
Molecular Dynamics Simulations of Mitochondrial Uncoupling Protein 2



Abstract
:
1. Introduction
2. Results
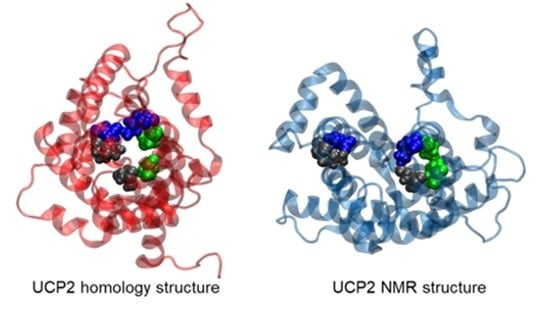
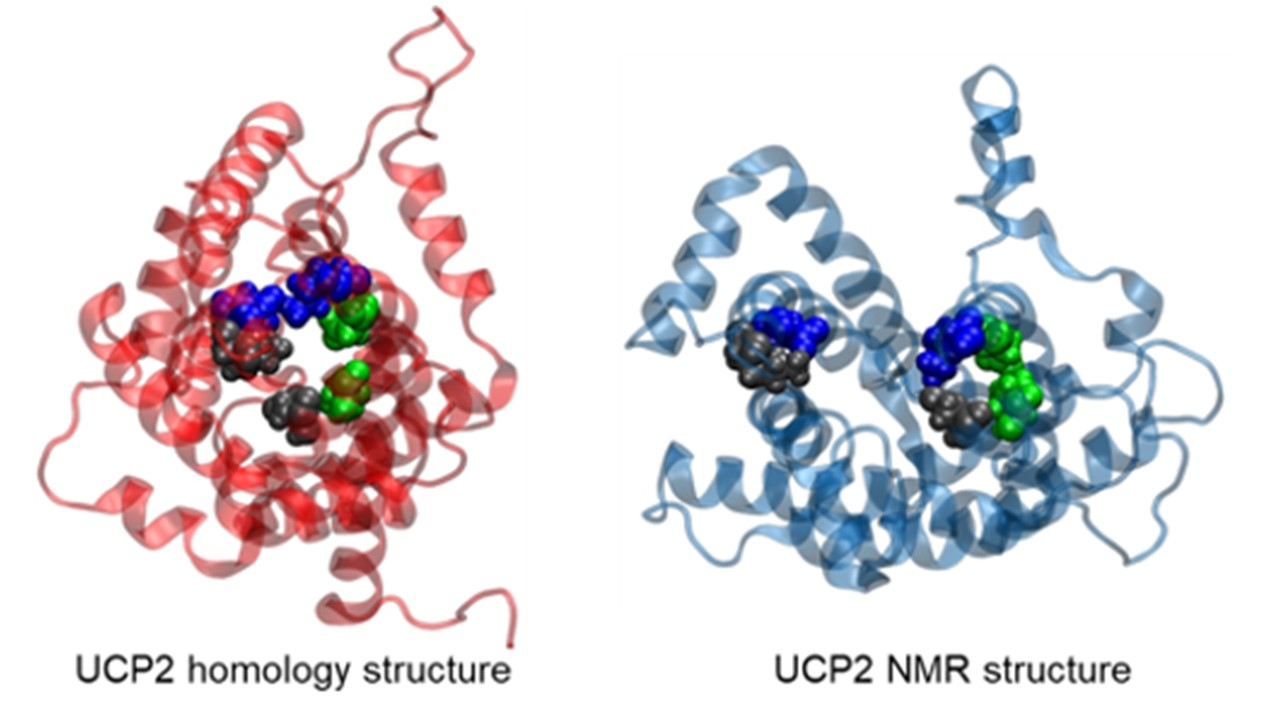
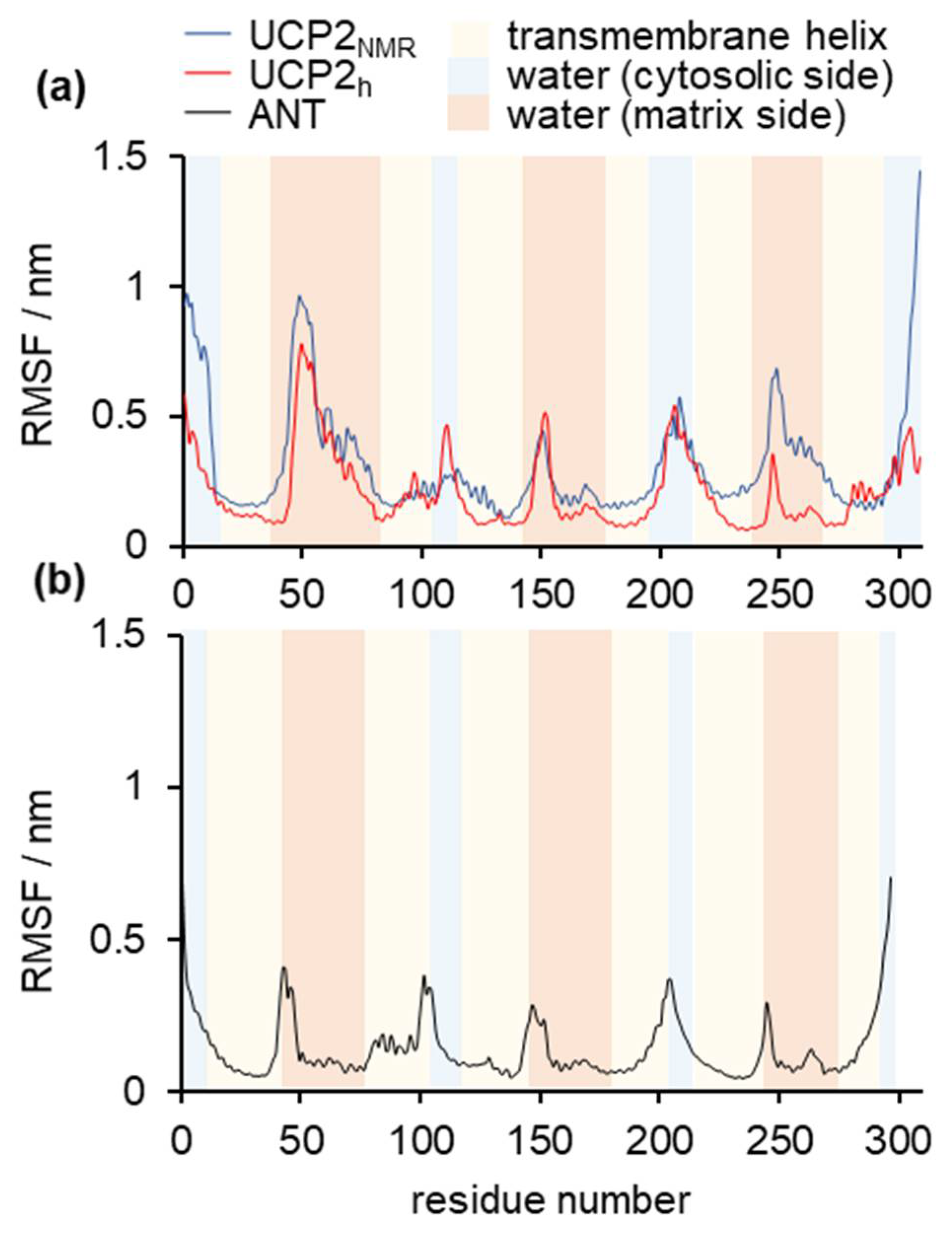
2.1. Structural Properties of Modeled Membrane Proteins
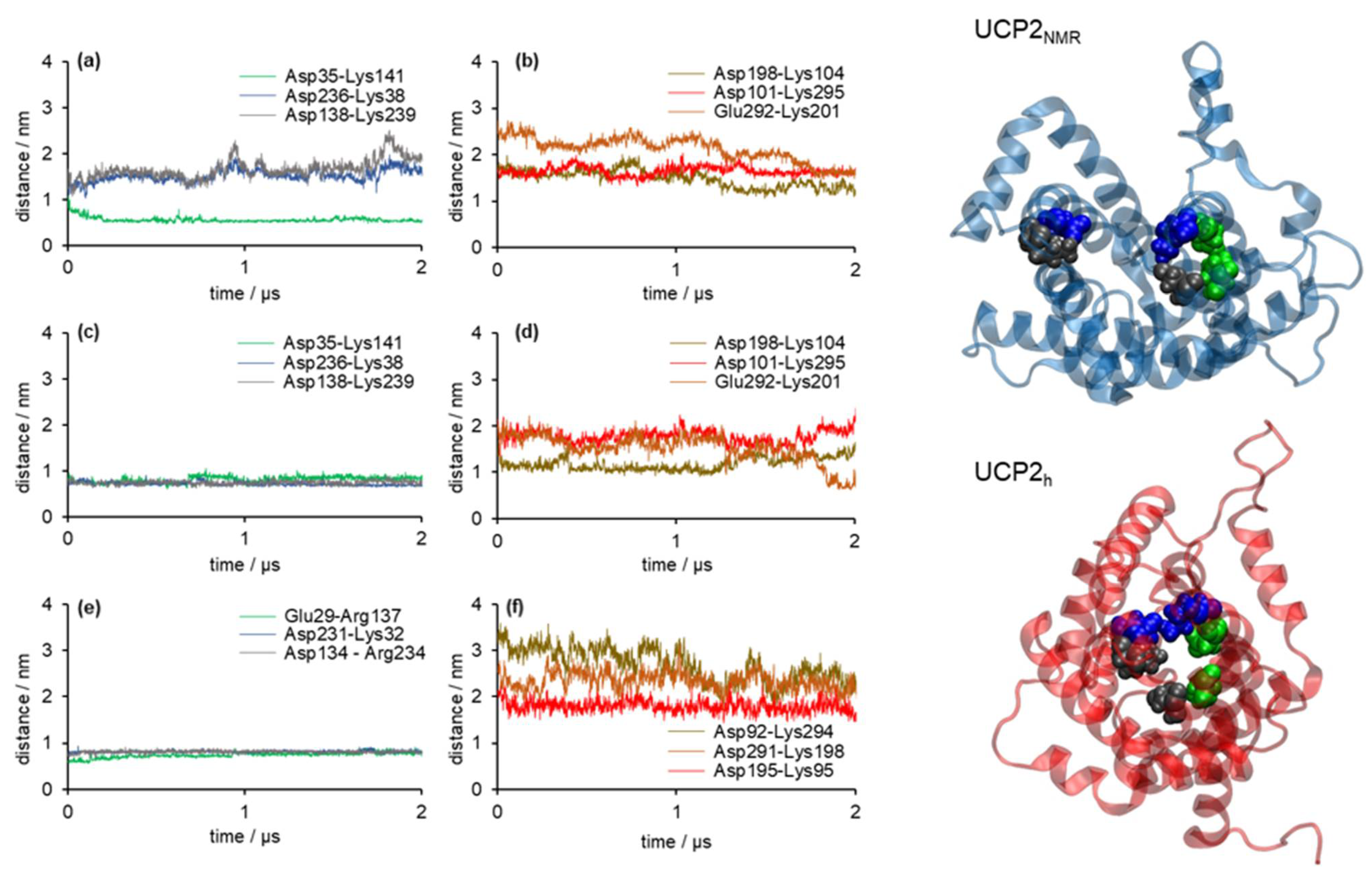
2.2. Stability of Salt Bridges Exposed to the Cytosolic and Matrix Side of the Inner Mitochondrial Membrane
2.3. Water Leakage across the Protein and Permeability Calculations
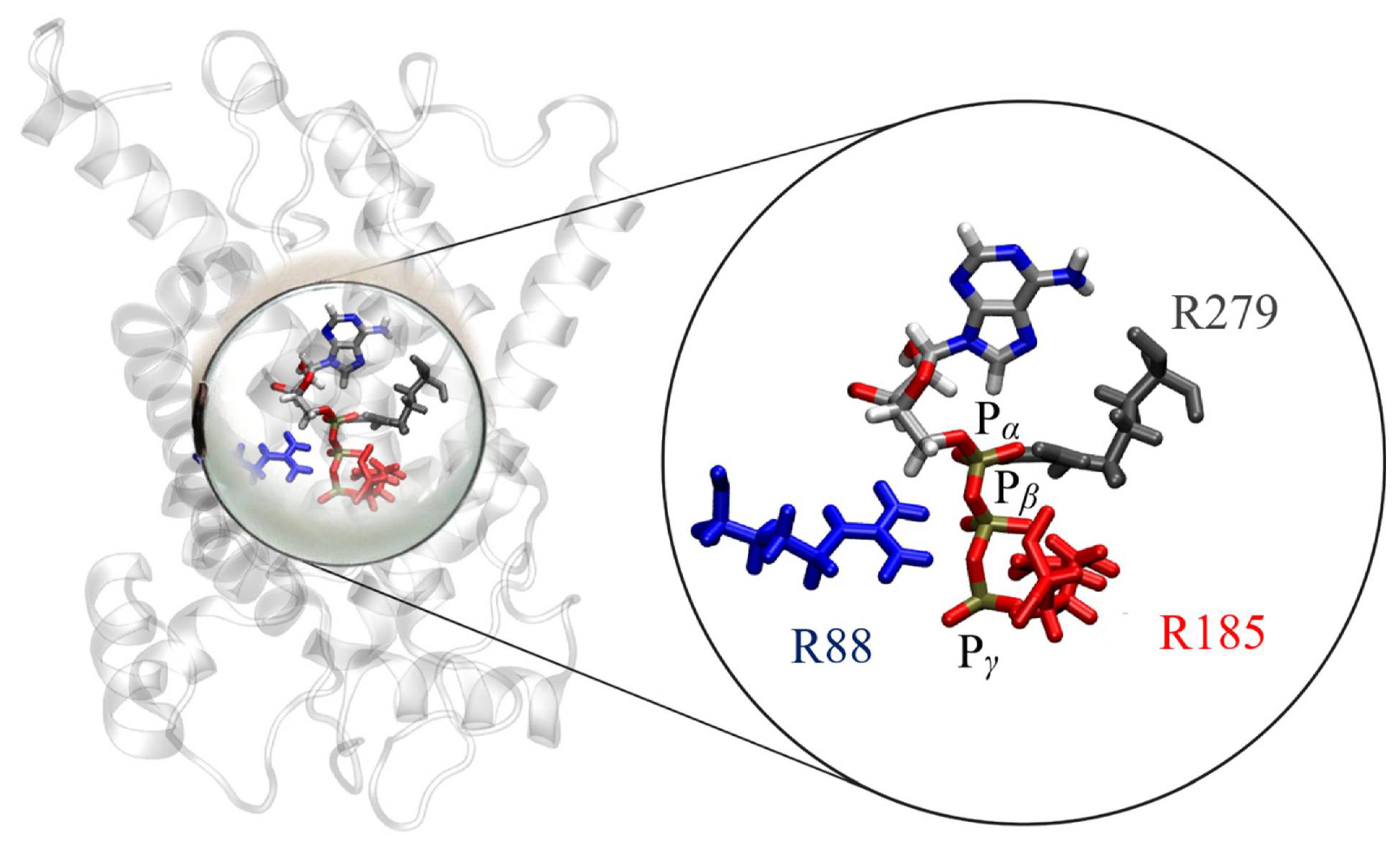
2.4. Binding of ATP in the UCP2 Cavity
2.5. Binding of Fatty Acid to UCP2
3. Discussion
4. Materials and Methods
4.1. Simulation Details
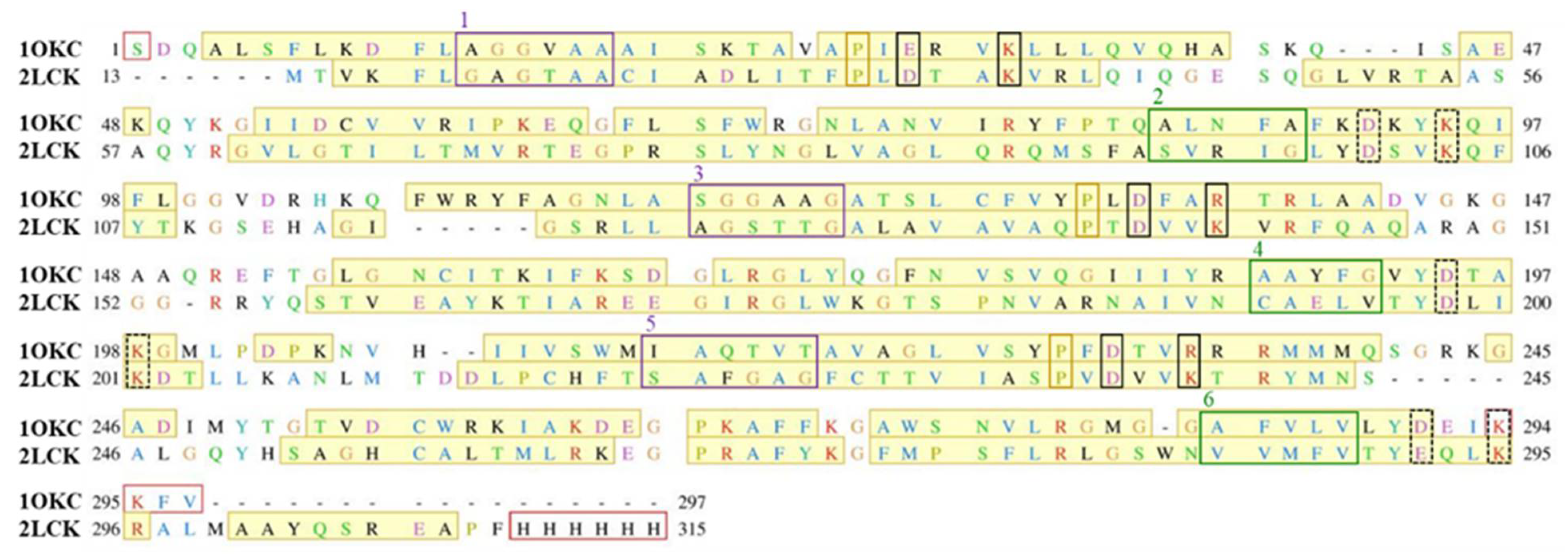
4.2. Homology Modeling
4.3. Permeability Calculations
4.4. Binding of ATP in the UCP2 Cavity
4.5. Chemicals
4.6. Cloning, Mutation and Expression of mUCP2 and Reconstitution into Liposomes
4.7. Measurements of Electrical Parameters of Membranes Reconstituted with mUCP2
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skulachev, V.P. Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett. 1991, 294, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Krauss, S.; Zhang, C.Y.; Lowell, B.B. The mitochondrial uncoupling-protein homologues. Nat. Rev. Mol. Cell Biol. 2005, 6, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Holendová, B.; Garlid, K.D.; Jabůrek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreyev, A.Y.; Bondareva, T.O.; Dedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Tsofina, L.M.; Volkov, N.I.; Vygodina, T.V. The ATP/ADP-antiporter is involved in the uncoupling effect of fatty acids on mitochondria. Eur. J. Biochem. 1989, 182, 585–592. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H+ transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Klingenberg, M. The reconstituted ADP/ATP carrier can mediate H+ transport by free fatty acids, which is further stimulated by mersalyl. J. Biol. Chem. 1994, 269, 27329–27336. [Google Scholar] [CrossRef]
- Garlid, K.D.; Orosz, D.E.; Modrianska, M.; Vassanelli, S.; Jezek, P. On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. J. Biol. Chem. 1996, 271, 2615–2620. [Google Scholar] [CrossRef] [Green Version]
- Beck, V.; Jaburek, M.; Demina, T.; Rupprecht, A.; Porter, R.K.; Jezek, P.; Pohl, E.E. Polyunsaturated fatty acids activate human uncoupling proteins 1 and 2 in planar lipid bilayers. FASEB J. 2007, 21, 1137–1144. [Google Scholar] [CrossRef] [Green Version]
- Malingriaux, E.A.; Rupprecht, A.; Gille, L.; Jovanovic, O.; Jezek, P.; Jaburek, M.; Pohl, E.E. Fatty Acids are Key in 4-Hydroxy-2-Nonenal-Mediated Activation of Uncoupling Proteins 1 and 2. PLoS ONE 2013, 8, e77786. [Google Scholar] [CrossRef] [Green Version]
- Kamp, F.; Hamilton, J.A. pH Gradients across Phospholipid Membranes Caused by Fast Flip-flop of Un-ionized Fatty Acids. Proc. Natl. Acad. Sci. USA 1992, 89, 11367–11370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, F.; Zakim, D.; Zhang, F.; Noy, N.; Hamilton, J.A. Fatty acid flip-flop in phospholipid bilayers is extremely fast. Biochemistry 1995, 34, 11928–11937. [Google Scholar] [CrossRef] [PubMed]
- Škulj, S.; Vazdar, M. Calculation of Apparent p K a Values of Saturated Fatty Acids with Different Lengths in DOPC Phospholipid Bilayers. Phys. Chem. Chem. Phys. 2019, 21, 10052–10060. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gale, P.A. Small-Molecule Uncoupling Protein Mimics: Synthetic Anion Receptors as Fatty Acid-Activated Proton Transporters. J. Am. Chem. Soc. 2016, 138, 16508–16514. [Google Scholar] [CrossRef]
- Jovanović, O.; Pashkovskaya, A.A.; Annibal, A.; Vazdar, M.; Burchardt, N.; Sansone, A.; Gille, L.; Fedorova, M.; Ferreri, C.; Pohl, E.E. The molecular mechanism behind reactive aldehyde action on transmembrane translocations of proton and potassium ions. Free Radic. Biol. Med. 2015, 89, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.; Klingenberg, M. Effect of fatty acids on H+ transport activity of the reconstituted uncoupling protein. J. Biol. Chem. 1994, 269, 2508–2515. [Google Scholar] [CrossRef]
- Klingenberg, M. UCP1-A sophisticated energy valve. Biochimie 2017, 134, 19–27. [Google Scholar] [CrossRef]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef] [Green Version]
- Berardi, M.J.; Shih, W.M.; Harrison, S.C.; Chou, J.J. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature 2011, 476, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Zoonens, M.; Comer, J.; Masscheleyn, S.; Pebay-Peyroula, E.; Chipot, C.; Miroux, B.; Dehez, F. Dangerous Liaisons between Detergents and Membrane Proteins. The Case of Mitochondrial Uncoupling Protein 2. J. Am. Chem. Soc. 2013, 135, 15174–15182. [Google Scholar] [CrossRef]
- Dehez, F.; Schanda, P.; King, M.S.; Kunji, E.R.S.; Chipot, C. Mitochondrial ADP/ATP Carrier in Dodecylphosphocholine Binds Cardiolipins with Non-native Affinity. Biophys. J. 2017, 113, 2311–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurauskas, V.; Hessel, A.; Ma, P.; Lunetti, P.; Weinhäupl, K.; Imbert, L.; Brutscher, B.; King, M.S.; Sounier, R.; Dolce, V.; et al. How Detergent Impacts Membrane Proteins: Atomic-Level Views of Mitochondrial Carriers in Dodecylphosphocholine. J. Phys. Chem. Lett. 2018, 9, 933–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurauskas, V.; Hessel, A.; Dehez, F.; Chipot, C.; Bersch, B.; Schanda, P. Dynamics and interactions of AAC3 in DPC are not functionally relevant. Nat. Struct. Mol. Biol. 2018, 25, 745–747. [Google Scholar] [CrossRef] [PubMed]
- King, M.S.; Crichton, P.G.; Ruprecht, J.J.; Kunji, E.R.S. Concerns with yeast mitochondrial ADP/ATP carrier’s integrity in DPC. Nat. Struct. Mol. Biol. 2018, 25, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Berardi, M.J.; Chou, J.J. Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria. Cell Metab. 2014, 20, 541–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüschweiler, S.; Yang, Q.; Run, C.; Chou, J.J. Substrate-modulated ADP/ATP-transporter dynamics revealed by NMR relaxation dispersion. Nat. Struct. Mol. Biol. 2015, 22, 636–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Brüschweiler, S.; Zhao, L.; Chou, J.J. Reply to ‘Concerns with yeast mitochondrial ADP/ATP carrier’s integrity in DPC’ and ‘Dynamics and interactions of AAC3 in DPC are not functionally relevant’. Nat. Struct. Mol. Biol. 2018, 25, 749–750. [Google Scholar] [CrossRef]
- Lindahl, E.; Sansom, M.S. Membrane proteins: Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008, 18, 425–431. [Google Scholar] [CrossRef]
- Weng, J.; Wang, W. Molecular dynamics simulation of membrane proteins. Adv. Exp. Med. Biol. 2014, 805, 305–329. [Google Scholar] [CrossRef]
- Dutagaci, B.; Heo, L.; Feig, M. Structure refinement of membrane proteins via molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 2018, 86, 738–750. [Google Scholar] [CrossRef]
- Almén, M.S.; Nordström, K.J.V.; Fredriksson, R.; Schiöth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newport, T.D.; Sansom, M.S.P.; Stansfeld, P.J. The MemProtMD database: A resource for membrane-embedded protein structures and their lipid interactions. Nucleic Acids Res. 2019, 47, D390–D397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipot, C.; Dehez, F.; Schnell, J.R.; Zitzmann, N.; Pebay-Peyroula, E.; Catoire, L.J.; Miroux, B.; Kunji, E.R.S.; Veglia, G.; Cross, T.A.; et al. Perturbations of Native Membrane Protein Structure in Alkyl Phosphocholine Detergents: A Critical Assessment of NMR and Biophysical Studies. Chem. Rev. 2018, 118, 3559–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromiha, M.M.; Nagarajan, R.; Selvaraj, S. Protein Structural Bioinformatics: An Overview. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 445–459. [Google Scholar]
- Abeln, S.; Feenstra, K.A.; Heringa, J. Protein Three-Dimensional Structure Prediction. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 497–511. [Google Scholar]
- Zhao, L.; Wang, S.; Zhu, Q.; Wu, B.; Liu, Z.; OuYang, B.; Chou, J.J. Specific Interaction of the Human Mitochondrial Uncoupling Protein 1 with Free Long-Chain Fatty Acid. Structure 2017, 25, 1371–1379.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardalan, A.; Sowlati-Hashjin, S.; Uwumarenogie, S.O.; Fish, M.; Mitchell, J.; Karttunen, M.; Smith, M.D.; Jelokhani-Niaraki, M. Functional Oligomeric Forms of Uncoupling Protein 2: Strong Evidence for Asymmetry in Protein and Lipid Bilayer Systems. J. Phys. Chem. B 2021, 125, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Monné, M.; Palmieri, F. Antiporters of the mitochondrial carrier family. In Current Topics in Membranes; Academic Press Inc.: Cambridge, MA, USA, 2014; Volume 73, pp. 289–320. [Google Scholar]
- Pietropaolo, A.; Pierri, C.L.; Palmieri, F.; Klingenberg, M. The switching mechanism of the mitochondrial ADP/ATP carrier explored by free-energy landscapes. Biochim. Biophys. Acta-Bioenerg. 2016, 1857, 772–781. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R. Structural changes in the transport cycle of the mitochondrial ADP/ATP carrier. Curr. Opin. Struct. Biol. 2019, 57, 135–144. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447.e15. [Google Scholar] [CrossRef] [Green Version]
- Pohl, E.E.; Rupprecht, A.; Macher, G.; Hilse, K.E. Important trends in UCP3 investigation. Front. Physiol. 2019, 10, 470. [Google Scholar] [CrossRef]
- Škulj, S.; Brkljača, Z.; Vazdar, M. Molecular Dynamics Simulations of the Elusive Matrix-Open State of Mitochondrial ADP/ATP Carrier. Isr. J. Chem. 2020, 60, 735–743. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Yang, L.-W.; Eyal, E. Global Dynamics of Proteins: Bridging Between Structure and Function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P. Anion carriers in fatty acid-mediated physiological uncoupling. J. Bioenerg. Biomembr. 1999, 31, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; Hellawell, A.M.; Harding, M.; Crichton, P.G.; McCoy, A.J.; Kunji, E.R.S. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, E426–E434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burykin, A.; Warshel, A. What Really Prevents Proton Transport through Aquaporin? Charge Self-Energy versus Proton Wire Proposals. Biophys. J. 2003, 85, 3696–3706. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, B. Why Can’t Protons Move through Water Channels? Biophys. J. 2003, 85, 3427–3428. [Google Scholar] [CrossRef] [Green Version]
- Aksimentiev, A.; Schulten, K. Imaging α-hemolysin with molecular dynamics: Ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys. J. 2005, 88, 3745–3761. [Google Scholar] [CrossRef] [Green Version]
- Ježek, P.; Modrianský, M.; Garlid, K.D. Inactive fatty acids are unable to flip-flop across the lipid bilayer. FEBS Lett. 1997, 408, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Willers, C.; Kunji, E.R.S.; Crichton, P.G. Uncoupling protein 1 binds one nucleotide per monomer and is stabilized by tightly bound cardiolipin. Proc. Natl. Acad. Sci. USA 2015, 112, 6973–6978. [Google Scholar] [CrossRef] [Green Version]
- Modrianský, M.; Murdza-Inglis, D.L.; Patel, H.V.; Freeman, K.B.; Garlid, K.D. Identification by site-directed mutagenesis of three arginines in uncoupling protein that are essential for nucleotide binding and inhibition. J. Biol. Chem. 1997, 272, 24759–24762. [Google Scholar] [CrossRef] [Green Version]
- Macher, G.; Koehler, M.; Rupprecht, A.; Kreiter, J.; Hinterdorfer, P.; Pohl, E.E. Inhibition of mitochondrial UCP1 and UCP3 by purine nucleotides and phosphate. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Jabůrek, M.; Ježek, P. The mechanism of proton transport mediated by mitochondrial uncoupling proteins. FEBS Lett. 1998, 438, 10–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tajkhorshid, E. Electrostatic funneling of substrate in mitochondrial inner membrane carriers. Proc. Natl. Acad. Sci. USA 2008, 105, 9598–9603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehez, F.; Pebay-Peyroula, E.; Chipot, C. Binding of ADP in the mitochondrial ADP/ATP carrier is driven by an electrostatic funnel. J. Am. Chem. Soc. 2008, 130, 12725–12733. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Kirichok, Y. UCP1: A transporter for H+ and fatty acid anions. Biochimie 2017, 134, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [Green Version]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Pundir, S.; Martin, M.J.; O’Donovan, C. UniProt protein knowledgebase. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1558, pp. 41–55. [Google Scholar]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2014, 47, 5.6.1–5.6.32. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; Tajkhorshid, E.; Schulten, K. Collective diffusion model for water permeation through microscopic channels. Phys. Rev. Lett. 2004, 93, 224501. [Google Scholar] [CrossRef]
- Rupprecht, A.; Sokolenko, E.A.; Beck, V.; Ninnemann, O.; Jaburek, M.; Trimbuch, T.; Klishin, S.S.; Jezek, P.; Skulachev, V.P.; Pohl, E.E. Role of the transmembrane potential in the membrane proton leak. Biophys. J. 2010, 98, 1503–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilse, K.E.; Rupprecht, A.; Kalinovich, A.; Shabalina, I.G.; Pohl, E.E. Quantification of Mitochondrial UCP3 Expression in Mouse Tissues. Biophys. J. 2014, 106, 592a. [Google Scholar] [CrossRef] [Green Version]
- Hilse, K.E.; Kalinovich, A.V.; Rupprecht, A.; Smorodchenko, A.; Zeitz, U.; Staniek, K.; Erben, R.G.; Pohl, E.E. The expression of UCP3 directly correlates to UCP1 abundance in brown adipose tissue. Biochim. Biophys. Acta-Bioenerg. 2016, 1857, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Beck, V.; Jabůrek, M.; Breen, E.P.; Porter, R.K.; Ježek, P.; Pohl, E.E. A new automated technique for the reconstitution of hydrophobic proteins into planar bilayer membranes. Studies of human recombinant uncoupling protein 1. Biochim. Biophys. Acta-Bioenerg. 2006, 1757, 474–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreiter, J.; Pohl, E.E. A Micro-agar Salt Bridge Electrode for Analyzing the Proton Turnover Rate of Recombinant Membrane Proteins. J. Vis. Exp. 2019, 143. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Permeability (cm3 s−1) |
|---|---|
| UCP2NMR after equilibration | (5.7 ± 0.4) × 10−13 |
| UCP2NMR after 200 ns | (3.2 ± 0.2) × 10−13 |
| UCP2NMR after 2 μs | (1.3 ± 0.1) × 10−13 |
| UCP2h after 2 μs | (2.0 ± 0.5) × 10−16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Škulj, S.; Brkljača, Z.; Kreiter, J.; Pohl, E.E.; Vazdar, M. Molecular Dynamics Simulations of Mitochondrial Uncoupling Protein 2. Int. J. Mol. Sci. 2021, 22, 1214. https://doi.org/10.3390/ijms22031214
Škulj S, Brkljača Z, Kreiter J, Pohl EE, Vazdar M. Molecular Dynamics Simulations of Mitochondrial Uncoupling Protein 2. International Journal of Molecular Sciences. 2021; 22(3):1214. https://doi.org/10.3390/ijms22031214
Chicago/Turabian StyleŠkulj, Sanja, Zlatko Brkljača, Jürgen Kreiter, Elena E. Pohl, and Mario Vazdar. 2021. "Molecular Dynamics Simulations of Mitochondrial Uncoupling Protein 2" International Journal of Molecular Sciences 22, no. 3: 1214. https://doi.org/10.3390/ijms22031214
APA StyleŠkulj, S., Brkljača, Z., Kreiter, J., Pohl, E. E., & Vazdar, M. (2021). Molecular Dynamics Simulations of Mitochondrial Uncoupling Protein 2. International Journal of Molecular Sciences, 22(3), 1214. https://doi.org/10.3390/ijms22031214