRef] [Green Version]
- Benninger, R.K.; Zhang, M.; Head, W.S.; Satin, L.S.; Piston, D.W. Gap junction coupling and calcium waves in the pancreatic islet. Biophys. J. 2008, 95, 5048–5061. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A.; Hodson, D.J. Beta cell connectivity in pancreatic islets: A type 2 diabetes target? Cell Mol. Life Sci. 2015, 72, 453–467. [Google Scholar] [CrossRef]
- Benninger, R.K.P.; Dorrell, C.; Hodson, D.J.; Rutter, G.A. The Impact of Pancreatic Beta Cell Heterogeneity on Type 1 Diabetes Pathogenesis. Curr. Diab. Rep. 2018, 18, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosak, M.; Stozer, A.; Markovic, R.; Dolensek, J.; Perc, M.; Rupnik, M.S.; Marhl, M. Critical and Supercritical Spatiotemporal Calcium Dynamics in Beta Cells. Front. Physiol. 2017, 8, 1106. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, S.; Tschen, S.I.; Zeng, C.; Guo, T.; Hebrok, M.; Matveyenko, A.; Bhushan, A. DNA methylation directs functional maturation of pancreatic beta cells. J. Clin. Investig. 2015, 125, 2851–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surani, M.A. Genomic imprinting: Control of gene expression by epigenetic inheritance. Curr. Opin. Cell. Biol. 1994, 6, 390–395. [Google Scholar] [CrossRef]
- Surani, M.A.; Barton, S.C. Development of gynogenetic eggs in the mouse: Implications for parthenogenetic embryos. Science 1983, 222, 1034–1036. [Google Scholar] [CrossRef]
- Surani, M.A.; Barton, S.C.; Norris, M.L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548–550. [Google Scholar] [CrossRef]
- McGrath, J.; Solter, D. Nuclear transplantation in mouse embryos. J. Exp. Zool. 1983, 228, 355–362. [Google Scholar] [CrossRef]
- McGrath, J.; Solter, D. Nuclear transplantation in the mouse embryo by microsurgery and cell fusion. Science 1983, 220, 1300–1302. [Google Scholar] [CrossRef]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Moore, T.; Haig, D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 1991, 7, 45–49. [Google Scholar] [CrossRef]
- Wolf, J.B.; Hager, R. A maternal-offspring coadaptation theory for the evolution of genomic imprinting. PLoS Biol. 2006, 4, e380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolomei, M.S.; Ferguson-Smith, A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011, 3, a002592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting, G. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson-Smith, A.C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011, 12, 565–575. [Google Scholar] [CrossRef]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Radford, E.J.; Ferron, S.R.; Ferguson-Smith, A.C. Genomic imprinting as an adaptative model of developmental plasticity. FEBS Lett. 2011, 585, 2059–2066. [Google Scholar] [CrossRef] [Green Version]
- Rampersaud, E.; Mitchell, B.D.; Naj, A.C.; Pollin, T.I. Investigating parent of origin effects in studies of type 2 diabetes and obesity. Curr. Diabetes Rev. 2008, 4, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Eggermann, T.; Perez de Nanclares, G.; Maher, E.R.; Temple, I.K.; Tumer, Z.; Monk, D.; Mackay, D.J.; Gronskov, K.; Riccio, A.; Linglart, A.; et al. Imprinting disorders: A group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin. Epigenetics 2015, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef]
- Soellner, L.; Begemann, M.; Mackay, D.J.; Gronskov, K.; Tumer, Z.; Maher, E.R.; Temple, I.K.; Monk, D.; Riccio, A.; Linglart, A.; et al. Recent Advances in Imprinting Disorders. Clin. Genet. 2017, 91, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, K.; Kagami, M.; Fukami, M. Uniparental disomy as a cause of pediatric endocrine disorders. Clin. Pediatr. Endocrinol. 2018, 27, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relkovic, D.; Doe, C.M.; Humby, T.; Johnstone, K.A.; Resnick, J.L.; Holland, A.J.; Hagan, J.J.; Wilkinson, L.S.; Isles, A.R. Behavioural and cognitive abnormalities in an imprinting centre deletion mouse model for Prader-Willi syndrome. Eur. J. Neurosci. 2010, 31, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Gurrieri, F.; Accadia, M. Genetic imprinting: The paradigm of Prader-Willi and Angelman syndromes. Endocr. Dev. 2009, 14, 20–28. [Google Scholar] [PubMed]
- Chamberlain, S.J.; Lalande, M. Angelman syndrome, a genomic imprinting disorder of the brain. J. Neurosci. 2010, 30, 9958–9963. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G. Prader-Willi Syndrome: Obesity due to Genomic Imprinting. Curr. Genom. 2011, 12, 204–215. [Google Scholar] [CrossRef]
- Chiesa, N.; De Crescenzo, A.; Mishra, K.; Perone, L.; Carella, M.; Palumbo, O.; Mussa, A.; Sparago, A.; Cerrato, F.; Russo, S.; et al. The KCNQ1OT1 imprinting control region and non-coding RNA: New properties derived from the study of Beckwith-Wiedemann syndrome and Silver-Russell syndrome cases. Hum. Mol. Genet. 2012, 21, 10–25. [Google Scholar] [CrossRef]
- Netchine, I.; Rossignol, S.; Azzi, S.; Brioude, F.; Le Bouc, Y. Imprinted anomalies in fetal and childhood growth disorders: The model of Russell-Silver and Beckwith-Wiedemann syndromes. Endocr. Dev. 2012, 23, 60–70. [Google Scholar]
- Ioannides, Y.; Lokulo-Sodipe, K.; Mackay, D.J.; Davies, J.H.; Temple, I.K. Temple syndrome: Improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: An analysis of 51 published cases. J. Med. Genet. 2014, 51, 495–501. [Google Scholar] [CrossRef]
- Temple, I.K.; Gardner, R.J.; Robinson, D.O.; Kibirige, M.S.; Ferguson, A.W.; Baum, J.D.; Barber, J.C.; James, R.S.; Shield, J.P. Further evidence for an imprinted gene for neonatal diabetes localised to chromosome 6q22-q23. Hum. Mol. Genet. 1996, 5, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Temple, I.K.; James, R.S.; Crolla, J.A.; Sitch, F.L.; Jacobs, P.A.; Howell, W.M.; Betts, P.; Baum, J.D.; Shield, J.P. An imprinted gene(s) for diabetes? Nat. Genet. 1995, 9, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.J.; Mackay, D.J.; Mungall, A.J.; Polychronakos, C.; Siebert, R.; Shield, J.P.; Temple, I.K.; Robinson, D.O. An imprinted locus associated with transient neonatal diabetes mellitus. Hum. Mol. Genet. 2000, 9, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, B.D.; Pollin, T.I. Genomic imprinting in diabetes. Genome Med. 2010, 2, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, D.J.; Temple, I.K. Transient neonatal diabetes mellitus type 1. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 335–342. [Google Scholar] [CrossRef]
- Kamiya, M.; Judson, H.; Okazaki, Y.; Kusakabe, M.; Muramatsu, M.; Takada, S.; Takagi, N.; Arima, T.; Wake, N.; Kamimura, K.; et al. The cell cycle control gene ZAC/PLAGL1 is imprinted—A strong candidate gene for transient neonatal diabetes. Hum. Mol. Genet. 2000, 9, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Varrault, A.; Bilanges, B.; Mackay, D.J.; Basyuk, E.; Ahr, B.; Fernandez, C.; Robinson, D.O.; Bockaert, J.; Journot, L. Characterization of the methylation-sensitive promoter of the imprinted ZAC gene supports its role in transient neonatal diabetes mellitus. J. Biol. Chem. 2001, 276, 18653–18656. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Shield, J.P.; Dean, W.; Leclerc, I.; Knauf, C.; Burcelin, R.R.; Rutter, G.A.; Kelsey, G. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J. Clin. Investig. 2004, 114, 339–348. [Google Scholar] [CrossRef]
- Millership, S.J.; Van de Pette, M.; Withers, D.J. Genomic imprinting and its effects on postnatal growth and adult metabolism. Cell Mol. Life Sci. 2019, 76, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Millership, S.J.; Da Silva Xavier, G.; Choudhury, A.I.; Bertazzo, S.; Chabosseau, P.; Pedroni, S.M.; Irvine, E.E.; Montoya, A.; Faull, P.; Taylor, W.R.; et al. Neuronatin regulates pancreatic beta cell insulin content and secretion. J. Clin. Investig. 2018, 128, 3369–3381. [Google Scholar] [CrossRef] [Green Version]
- Joe, M.K.; Lee, H.J.; Suh, Y.H.; Han, K.L.; Lim, J.H.; Song, J.; Seong, J.K.; Jung, M.H. Crucial roles of neuronatin in insulin secretion and high glucose-induced apoptosis in pancreatic beta-cells. Cell. Signal. 2008, 20, 907–915. [Google Scholar] [CrossRef]
- Hoffmann, A.; Spengler, D. Transient neonatal diabetes mellitus gene Zac1 impairs insulin secretion in mice through Rasgrf1. Mol. Cell. Biol. 2012, 32, 2549–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avrahami, D.; Li, C.; Yu, M.; Jiao, Y.; Zhang, J.; Naji, A.; Ziaie, S.; Glaser, B.; Kaestner, K.H. Targeting the cell cycle inhibitor p57Kip2 promotes adult human beta cell replication. J. Clin. Investig. 2014, 124, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Ou, K.; Yu, M.; Moss, N.G.; Wang, Y.J.; Wang, A.W.; Nguyen, S.C.; Jiang, C.; Feleke, E.; Kameswaran, V.; Joyce, E.F.; et al. Targeted demethylation at the CDKN1C/p57 locus induces human beta cell replication. J. Clin. Investig. 2019, 129, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, M.; Lee, S.H.; Kim, J.W.; Ham, D.S.; Park, H.S.; Yang, H.K.; Shin, J.Y.; Cho, J.H.; Kim, Y.B.; Youn, B.S.; et al. Preadipocyte factor 1 induces pancreatic ductal cell differentiation into insulin-producing cells. Sci. Rep. 2016, 6, 23960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sojoodi, M.; Stradiot, L.; Tanaka, K.; Heremans, Y.; Leuckx, G.; Besson, V.; Staels, W.; Van de Casteele, M.; Marazzi, G.; Sassoon, D.; et al. The zinc finger transcription factor PW1/PEG3 restrains murine beta cell cycling. Diabetologia 2016, 59, 1474–1479. [Google Scholar] [CrossRef] [Green Version]
- Prokopenko, I.; Poon, W.; Magi, R.; Prasad, B.R.; Salehi, S.A.; Almgren, P.; Osmark, P.; Bouatia-Naji, N.; Wierup, N.; Fall, T.; et al. A central role for GRB10 in regulation of islet function in man. PLoS Genet. 2014, 10, e1004235. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, N.; Liu, M.; Li, X.; Zhou, L.; Huang, W.; Xu, Z.; Liu, J.; Musi, N.; DeFronzo, R.A.; et al. Disruption of growth factor receptor-binding protein 10 in the pancreas enhances beta-cell proliferation and protects mice from streptozotocin-induced beta-cell apoptosis. Diabetes 2012, 61, 3189–3198. [Google Scholar] [CrossRef] [Green Version]
- Font de Mora, J.; Esteban, L.M.; Burks, D.J.; Nunez, A.; Garces, C.; Garcia-Barrado, M.J.; Iglesias-Osma, M.C.; Moratinos, J.; Ward, J.M.; Santos, E. Ras-GRF1 signaling is required for normal beta-cell development and glucose homeostasis. EMBO J. 2003, 22, 3039–3049. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, Y.; Xie, M.; Wang, L.; Jin, F.; Li, Y.; Yuan, Q.; De, W. Long Noncoding RNA Meg3 Regulates Mafa Expression in Mouse Beta Cells by Inactivating Rad21, Smc3 or Sin3alpha. Cell Physiol. Biochem. 2018, 45, 2031–2043. [Google Scholar] [CrossRef]
- Kameswaran, V.; Golson, M.L.; Ramos-Rodriguez, M.; Ou, K.; Wang, Y.J.; Zhang, J.; Pasquali, L.; Kaestner, K.H. The Dysregulation of the DLK1-MEG3 Locus in Islets from Patients With Type 2 Diabetes is Mimicked by Targeted Epimutation of Its Promoter with TALE-DNMT Constructs. Diabetes 2018, 67, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Parra, C.; Jacovetti, C.; Dumortier, O.; Lee, K.; Peyot, M.L.; Guay, C.; Prentki, M.; Laybutt, D.R.; Van Obberghen, E.; Regazzi, R. Contribution of the Long Noncoding RNA H19 to beta-Cell Mass Expansion in Neonatal and Adult Rodents. Diabetes 2018, 67, 2254–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamato, E.; Tashiro, F.; Miyazaki, J. Microarray analysis of novel candidate genes responsible for glucose-stimulated insulin secretion in mouse pancreatic beta cell line MIN6. PLoS ONE 2013, 8, e61211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadista, J.; Vikman, P.; Laakso, E.O.; Mollet, I.G.; Esguerra, J.L.; Taneera, J.; Storm, P.; Osmark, P.; Ladenvall, C.; Prasad, R.B.; et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 13924–13929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, N.; George, J.; Bolisetty, M.; Kursawe, R.; Sun, L.; Sivakamasundari, V.; Kycia, I.; Robson, P.; Stitzel, M.L. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 2017, 27, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Dayeh, T.; Volkov, P.; Salo, S.; Hall, E.; Nilsson, E.; Olsson, A.H.; Kirkpatrick, C.L.; Wollheim, C.B.; Eliasson, L.; Ronn, T.; et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014, 10, e1004160. [Google Scholar] [CrossRef]
- Kong, A.; Steinthorsdottir, V.; Masson, G.; Thorleifsson, G.; Sulem, P.; Besenbacher, S.; Jonasdottir, A.; Sigurdsson, A.; Kristinsson, K.T.; Jonasdottir, A.; et al. Parental origin of sequence variants associated with complex diseases. Nature 2009, 462, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Miyake, K.; Horikawa, Y.; Hara, K.; Osawa, H.; Furuta, H.; Hirota, Y.; Mori, H.; Jonsson, A.; Sato, Y.; et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 2008, 40, 1092–1097. [Google Scholar] [CrossRef]
- Unoki, H.; Takahashi, A.; Kawaguchi, T.; Hara, K.; Horikoshi, M.; Andersen, G.; Ng, D.P.; Holmkvist, J.; Borch-Johnsen, K.; Jorgensen, T.; et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 2008, 40, 1098–1102. [Google Scholar] [CrossRef]
- Rampersaud, E.; Damcott, C.M.; Fu, M.; Shen, H.; McArdle, P.; Shi, X.; Shelton, J.; Yin, J.; Chang, Y.P.; Ott, S.H.; et al. Identification of novel candidate genes for type 2 diabetes from a genome-wide association scan in the Old Order Amish: Evidence for replication from diabetes-related quantitative traits and from independent populations. Diabetes 2007, 56, 3053–3062. [Google Scholar] [CrossRef] [Green Version]
- Smith, F.M.; Holt, L.J.; Garfield, A.S.; Charalambous, M.; Koumanov, F.; Perry, M.; Bazzani, R.; Sheardown, S.A.; Hegarty, B.D.; Lyons, R.J.; et al. Mice with a disruption of the imprinted Grb10 gene exhibit altered body composition, glucose homeostasis, and insulin signaling during postnatal life. Mol. Cell. Biol. 2007, 27, 5871–5886. [Google Scholar] [CrossRef] [Green Version]
- Salomon, D.; Meda, P. Heterogeneity and contact-dependent regulation of hormone secretion by individual B cells. Exp. Cell Res. 1986, 162, 507–520. [Google Scholar] [CrossRef]
- Bosco, D.; Meda, P. Actively synthesizing beta-cells secrete preferentially after glucose stimulation. Endocrinology 1991, 129, 3157–3166. [Google Scholar] [CrossRef] [PubMed]
- Kiekens, R.; In’t Veld, P.; Mahler, T.; Schuit, F.; Van De Winkel, M.; Pipeleers, D. Differences in glucose recognition by individual rat pancreatic B cells are associated with intercellular differences in glucose-induced biosynthetic activity. J. Clin. Investig. 1992, 89, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Van Schravendijk, C.F.; Kiekens, R.; Pipeleers, D.G. Pancreatic beta cell heterogeneity in glucose-induced insulin secretion. J. Biol. Chem. 1992, 267, 21344–21348. [Google Scholar] [CrossRef]
- Giordano, E.; Bosco, D.; Cirulli, V.; Meda, P. Repeated glucose stimulation reveals distinct and lasting secretion patterns of individual rat pancreatic B cells. J. Clin. Investig. 1991, 87, 2178–2185. [Google Scholar] [CrossRef] [Green Version]
- Wojtusciszyn, A.; Armanet, M.; Morel, P.; Berney, T.; Bosco, D. Insulin secretion from human beta cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia 2008, 51, 1843–1852. [Google Scholar] [CrossRef] [Green Version]
- Soria, B.; Chanson, M.; Giordano, E.; Bosco, D.; Meda, P. Ion channels of glucose-responsive and -unresponsive beta-cells. Diabetes 1991, 40, 1069–1078. [Google Scholar] [CrossRef]
- Holz, G.G.t.; Kuhtreiber, W.M.; Habener, J.F. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7-37). Nature 1993, 361, 362–365. [Google Scholar] [CrossRef] [Green Version]
- Jetton, T.L.; Magnuson, M.A. Heterogeneous expression of glucokinase among pancreatic beta cells. Proc. Natl. Acad. Sci. USA 1992, 89, 2619–2623. [Google Scholar] [CrossRef] [Green Version]
- Heimberg, H.; De Vos, A.; Vandercammen, A.; Van Schaftingen, E.; Pipeleers, D.; Schuit, F. Heterogeneity in glucose sensitivity among pancreatic beta-cells is correlated to differences in glucose phosphorylation rather than glucose transport. EMBO J. 1993, 12, 2873–2879. [Google Scholar] [CrossRef]
- Katsuta, H.; Aguayo-Mazzucato, C.; Katsuta, R.; Akashi, T.; Hollister-Lock, J.; Sharma, A.J.; Bonner-Weir, S.; Weir, G.C. Subpopulations of GFP-marked mouse pancreatic beta-cells differ in size, granularity, and insulin secretion. Endocrinology 2012, 153, 5180–5187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrell, C.; Schug, J.; Canaday, P.S.; Russ, H.A.; Tarlow, B.D.; Grompe, M.T.; Horton, T.; Hebrok, M.; Streeter, P.R.; Kaestner, K.H.; et al. Human islets contain four distinct subtypes of beta cells. Nat. Commun. 2016, 7, 11756. [Google Scholar] [CrossRef] [PubMed]
- Karaca, M.; Castel, J.; Tourrel-Cuzin, C.; Brun, M.; Geant, A.; Dubois, M.; Catesson, S.; Rodriguez, M.; Luquet, S.; Cattan, P.; et al. Exploring functional beta-cell heterogeneity in vivo using PSA-NCAM as a specific marker. PLoS ONE 2009, 4, e5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.B.; Groop, L. Single-Cell Sequencing of Human Pancreatic Islets-New Kids on the Block. Cell Metab. 2016, 24, 523–524. [Google Scholar] [CrossRef] [Green Version]
- Carrano, A.C.; Mulas, F.; Zeng, C.; Sander, M. Interrogating islets in health and disease with single-cell technologies. Mol. Metab. 2017, 6, 991–1001. [Google Scholar] [CrossRef]
- Wang, Y.J.; Kaestner, K.H. Single-Cell RNA-Seq of the Pancreatic Islets—A Promise not yet Fulfilled? Cell Metab. 2019, 29, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Klughammer, J.; Farlik, M.; Penz, T.; Spittler, A.; Barbieux, C.; Berishvili, E.; Bock, C.; Kubicek, S. Single-cell transcriptomes reveal characteristic features of human pancreatic islet cell types. EMBO Rep. 2016, 17, 178–187. [Google Scholar] [CrossRef]
- Wang, Y.J.; Schug, J.; Won, K.J.; Liu, C.; Naji, A.; Avrahami, D.; Golson, M.L.; Kaestner, K.H. Single-Cell Transcriptomics of the Human Endocrine Pancreas. Diabetes 2016, 65, 3028–3038. [Google Scholar] [CrossRef] [Green Version]
- Segerstolpe, A.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andreasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016, 3, 346–360.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grun, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camunas-Soler, J.; Dai, X.Q.; Hang, Y.; Bautista, A.; Lyon, J.; Suzuki, K.; Kim, S.K.; Quake, S.R.; MacDonald, P.E. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab. 2020, 31, 1017–1031.e4. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, G.D.; Gromada, J.; Sussel, L. Heterogeneity of the Pancreatic Beta Cell. Front. Genet. 2017, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Benninger, R.K.P.; Hodson, D.J. New Understanding of beta-Cell Heterogeneity and In Situ Islet Function. Diabetes 2018, 67, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Xavier, G.; Rutter, G.A. Metabolic and Functional Heterogeneity in Pancreatic beta Cells. J. Mol. Biol. 2020, 432, 1395–1406. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Migliorini, A.; Gegg, M.; Lickert, H. Impact of islet architecture on beta-cell heterogeneity, plasticity and function. Nat. Rev. Endocrinol. 2016, 12, 695–709. [Google Scholar] [CrossRef]
- Benninger, R.K.; Hutchens, T.; Head, W.S.; McCaughey, M.J.; Zhang, M.; Le Marchand, S.J.; Satin, L.S.; Piston, D.W. Intrinsic islet heterogeneity and gap junction coupling determine spatiotemporal Ca2+ wave dynamics. Biophys. J. 2014, 107, 2723–2733. [Google Scholar] [CrossRef] [Green Version]
- Serre-Beinier, V.; Le Gurun, S.; Belluardo, N.; Trovato-Salinaro, A.; Charollais, A.; Haefliger, J.A.; Condorelli, D.F.; Meda, P. Cx36 preferentially connects beta-cells within pancreatic islets. Diabetes 2000, 49, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Bavamian, S.; Klee, P.; Britan, A.; Populaire, C.; Caille, D.; Cancela, J.; Charollais, A.; Meda, P. Islet-cell-to-cell communication as basis for normal insulin secretion. Diabetes Obes. Metab. 2007, 9 (Suppl. 2), 118–132. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.; Haefliger, J.A.; Meda, P. Connexins: Key mediators of endocrine function. Physiol. Rev. 2011, 91, 1393–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnsworth, N.L.; Hemmati, A.; Pozzoli, M.; Benninger, R.K. Fluorescence recovery after photobleaching reveals regulation and distribution of connexin36 gap junction coupling within mouse islets of Langerhans. J. Physiol. 2014, 592, 4431–4446. [Google Scholar] [CrossRef] [PubMed]
- Gosak, M.; Markovic, R.; Dolensek, J.; Slak Rupnik, M.; Marhl, M.; Stozer, A.; Perc, M. Network science of biological systems at different scales: A review. Phys. Life Rev. 2018, 24, 118–135. [Google Scholar] [CrossRef]
- Hodson, D.J.; Schaeffer, M.; Romano, N.; Fontanaud, P.; Lafont, C.; Birkenstock, J.; Molino, F.; Christian, H.; Lockey, J.; Carmignac, D.; et al. Existence of long-lasting experience-dependent plasticity in endocrine cell networks. Nat. Commun. 2012, 3, 605. [Google Scholar] [CrossRef]
- Salem, V.; Silva, L.D.; Suba, K.; Georgiadou, E.; Neda Mousavy Gharavy, S.; Akhtar, N.; Martin-Alonso, A.; Gaboriau, D.C.A.; Rothery, S.M.; Stylianides, T.; et al. Leader beta-cells coordinate Ca2+ dynamics across pancreatic islets in vivo. Nat. Metab. 2019, 1, 615–629. [Google Scholar] [CrossRef] [Green Version]
- Westacott, M.J.; Ludin, N.W.F.; Benninger, R.K.P. Spatially Organized beta-Cell Subpopulations Control Electrical Dynamics across Islets of Langerhans. Biophys. J. 2017, 113, 1093–1108. [Google Scholar] [CrossRef] [Green Version]
- Hodson, D.J.; Mitchell, R.K.; Bellomo, E.A.; Sun, G.; Vinet, L.; Meda, P.; Li, D.; Li, W.H.; Bugliani, M.; Marchetti, P.; et al. Lipotoxicity disrupts incretin-regulated human beta cell connectivity. J. Clin. Investig. 2013, 123, 4182–4194. [Google Scholar] [CrossRef] [Green Version]
- Arrojo, E.D.R.; Jacob, S.; Garcia-Prieto, C.F.; Zheng, X.; Fukuda, M.; Nhu, H.T.T.; Stelmashenko, O.; Pecanha, F.L.M.; Rodriguez-Diaz, R.; Bushong, E.; et al. Structural basis for delta cell paracrine regulation in pancreatic islets. Nat. Commun. 2019, 10, 3700. [Google Scholar] [CrossRef]
- Satin, L.S.; Zhang, Q.; Rorsman, P. “Take Me To Your Leader”: An Electrophysiological Appraisal of the Role of Hub Cells in Pancreatic Islets. Diabetes 2020, 69, 830–836. [Google Scholar] [CrossRef]
- Satin, L.S.; Rorsman, P. Response to Comment on Satin et al. “Take Me To Your Leader”: An Electrophysiological Appraisal of the Role of Hub Cells in Pancreatic Islets. Diabetes 2020, 69, e12–e13. [Google Scholar] [CrossRef] [PubMed]
- Dwulet, J.M.; Briggs, J.K.; Benninger, R.K.P. Small subpopulations of beta cells do not drive islet oscillatory [Ca2+] dynamics via gap junction communication. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lu, T.T.-H.; Heyne, S.; Dror, E.; Casas, E.; Leonhardt, L.; Boenke, T.; Yang, C.-H.; Arrigoni, L.; Dalgaard, K.; Teperino, R.; et al. The Polycomb-Dependent Epigenome Controls beta Cell Dysfunction, Dedifferentiation, and Diabetes. Cell Metab. 2018, 27, 1294–1308.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hajj, N.; Schneider, E.; Lehnen, H.; Haaf, T. Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment. Reproduction 2014, 148, R111–R120. [Google Scholar] [CrossRef] [PubMed]
- Soubry, A.; Schildkraut, J.M.; Murtha, A.; Wang, F.; Huang, Z.; Bernal, A.; Kurtzberg, J.; Jirtle, R.L.; Murphy, S.K.; Hoyo, C. Paternal obesity is associated with IGF2 hypomethylation in newborns: Results from a Newborn Epigenetics Study (NEST) cohort. BMC Med. 2013, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Soubry, A.; Murphy, S.K.; Wang, F.; Huang, Z.; Vidal, A.C.; Fuemmeler, B.F.; Kurtzberg, J.; Murtha, A.; Jirtle, R.L.; Schildkraut, J.M.; et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int. J. Obes. (Lond.) 2015, 39, 650–657. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef]
- Van de Pette, M.; Abbas, A.; Feytout, A.; McNamara, G.; Bruno, L.; To, W.K.; Dimond, A.; Sardini, A.; Webster, Z.; McGinty, J.; et al. Visualizing Changes in Cdkn1c Expression Links Early-Life Adversity to Imprint Mis-regulation in Adults. Cell Rep. 2017, 18, 1090–1099. [Google Scholar] [CrossRef] [Green Version]
- Van de Pette, M.; Galvao, A.; Millership, S.J.; To, W.K.; Dimond, A.; Prodani, C.; McNamara, G.; Bruno, L.; Sardini, A.; Webster, Z.; et al. Epigenetic change induced by utero dietary challenge provokes phenotypic variability across multiple generations of mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Buklijas, T.; Low, F.M.; Beedle, A.S. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat. Rev. Endocrinol. 2009, 5, 401–408. [Google Scholar] [CrossRef]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.F.; Lin, R.C.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 2010, 467, 963–966. [Google Scholar] [CrossRef] [PubMed]

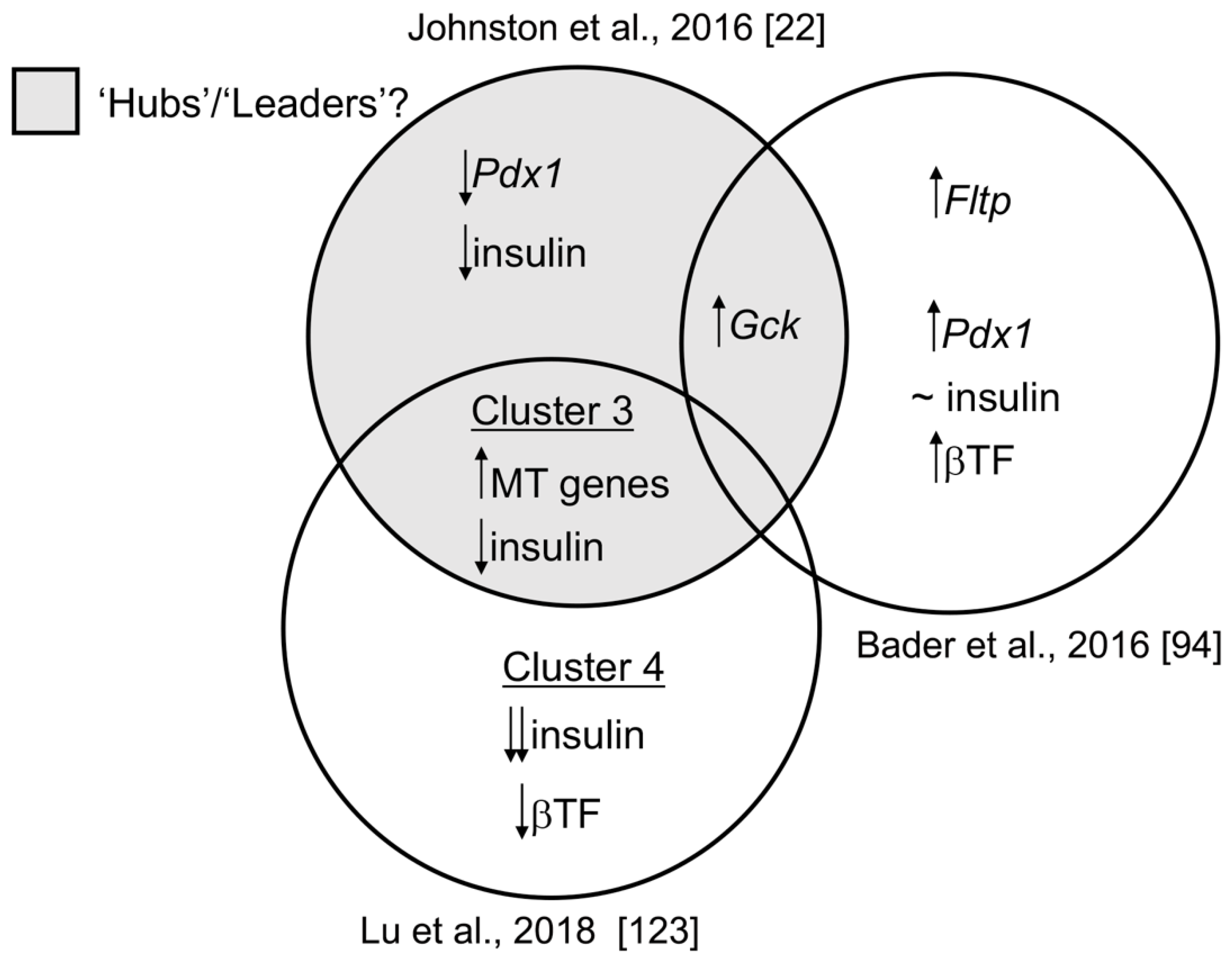
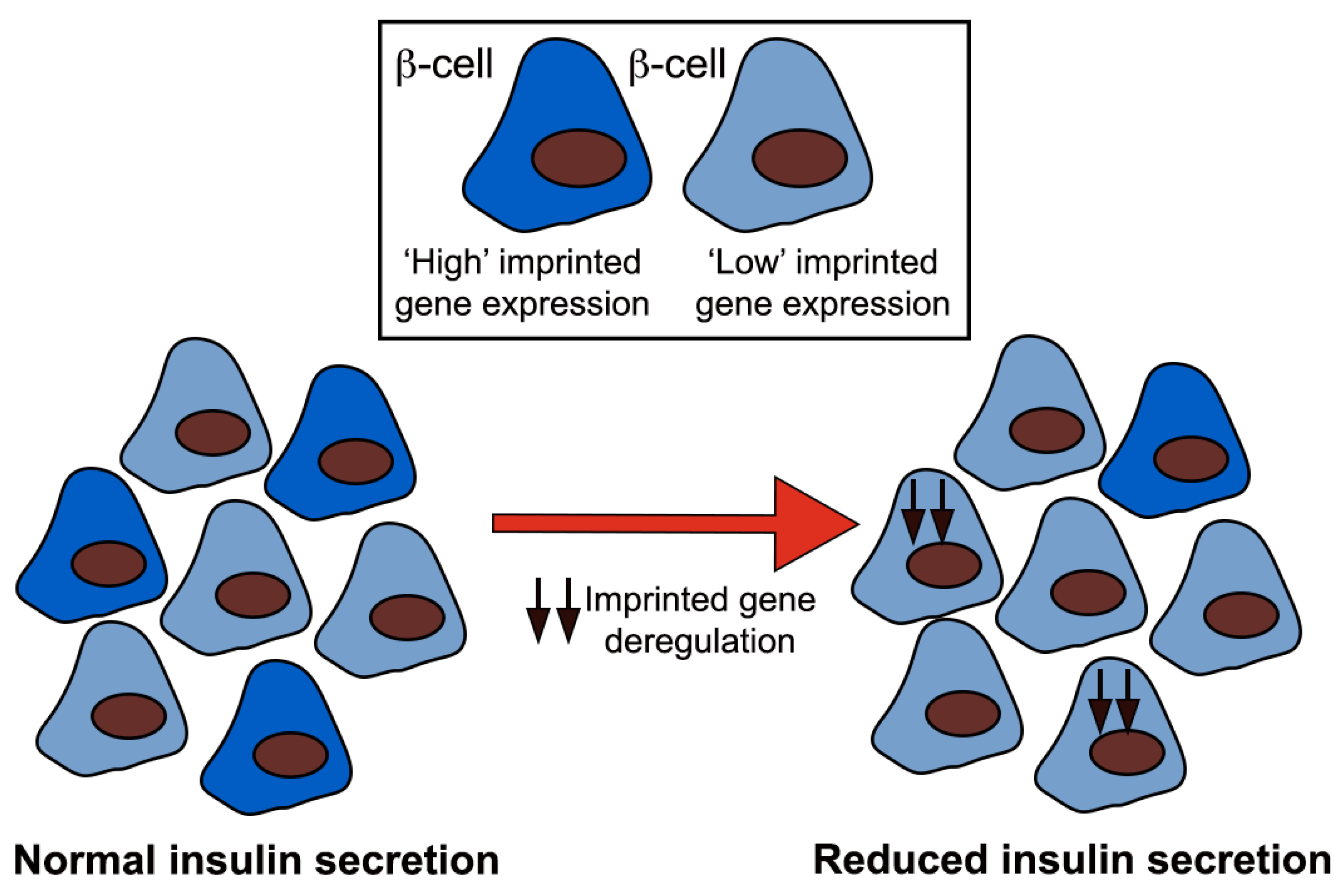

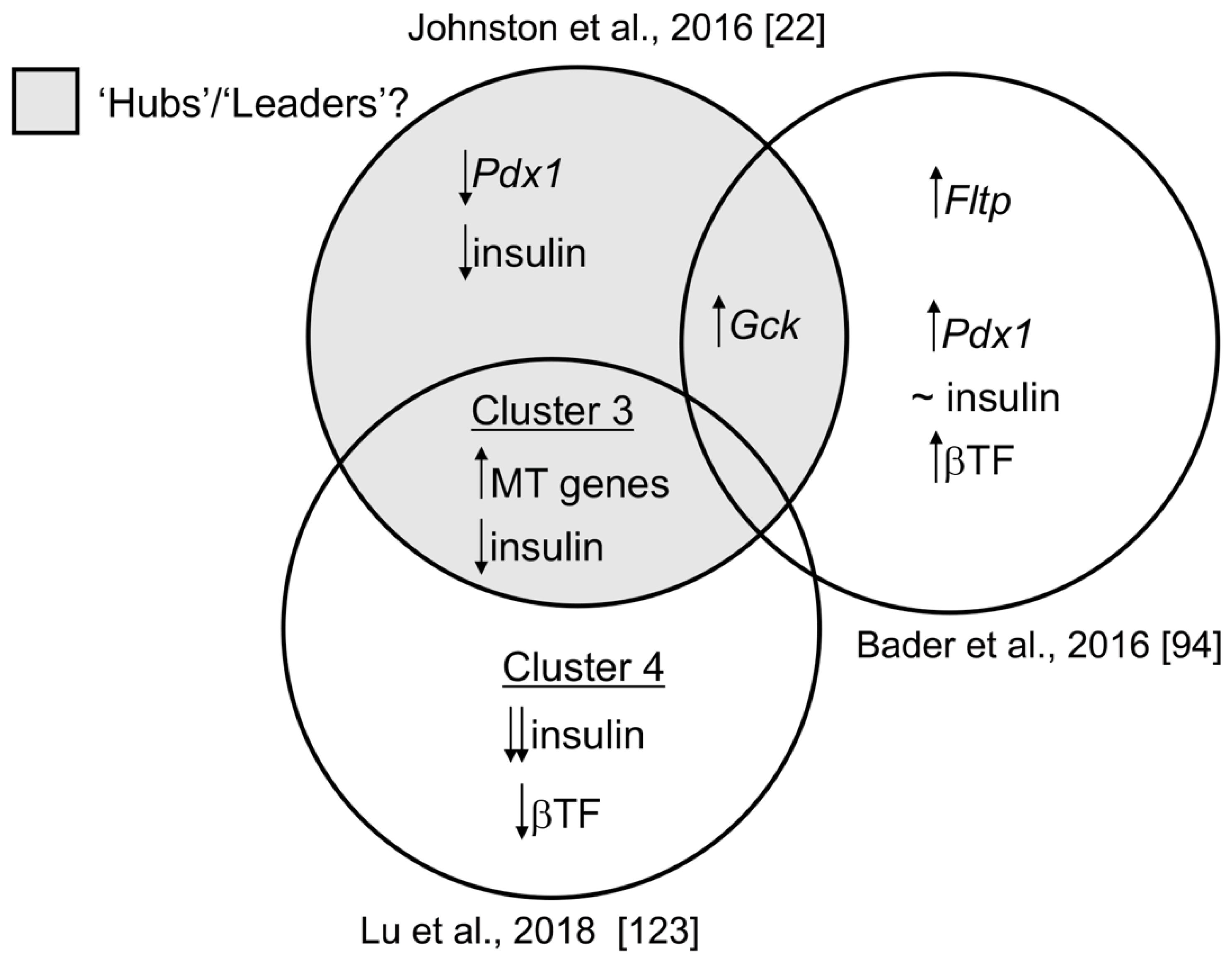
{kind=link}
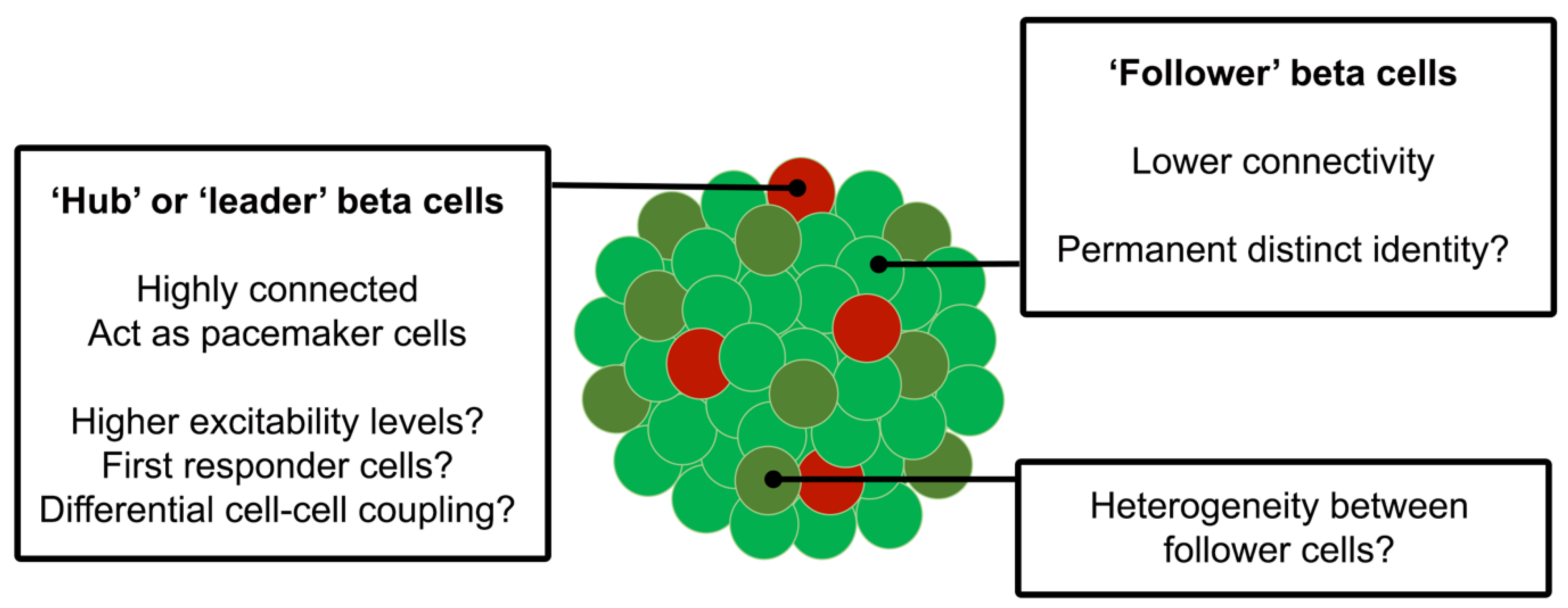
{kind=link}
{kind=link}
| Imprinted Gene | Proposed Functional Role in Beta Cells | Effect of Altered Expression in Beta Cells | References |
|---|---|---|---|
| Cdkn1c | Cell cycle control | Increased beta cell replication upon knockdown in human islets | [62,63] |
| Dlk1 | Cellular differentiation | Overexpression resulted in differentiation of human pancreatic ductal cells into beta-like cells and an increase in insulin secretion | [64] |
| Grb10 | Receptor tyrosine kinase adaptor protein | Knockdown in human islets reduced insulin secretion. However, increased beta cell mass, insulin secretion and improved whole body glucose tolerance in knockout mice | [66,67,80] |
| Gtl2 | Long non-coding RNA | Knockdown in stable mouse beta cells increased sensitivity to cytokine-mediated oxidative stress | [70] |
| H19 | Long non-coding RNA | Knockdown decreased rat beta cell expansion | [71] |
| Nnat | Mediator of preproinsulin processing | Knockout in mice leads to reduced beta cell insulin content, glucose-stimulated insulin secretion (GSIS) and glucose tolerance | [59,60] |
| Peg3 | Zinc finger protein, regulates apoptosis | Viral-mediated knockdown in vitro activates beta cell cycling | [65] |
| Plagl1 | Zinc finger protein, suppresses cell growth | Transient neonatal diabetes upon overexpression of Plagl1 in mice | [52,57] |
| Rasgrf1 | Guanine nucleotide exchange factor | Knockout in mice leads to reduced beta cell mass, hypoinsulinaemia and impaired glucose tolerance | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chabosseau, P.; Rutter, G.A.; Millership, S.J. Importance of Both Imprinted Genes and Functional Heterogeneity in Pancreatic Beta Cells: Is There a Link? Int. J. Mol. Sci. 2021, 22, 1000. https://doi.org/10.3390/ijms22031000
Chabosseau P, Rutter GA, Millership SJ. Importance of Both Imprinted Genes and Functional Heterogeneity in Pancreatic Beta Cells: Is There a Link? International Journal of Molecular Sciences. 2021; 22(3):1000. https://doi.org/10.3390/ijms22031000
Chicago/Turabian StyleChabosseau, Pauline, Guy A. Rutter, and Steven J. Millership. 2021. "Importance of Both Imprinted Genes and Functional Heterogeneity in Pancreatic Beta Cells: Is There a Link?" International Journal of Molecular Sciences 22, no. 3: 1000. https://doi.org/10.3390/ijms22031000
APA StyleChabosseau, P., Rutter, G. A., & Millership, S. J. (2021). Importance of Both Imprinted Genes and Functional Heterogeneity in Pancreatic Beta Cells: Is There a Link? International Journal of Molecular Sciences, 22(3), 1000. https://doi.org/10.3390/ijms22031000