
Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
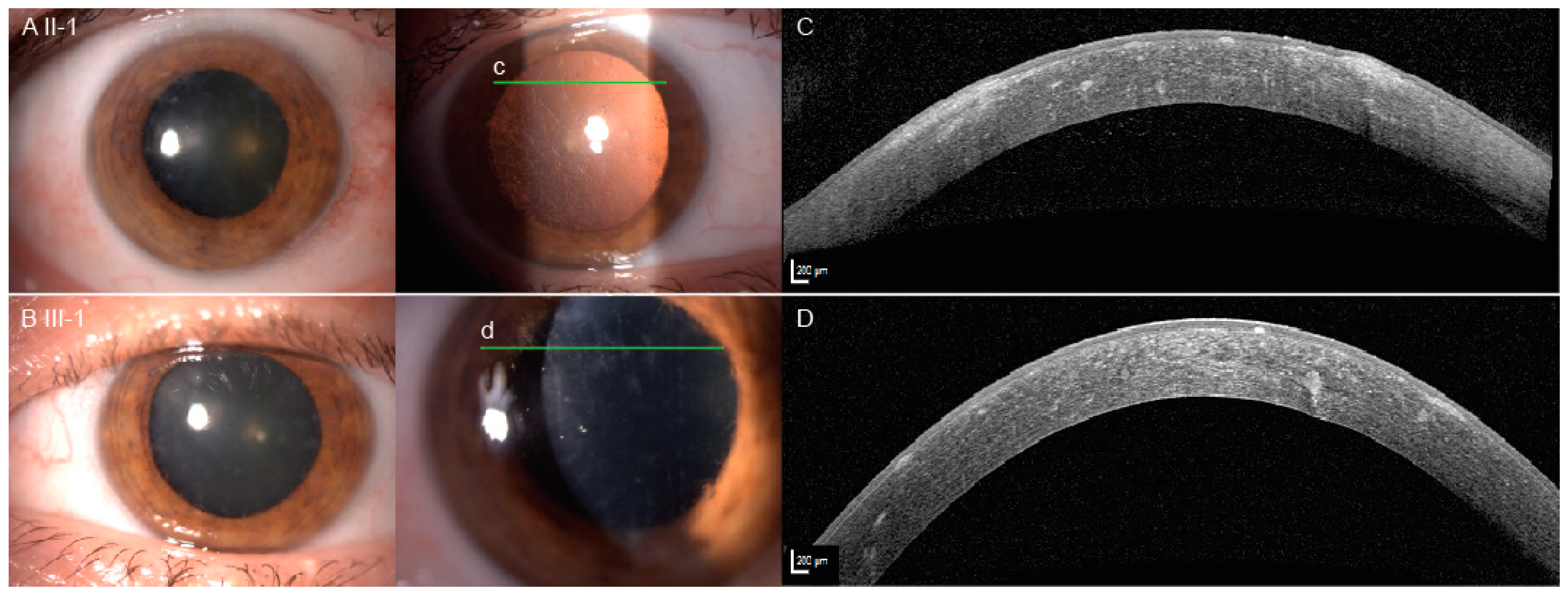
2.1. Clinical Presentation
2.2. Molecular Analysis
2.3. Pathological Findings
2.4. Review of the Phenotypes Associated with GSN Mutations
3. Discussion
3.1. Optic Nerve Involvement
3.2. Pathogenetic Mechanisms and Genotype-Phenotype Correlations
4. Materials and Methods
4.1. Patients
4.2. Clinical Examination
4.3. Genetic and Bioinformatic Analysis
4.4. In Silico Analysis
4.5. Pathological Analysis
4.6. Review of the Literature
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GSN | Gelsolin gene |
| RE | Right eye |
| LE | Left eye |
| OCT | Optical coherence tomography |
| RNFL | Retinal nerve fiber layer |
| ID | Identity document |
| DOB | Date of birth |
| N/A | Not available |
References
- Solomon, J.P.; Page, L.J.; Balch, W.E.; Kelly, J.W. Gelsolin amyloidosis: Genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 282–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, H.; Burtnick, L.D.; Mejillano, M.; Yin, H.L.; Robinson, R.C.; Choe, S. The calcium activation of gelsolin: Insights from the 3A structure of the G4-G6/actin complex. J. Mol. Biol. 2002, 324, 691–702. [Google Scholar] [CrossRef]
- Meretoja, J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann. Clin. Res. 1969, 1, 314–324. [Google Scholar] [PubMed]
- de la Chapelle, A.; Tolvanen, R.; Boysen, G.; Santavy, J.; Bleeker-Wagemakers, L.; Maury, C.P.; Kere, J. Gelsolin-derived familial amyloidosis caused by asparagine or tyrosine substitution for aspartic acid at residue 187. Nat. Genet. 1992, 2, 157–160. [Google Scholar] [CrossRef]
- Maury, C.P.; Liljeström, M.; Boysen, G.; Törnroth, T.; de la Chapelle, A.; Nurmiaho-Lassila, E.L. Danish type gelsolin related amyloidosis: 654G-T mutation is associated with a disease pathogenetically and clinically similar to that caused by the 654G-A mutation (familial amyloidosis of the Finnish type). J. Clin. Pathol. 2000, 53, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Giorgino, T.; Mattioni, D.; Hassan, A.; Milani, M.; Mastrangelo, E.; Barbiroli, A.; Verhelle, A.; Gettemans, J.; Barzago, M.M.; Diomede, L.; et al. Nanobody interaction unveils structure, dynamics and proteotoxicity of the Finnish-type amyloidogenic gelsolin variant. Biochim. Biophys. Acta Mol.r Basis Dis. 2019, 1865, 648–660. [Google Scholar] [CrossRef]
- Zorgati, H.; Larsson, M.; Ren, W.; Sim, A.Y.L.; Gettemans, J.; Grimes, J.M.; Li, W.; Robinson, R.C. The role of gelsolin domain 3 in familial amyloidosis (Finnish type). Proc. Natl. Acad. Sci. USA 2019, 116, 13958–13963. [Google Scholar] [CrossRef] [Green Version]
- Kiuru, S. Familial amyloidosis of the Finnish type (FAF) A clinical study of 30 patients. Acta Neurol. Scand. 1992, 86, 346–353. [Google Scholar] [CrossRef]
- Dammacco, R.; Merlini, G.; Lisch, W.; Kivela, T.T.; Giancipoli, E.; Vacca, A.; Dammacco, F. Amyloidosis and Ocular Involvement: An Overview. Semin. Ophthalmol. 2020, 35, 7–26. [Google Scholar] [CrossRef]
- Casal, I.; Monteiro, S.; Abreu, C.; Neves, M.; Oliveira, L.; Beirao, M. Meretoja’s Syndrome: Lattice Corneal Dystrophy, Gelsolin Type. Case Rep. Med. 2017, 2017, 2843417. [Google Scholar] [CrossRef] [Green Version]
- Kivela, T.; Tarkkanen, A.; Frangione, B.; Ghiso, J.; Haltia, M. Ocular amyloid deposition in familial amyloidosis, Finnish: An analysis of native and variant gelsolin in Meretoja’s syndrome. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3759–3769. [Google Scholar]
- Carrwik, C.; Stenevi, U. Lattice corneal dystrophy, gelsolin type (Meretoja’s syndrome). Acta Ophthalmol. 2009, 87, 813–819. [Google Scholar] [CrossRef]
- Feng, X.; Zhu, H.; Zhao, T.; Hou, Y.; Liu, J. A new heterozygous G duplicate in exon1 (c.100dupG) of gelsolin gene causes Finnish gelsolin amyloidosis in a Chinese family. Brain Behav. 2018, 8, e01151. [Google Scholar] [CrossRef] [Green Version]
- Sethi, S.; Theis, J.D.; Quint, P.; Maierhofer, W.; Kurtin, P.J.; Dogan, A.; Highsmith, E.W. Renal amyloidosis associated with a novel sequence variant of gelsolin. Am. J. Kidney Dis. 2013, 61, 161–166. [Google Scholar] [CrossRef]
- Efebera, Y.A.; Sturm, A.; Baack, E.C.; Hofmeister, C.C.; Satoskar, A.; Nadasdy, T.; Nadasdy, G.; Benson, D.M.; Gillmore, J.D.; Hawkins, P.N.; et al. Novel gelsolin variant as the cause of nephrotic syndrome and renal amyloidosis in a large kindred. Amyloid 2014, 21, 110–112. [Google Scholar] [CrossRef]
- Oregel, K.Z.; Shouse, G.P.; Oster, C.; Martinez, F.; Wang, J.; Rosenzweig, M.; Deisch, J.K.; Chen, C.S.; Nagaraj, G. Atypical Presentation of Gelsolin Amyloidosis in a Man of African Descent with a Novel Mutation in the Gelsolin Gene. Am. J. Case Rep. 2018, 19, 374–381. [Google Scholar] [CrossRef]
- Cabral-Macias, J.; Garcia-Montaño, L.A.; Pérezpeña-Díazconti, M.; Aguilar, M.C.; Garcia, G.; Vencedor-Meraz, C.I.; Graue-Hernandez, E.O.; Chacón-Camacho, O.F.; Zenteno, J.C. Clinical, histopathological, and in silico pathogenicity analyses in a pedigree with familial amyloidosis of the Finnish type (Meretoja syndrome) caused by a novel gelsolin mutation. Mol. Vis. 2020, 26, 345–354. [Google Scholar]
- Sridharan, M.; Highsmith, W.E.; Kurtin, P.J.; Zimmermann, M.T.; Theis, J.D.; Dasari, S.; Dingli, D. A Patient with Hereditary ATTR and a Novel AGel p.Ala578Pro Amyloidosis. Mayo Clin. Proc. 2018, 93, 1678–1682. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- de la Chapelle, A.; Kere, J.; Sack, G.H.; Tolvanen, R.; Maury, C.P. Familial amyloidosis, Finnish type: G654—A mutation of the gelsolin gene in Finnish families and an unrelated American family. Genomics 1992, 13, 898–901. [Google Scholar] [CrossRef]
- Neri, A.; Rubino, P.; Macaluso, C.; Gandolfi, S.A. Light-chain amyloidosis mimicking giant cell arteritis in a bilateral anterior ischemic optic neuropathy case. BMC Ophthalmol. 2013, 13, 82. [Google Scholar] [CrossRef] [Green Version]
- Kanaan, M.Z.; Lorenzi, A.R.; Thampy, N.; Pandit, R.; Dayan, M. Bilateral Non-arteritic Anterior Ischaemic Optic Neuropathy as the Presentation of Systemic Amyloidosis. Neuro Ophthalmol. 2017, 41, 330–334. [Google Scholar] [CrossRef]
- Burtnick, L.D.; Koepf, E.K.; Grimes, J.; Jones, E.Y.; Stuart, D.I.; McLaughlin, P.J.; Robinson, R.C. The crystal structure of plasma gelsolin: Implications for actin severing, capping, and nucleation. Cell 1997, 90, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Boni, F.; Milani, M.; Porcari, R.; Barbiroli, A.; Ricagno, S.; de Rosa, M. Molecular basis of a novel renal amyloidosis due to N184K gelsolin variant. Sci. Rep. 2016, 6, 33463. [Google Scholar] [CrossRef] [Green Version]
- Boni, F.; Milani, M.; Barbiroli, A.; Diomede, L.; Mastrangelo, E.; de Rosa, M. Gelsolin pathogenic Gly167Arg mutation promotes domain-swap dimerization of the protein. Hum. Mol. Genet. 2018, 27, 53–65. [Google Scholar] [CrossRef]
- Stefl, S.; Nishi, H.; Petukh, M.; Panchenko, A.R.; Alexov, E. Molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013, 425, 3919–3936. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Meynert, A.M.; Bicknell, L.S.; Hurles, M.E.; Jackson, A.P.; Taylor, M.S. Quantifying single nucleotide variant detection sensitivity in exome sequencing. BMC Bioinform. 2013, 14, 195. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Ma, Q.; Wang, H.; Chumnarnsilpa, S.; Lee, W.L.; Larsson, M.; Kannan, B.; Hernandez-Valladares, M.; Burtnick, L.D.; Robinson, R.C. Ca2+ binding by domain 2 plays a critical role in the activation and stabilization of gelsolin. Proc. Natl. Acad. Sci. USA 2009, 106, 13713–13718. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.L.; Kwiatkowski, D.J.; Mole, J.E.; Cole, F.S. Structure and biosynthesis of cytoplasmic and secreted variants of gelsolin. J. Biol. Chem. 1984, 259, 5271–5276. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | DOB (Age) | Genotype | Clinical Presentation | |||||
|---|---|---|---|---|---|---|---|---|---|
| Corneal Lattice Dystrophy | Loose Skin | Cranial Neuropathy | Heart Arrhythmia | Renal Involvement | Other Phenotypic Features | ||||
| II-1 * | F | 1956 (64 y) | c.1738G>A (p.Glu580Lys) | +(diagnosed at 56 y) | +(dermatochalasis) | +(optic neuropathy) | - | cataract, brain calcifications, sensory ataxia, carpal tunnel syndrome, white matter lesions | |
| II-2 | F | 1948 (72 y) | c.1738G>A (p.Glu580Lys) | +(diagnosed at 50 y) | +(not specified) | - | ICD (63 y) | - | diabetes mellitus, insomnia |
| II-3 | F | 1951 (69 y) | c.1738G>A (p.Glu580Lys) | +(diagnosed at 44 y) | +(dermatochalasis) | - | ICD (65 y) | urolithiasis, renal cysts (diagnosed at 40 y) | angina pectoris, coronary angioplasty and stenting (50 y), hypertension |
| II-8 | M | 1954 (66 y) | c.1738G>A (p.Glu580Lys) | +(diagnosed at 60 y) | +(not specified) | - | ICD (62 y) | - | cataract, open angle glaucoma |
| III-1 * | F | 1974 (46 y) | c.1738G>A (p.Glu580Lys) | +(diagnosed at 40 years) | - | - | - | - | torticollis, tremor with sensory tic, blepharospasm, oromandibular dystonia |
| III-3 | M | 1981 (39 y) | c.1738G>A (p.Glu580Lys) | - | +(dermatochalasis) | - | - | urolithiasis (diagnosed at 28 y) | |
| II-5 | F | 1939 (81 y) | Wild type | - | - | - | - | - | hypertension, dilated cardiomyopathy, aortic valve stenosis with mechanical valve replacement |
| Base Change (NM_000177.4) | Amino Acid Change | Mature Plasma Protein Numbering (Devoid of the 27-aa Signal Peptide) | GnomAD Allele Frequency | Polyphen (Uniprot ID P06396) | Corneal Lattice Dystrophy | Other Clinical Features | Reference | Localization: Protein Domain (G1-6) * | Protein Destabilization | Susceptibility to Furin Proteolysis | Molecular Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| c.100dupG | p.Ala34fs | p.Ala7fs | 0 | / | not reported | seizures, brain lesions | Feng et al. 2018 [13] | / | N/A | N/A | |
| c.580G>A | p.Gly194Arg | p.Gly167Arg | 0.000039 | Probably damaging | not reported | renal involvement | Sethi et al. 2013 [14] | G2 | + | + | Bonì 2018 |
| c.633C>A | p.Asn211Lys | p.Asn184Lys | 0 | Probably damaging | not reported | renal involvement | Efebera et al. 2014 [15] | G2 | + | + | Bonì 2016 |
| c.640G>A | p.Asp214Asn+ | p.Asp187Asn# | 0.000007 | Probably damaging | yes | cutis laxa, cranial neuropathy, heart arrhythmia, renal involvement | Meretoja, Ann Clin Res. 1969 [3] | G2 (Ca-binding site) | + | + | Isaacson 1999 |
| c.640G>T | p.Asp214Tyr | p.Asp187Tyr | 0 | Probably damaging | yes | cutis laxa, cranial neuropathy | de la Chapelle et al. 1992 [20] | G2 (Ca-binding site) | + | + | Isaacson 1999 |
| c.1375C>G | p.Pro459Arg | p.Pro432Arg | 0.000004 | Possibly damaging | not reported | dermatomyositis -like | Oregel et al. 2018 [16] | G4 | N/A | N/A | |
| c.1631T>G | p.Met544Arg | p.Met517Arg | 0 | Probably damaging | yes | cutis laxa, peripheral neuropathy | Cabral-Macias et al. 2020 [17] | G4 (G4-G5 interface) | N/A | N/A | |
| c.1476del | p.Trp493GlyfsTer17 | p.Trp466fsTer17 | 0 | Probably damaging (GERP score 5.25) | N/A | N/A | ClinVar ID: 493491 https://www.ncbi.nlm.nih.gov/clinvar/(accessed on 5 December 2020) | G4 | N/A | N/A | |
| c.1732G>C | p.Ala578Pro | p.Ala551Pro | 0 | Probably damaging | not reported | cardiac involvement (additional mutation in TTR) | Sridharan et al. 2018 [18] | G5 (G4:G5 interface) | N/A | N/A | |
| c.1738G>A | p.Glu580Lys | p.Glu553Lys | 0 | Probably damaging | yes | cutis laxa, cranial neuropathy including the optic nerve, cardiac involvement | This report | G5 (G4:G5 interface) | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potrč, M.; Volk, M.; de Rosa, M.; Pižem, J.; Teran, N.; Jaklič, H.; Maver, A.; Drnovšek-Olup, B.; Bollati, M.; Vogelnik, K.; et al. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int. J. Mol. Sci. 2021, 22, 1084. https://doi.org/10.3390/ijms22031084
Potrč M, Volk M, de Rosa M, Pižem J, Teran N, Jaklič H, Maver A, Drnovšek-Olup B, Bollati M, Vogelnik K, et al. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. International Journal of Molecular Sciences. 2021; 22(3):1084. https://doi.org/10.3390/ijms22031084
Chicago/Turabian StylePotrč, Maja, Marija Volk, Matteo de Rosa, Jože Pižem, Nataša Teran, Helena Jaklič, Aleš Maver, Brigita Drnovšek-Olup, Michela Bollati, Katarina Vogelnik, and et al. 2021. "Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys" International Journal of Molecular Sciences 22, no. 3: 1084. https://doi.org/10.3390/ijms22031084