Abstract
MTH1 is an enzyme that hydrolyzes 8-oxo-dGTP, which is an oxidatively damaged nucleobase, into 8-oxo-dGMP in nucleotide pools to prevent its mis-incorporation into genomic DNA. Selective and potent MTH1-binding molecules have potential as biological tools and drug candidates. We recently developed 8-halogenated 7-deaza-dGTP as an 8-oxo-dGTP mimic and found that it was not hydrolyzed, but inhibited enzyme activity. To further increase MTH1 binding, we herein designed and synthesized 7,8-dihalogenated 7-deaza-dG derivatives. We successfully synthesized multiple derivatives, including substituted nucleosides and nucleotides, using 7-deaza-dG as a starting material. Evaluations of the inhibition of MTH1 activity revealed the strong inhibitory effects on enzyme activity of the 7,8-dihalogenated 7-deaza-dG derivatives, particularly 7,8-dibromo 7-daza-dGTP. Based on the results obtained on kinetic parameters and from computational docking simulating studies, these nucleotide analogs interacted with the active site of MTH1 and competitively inhibited the substrate 8-oxodGTP. Therefore, novel properties of repair enzymes in cells may be elucidated using new compounds.
1. Introduction
Reactive oxygen species (ROS) are a product or byproduct of cellular metabolism; some are generated in the mitochondria of cells, while others are exogenously produced, such as air and water pollution, cigarette smoke, alcohol, heavy or transition metals, and certain drugs [1,2]. ROS include oxygen-derived radicals and non-radical species, such as the hydroxyl radical, superoxide ion, hydrogen peroxide, and peroxynitrite. These ROS are involved in several signaling pathways and affect cellular function. However, they also have the potential to induce damage to lipids, proteins, and DNA [3]. Free nucleotides are particularly vulnerable to oxidative damage by ROS. The major forms of oxidative damage induced by ROS in the nucleotide pool are 8-oxo-dGTP and 2-OH-dATP [4,5,6,7]. 8-oxo-dGTP prefers the syn-conformation at glycosidic bonds and is incorporated into the duplex DNA strand not only for the dC-template, but also dA-template at the stage of DNA replication steps [8,9,10,11]. This property of 8-oxo-dGTP for the dA-template leads to a genetic mutation known as the TA to GC transversion mutation [12,13,14]. The nucleotide repair enzyme, MTH1 (human mutt homolog 1, also called NUDT1), may hydrolyze 8-oxo-dGTP to its monophosphate, 8-oxo-dGMP, to prevent its mis-incorporation into duplex DNA during DNA replication steps [15,16,17].
In 2014, Gad and Huber et al. reported that the inhibition of MTH1 function by the RNAi-mediated knockdown of expression or by novel compounds (including TH588, TH287, and Crizotinib) exerted strong anti-proliferation effects in various cancer cells, but not normal cells [18,19,20]. The inhibition of MTH1 has been proposed to increase the incorporation of 8-oxo-dGTP in cells, thereby enhancing DNA damage and, ultimately, resulting in cancer cell death [21,22,23]. In contrast, recent studies suggested that MTH1 does not affect the survival of cancer cells [24,25,26].
To address this discrepancy and reveal its functions or properties in living cells, we developed 8-halogenated 7-deaza-dGTP, an 8-oxo-dGTP analogue [27,28,29], and γ-N-modified 8-oxo-dGTP [30] as a MTH1-binding compound. This structural mimicry of a natural product is important for elucidating the functions or mechanisms of intracellular events. We demonstrated that 8-iodo-7-deaza-dGTP inhibited the hydrolytic activity of MTH1, the IC50 value of which was 0.42 μM and Ki value was 61.7 nM. This was the first example of the development of a MHT1 inhibitor using nucleotide derivatives, which prompted us to conduct further experiments. Based on the predicted complex structure between 8-iodo-7-deaza-dGTP and MTH1, a space exists between the active site of MTH1 and the 8-position of the nucleobase. Therefore, in the present study, we designed 7,8-dihalogenated 7-deaza-dG derivatives as novel MTH1-binding compounds to increase binding affinity (Figure 1).
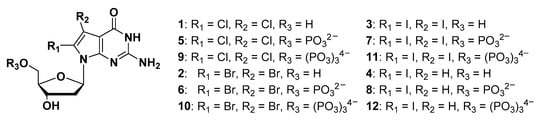
Figure 1.
Structures of 8-substituted 7-deaza-dG derivatives and 7,8-disubstituted 7-deaza-dG derivatives.
2. Results and Discussion
2.1. Synthesis of New 7-Deaza-dG Derivatives
7-Deaza-2′-deoxyguanosine (7-deaza-dG) was used as the starting material to synthesize newly designed substituted derivatives. The synthetic schemes of these derivatives are summarized in Scheme 1. The 3′- and 5′-hydroxyl groups of 7-deaza-dG were protected with an acetyl group by using acetic anhydride in pyridine. The acetylated compound (13) was then treated with the 2.0 equivalent of N-chlorosuccinimide (NCS) to modify the 7- and 8-positions of 7-deazaguanine using a chlorination reaction. In contrast to the previously reported regioselective reaction at the 8-position, a dihalogen-introduced compound was obtained at the 7- and 8-positions under this condition in 49% yield. Although the solvent, reaction time, and temperature were changed to improve yield, satisfactory yield was not obtained. 7,8-Dibromo-7-deaza-dG and 7,8-diiodo-7-deaza-dG were also obtained using a 2.0 equivalent of N-bromosuccinimide (NBS) and 2.5 equivalent of N-iodosuccinimide (NIS) in 51% and 51% yield, respectively. After removing the acetyl groups at the 5′- and 3′-hydroxyl groups, the corresponding 7,8-dihalogenated 7-deaza-dGs (1, 2, and 3) were obtained.
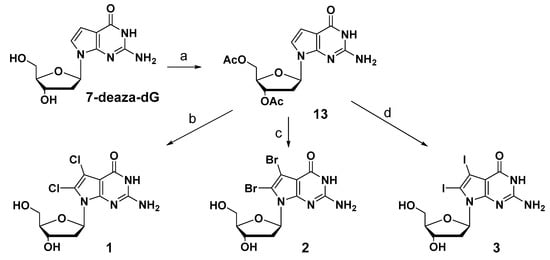
Scheme 1.
Reagents and conditions: (a) acetic anhydride, anhydrous pyridine, r.t. 91%; (b) (1) N-chlorosuccinimide (NCS) (2.0 eq), anhydrous DMF, r.t., 1 h; (2) 7N ammonium in methanol, r.t., 18 h, 36% for two steps; (c) (1) NBS (2.0 eq), anhydrous DMF, r.t., 1 h; (2) 7N ammonium in methanol, r.t., 18 h, 35% for two steps. (d) (1) N-iodosuccinimide (NIS) (2.5 eq), anhydrous DMF, r.t., 1 h; (2) 7N ammonium in methanol, r.t., 18 h, 18% for two steps.
The synthesis of monophosphate and triphosphate derivatives is depicted in Scheme 2. Regarding the synthesis of a monophosphate compound, when treated with POCl3, which is a general method, the dehalogenation reaction proceeded and the desired product was not obtained. Therefore, to prevent the decomposition of halogen substituents under strong acidic conditions [31], the diol compounds were converted into the corresponding 3’-acetylated compounds (14, 15, and 16) in three steps. These 3’-acetylated compounds (14, 15, and 16) and 2-N-phenoxylacetyl-3′-O-acetyl-8-iodo-7-deaza-dG (17) were treated with 2-cyanoethylbarium phosphate hydrate under mild phosphorylation reaction conditions, followed by ammonia solution to give the corresponding monophosphate derivatives (5, 6, 7, and 8). The synthesis of triphosphate derivatives was performed in a one-step reaction [32,33]. 2-Chloro-4-H-1,3,2-benzodioxaphosphoryl-4-one and pyrophosphate were used to prepare a bulky phosphorylation reagent, which selectively phosphorylates at the 5′-hydroxyl group of the diol compound under mild conditions. After the oxidation of phosphite and the hydrolysis of the cyclic structure, the corresponding triphosphate derivatives (9, 10, and 11) were successfully synthesized in moderate yield.
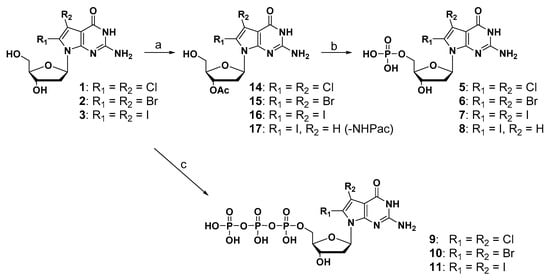
Scheme 2.
Reagents and conditions: (a) (1) TBSCl, dry pyridine, imidazole, DMAP, r.t., overnight, 37–71%; (2) acetic anhydride, dry pyridine, r.t., overnight, 49–92%; (3) Et3N-3HF, TEA, dry THF, r.t., 5 h, 91–98%; (b) (1) barium-2-cyanoethylphosphate hydrate, dowex (50) H+, DCC; (2) 28% ammonia solution, r.t., 48 h, 9–31%; (c) (1). 2-Chloro-4H-1,3,2-benzodioxa-phosphorin-4-one, tributyl-ammonium pyrophosphate, tributylamine, dry DMF; (2). I2 in pyridine/H2O = 98:2 solution; (3) 28% ammonia solution, r.t., 14–30%.
2.2. Binding Properties of Dihalogenated-deaza-dG Derivatives for MTH1
2.2.1. Hydrolytic Properties of New 7-Deaza-dGTP Derivatives by MTH1
Since the shapes of synthesized triphosphate derivatives were similar to 8-oxo-dGTP, we investigated whether these triphosphate derivatives are hydrolyzed by MTH1. MTH1 and these triphosphate derivatives were mixed and incubated at 37 °C for various times, and hydrolysis reactions were analyzed by HPLC. Hydrolyzed ratios were calculated from the peak area, which was summarized in Figure 2 as a time course plot. 8-Oxo-dGTP was rapidly hydrolyzed to its monophosphate, 8-oxo-dGMP, by MTH1 (Figure 2. sky-blue line). The triphosphate group of 7-deaza-dGTP was also hydrolyzed, but more slowly than 8-oxo-dGTP (Figure 2. orange line). However, in the case of 7,8-dihalogenated triphosphate derivatives, the peak of the corresponding monophosphate was not detected in HPLC following the treatment with MTH1 (Figure 2. 9: purple, 10: red and 11: blue lines). Although these results were similar to those obtained for 8-iodo-7-deaza-dGTP, they indicated that these triphosphates were poor substrates or had no binding property for the active site of MTH1.
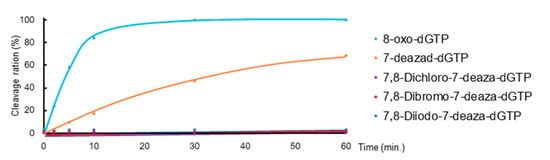
Figure 2.
Time courses of the hydrolysis of 8-oxo-dGTP, 7-deaza-dGTP, and 7,8-dihalogenated-7-deaza-dGTP (9, 10, and 11) by MTH1. Reaction conditions: 20 mM Tris-HCl pH 7.5, 4 mM MgCl2, 40 mM NaCl, 80 μg/mL bovine serum albumin (BSA), 8 mM dithiothreitol (DTT), 10% glycerol; 50 μM 8-oxo-dGTP, 5 nM MTH1; 50 μM dNTPs; incubated at 37 °C for various times. Hydrolysis reactions were analyzed by HPLC, and the cleavage rate was calculated from the peak area.
2.2.2. Inhibitory Effects of New 7-Deaza-dG Derivatives on the Hydrolytic Activity of MTH1
We examined whether the nucleoside diols, nucleoside monophosphate (NMP), and nucleoside triphosphate (NTP) derivatives inhibit the MTH1 hydrolysis of 8-oxo-dGTP. MTH1 and 8-oxo-dGTP were mixed with various concentrations of synthesized derivatives, and this mixture was incubated and analyzed by HPLC. The inhibition efficiencies of IC50 values were calculated and summarized in Table 1. In the case of diol compounds, 7,8-dichloro-7-deaza-dG (1), 7,8-dibromo-7-deaza-dG (2), and 7,8-diiodo-7-deaza-dG (3) exerted inhibitory effects on the hydrolytic activity of MHT1 at a micromolar level (Table 1, Entries 1–3). Their IC50 values were better than that of 8-iodo-7-deaza-dG (4) (Table 1, Entry 4).

Table 1.
IC50 values of diol, monophosphate, and triphosphate derivatives 1.
We then examined monophosphate compounds, the structures of which were products of a hydrolyzed reaction. The IC50 values of these compounds were markedly smaller than that of the diol compound (IC50 values: 5 is 0.13 μM, 6 is 0.26 μM, 7 is 0.13 μM, and 8 is 0.84 μM in Table 1, Entries 5–8.). These IC50 values were approximately 100-fold smaller for each corresponding diol compound. These results indicated that the monophosphate moiety strongly interacted with the enzyme, and this interaction was also observed in 8-oxo-dGMP. We then assessed the inhibitory effects of triphosphate derivatives (Entries 9–12) on MTH1 activity. The IC50 values of dihalogenated triphosphate analogs were not better than those of the corresponding monophosphate compounds (Entries 9–11 vs. 5–7); however, in many cases of triphosphate modifications, the value was markedly better than those of monophosphates. These results indicated that the phosphate group at the α-position was very important for the interaction with the enzyme, and modifying not only the 8-position, but also the 7-position with a halogen atom further contributed to binding to the enzyme and the inhibition of enzyme activity. Since the influence of the size of the halogen atom was small, these effects were attributed more to the electronic change of the nucleobase position, i.e., stacking interaction, than to the direct interaction of the halogen atom with the enzyme. Compared with the IC50 value of TH287 (Table 1, Entry 13) [28], which is the strongest class inhibitor as a small molecule, the IC50 value of our triphosphate compounds were about 10-fold different. These results were also shown to be very interesting as an inhibitory effect on MTH1 activity.
2.2.3. Assessment of Inhibition Modes of New 7-Deaza-dG Derivatives
We made a Lineweaver-Burk plot to calculate kinetic parameters (KM and kcat) in order to reveal the types of inhibition modes for MTH1. Kinetic parameters were summarized in Table 2. In the absence of inhibitors, kcat was 10.6 s−1, and KM was 17.6 μM (Entry 1). In the presence of new 7-deaza-dG monophosphate and triphosphate, kcat values were only slightly altered (Entries 2–7). On the other hand, all KM values increased (Entries 2–7). These results clearly demonstrated that the inhibition modes of new compounds involve competitive inhibition with the substrate of 8-oxo-dGTP. Among these derivatives, 7,8-dibromo-7-deaza-dGTP (10) showed the best Ki value (24.2 nM) in this experiment (Entry 5).
Table 2.
Steady-state kinetic parameters for the MTH1 hydrolysis of 8-oxo-dGTP in the presence and absence of new 7-deaza-dG derivatives as inhibitors 1.
2.2.4. Prediction of Binding Properties of New 7-Deaza-dG Derivatives with MTH1
We predicted the complex structure between 8-dihalogenated-7-deaza-dGTP (7, 10, and 13) and MTH1 using a docking study. The best docking conformations were shown in Figure 3 and the corresponding 2D-interaction diagrams were depicted in Figure 4. The docking results showed that the positions of the nucleobase part were similar to the substrate, 8-oxo-dGTP. Since these derivatives, such as 8-oxo-dGTP, have the ability to form a hydrogen bond at the Watson-Click phase of them (Figure 3, thin stick vs. thick stick structures). On the other hand, their sugar and triphosphate parts slightly shifted from those of 8-oxo-dGTP in the MTH1 active site. Although the underlying mechanisms currently remain unknown, this shift may be one reason why these 7,8-dihalogenated-7-deaza-dGTP derivatives were not hydrolyzed by MTH1. In addition, the binding characteristics of these 7,8-dihalogenated-7-deaza-dGTP derivatives to MTH1 revealed that their nucleobases and phosphate groups mainly and consistently interacted with Asp120, Trp117, and Lys38, respectively, with these amino acid showing identical interactions to those in 8-oxo-dGTP (Figure 4A–D). However, these 7,8-dihalogenated-7-deaza-dGTP derivatives showed better binding profiles in terms of binding energy and percentage binding than 8-oxo-dGTP (Figure 4E). Therefore, the introduction of a halogen atom into the nucleobase portion may increase binding due to changes in the surrounding electric environment, which may further enhance the pi-pi stacking interaction with the amino acid residue, Trp117 at the active site of MTH1.
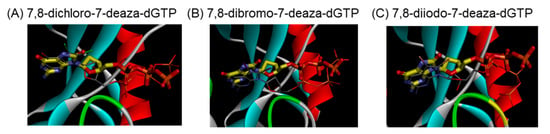
Figure 3.
Images of optimal docking conformations of (A) 7,8-dichloro-7-deaza-dGTP (7) (thin stick with nitrogen colored in blue, oxygen colored in red, phosphorus colored in orange and chlorine colored in green), (B) 7,8-dibromo-7-deaza-dGTP (10) (thin stick with bromine colored in reddish brown), and (C) 7,8-diiodo-7-deazadGTP (13) (thin stick with iodine colored in light purple) in the binding pocket of MTH1 relative to 8-oxo-dGTP (thick stick with carbon colored in yellow).
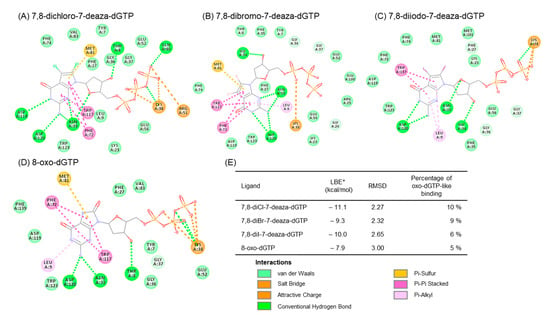
Figure 4.
2D-Interaction diagram between docked MTH1 and (A) 8-oxo-dGTP (PDB: 5FSI), (B) 7,8-dichloro-7-deaza-dGTP (7), (C) 7,8-dibromo-7-deaza-dGTP (10), (D) 7,8-diI-7-deazadGTP (13), and (E) and binding properties. * LBE (Lowest binding energy) value for the conformation with RMSD < 5 Å compared to the 8-oxo-dGTP moiety in the crystal structure 5FSI. Percentage of conformations with the ability to bind to the active site of hMTH1 with RMSD < 5 Å. One hundred conformations (n-100) will be generated as the docking resulting.
Although these compounds that showed very interesting binding properties, these triphosphates are expected to be very high water solubility and polarity. These are also supported by the ADME prediction results (Supplementary Figures S1–S4). Since the γ-position of triphosphate group placed the outside of binding pocket of the enzyme from the docking study, the further chemical modifications of this part are possible.
3. Materials and Methods
3.1. Synthesis of New 7-Deaza-dG Derivatives
General: 1H NMR, 13C NMR, and 31P-NMR spectra was measured with 500 MHz (Ascend 500, Bruker, Billerica, MA, USA), 400 MHz (UNITY 400, Agilent, Santa Clara, CA, USA) nuclear magnetic resonance instruments. Merck TLC Plate Silica Gel 60 F254 was used to detect reactions and column chromatography was conducted with silica gel 60N (Kanto Chemical Co., Inc., Tokyo, Japan). YAMAZEN AI-580S (Yamazen Co., Osaka, Japan) was used for flash column chromatography. MALDI-TOF mass spectra were recorded in the negative mode by Bruker Daltonics Microflex. HPLC purification (JASCO Co., Tokyo, Japan) and analyses were performed with reverse-phase C18 columns, including SHISEIDO CAPCELL PAK C18 MG (4.6 × 250 mm) and COSMOSIL 5C18-AR-II (4.6 × 250 mm) (Nacalai Tesque, Kyoto, Japan).
3.1.1. General Procedure for the Synthesis of 7,8-Dihalogeneted-7-deaza-2′-deoxy-guanosine
3′,5′-di-O-Acetyl-7-deaza-2′-deoxyguanosine (13) (1 g, 2.86 mmol) was dissolved in 48 mL anhydrous DMF under an argon atmosphere at room temperature. After covering the flask, the 2 equivalent of NCS or NBS or 2.5 equivalent of NIS was added in two portions over one hour. The organic solvent was concentrated, and the resulting residue was purified by column chromatography (silica gel, CHCl3/MeOH = 30/1) to give a white solid. This solid was suspended in 20 mL of 7 N ammonium in methanol solution. The mixture was stirred at room temperature overnight under argon, concentrated, and the residue was purified by column chromatography (silica gel, CHCl3/MeOH = 10/1) to give a white solid.
7,8-di-Chloro-7-deaza-2′-deoxy-guanosine (1): Yield was 36% for two steps. 1H-NMR (500 MHz, CD3OD) δ (ppm): 6.41 (t, 1H, J = 1.8 Hz), 4.57–4.55 (m, 1H), 3.99–3.95 (m, 1H), 3.95–3.81 (m, 1H), 3.76–3.72 (m, 1H), 3.14–3.08 (m, 1H), 2.19–2.14 (m, 1H). HR-ESI-MS (m/z) calcd. for C11H12N4O4Cl2Na [M + Na]+: 357.0128, 359.0099, 361.0089, Found: 357.0159, 359.0138, 361.0130.
7,8-di-Bromo-7-deaza-2′-deoxy-guanosine (2): Yield was 35% for two steps. 1H-NMR (500 MHz, CD3OD) δ (ppm): 6.45–6.42 (m, 1H), 4.59–4.56 (m, 1H), 3.98–3.96 (m, 1H), 3.86–3.82 (m, 1H), 3.77–3.74 (m, 1H), 3.17–3.11 (m, 1H), 2.17–2.13 (m, 1H). HR-ESI-MS (m/z) calcd. for C11H12N4O4Br2Na [M + Na]+: 444.9118, 446.9097, 448.9077, Found: 444.9112, 446.9078, 448.9067.
7,8-di-Iodo-7-deaza-2′-deoxy-guanosine (3): Yield was 18% for two steps. 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 10.62 (bs, 1H), 6.29 (t, 1H, J = 2.0 Hz), 6.24 (bs, 2H), 5.19 (d, 1H, J = 4.5 Hz), 4.94–4.92 (m, 1H), 4.39–4.36 (m, 1H), 3.76–3.75 (m, 1H), 3.65–3.64 (m, 1H), 3.57–3.52 (m, 1H), 3.16–3.08 (m, 1H), 2.01–1.98 (m, 1H). HR-ESI-MS (m/z) calcd. for C11H13N4O4I2Na [M + Na]+: 540.8841, Found: 540.8844.
3.1.2. General Procedure for the Synthesis of 3′-O-Acetylated Compounds
The corresponding diol compounds (1, 2, and 3) (0.24 mmol) were dissolved in anhydrous pyridine (1.2 mL) under an argon atmosphere at room temperature, and tert-butyldimethylsilyl chloride (0.34 mmol), imidazole (0.48 mmol), and dimethylaminopyridine (DMAP) (0.05 mmol) were then added to the reaction mixture. The resulting mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure, and the residue was purified by flash silica-gel chromatography (Flash column, 10 g, CHCl3/MeOH = 100/0 to 86/14) to give a 5′-OTBS protected compound as a white powder. Acetic anhydride (0.95 mmol) was dropped into the solution of the above compound (0.24 mmol) in anhydrous pyridine (2.4 mL), and the reaction mixture was stirred overnight. The solvent was concentrated under reduced pressure, and the residue was purified by flash silica-gel chromatography (Flash column, 10 g, CHCl3/MeOH = 100/0 to 86/14) to give 3′-OAc and 5′-OTBS-protected compounds as a white powder. This compound (0.24 mmol) was dissolved in dry THF (2 mL). Triethylamine trihydrofluoride (0.33 mmol) and triethylamine (0.19 mmol) were then dropped into the reaction mixture at room temperature under an argon atmosphere. The reaction solution was stirred at room temperature for 5 h. After the removal of the solvent under reduced pressure, the residue was purified by flash silica-gel chromatography (Flash column, 10 g, CHCl3/MeOH = 100/0 to 95/5) to give 14, 15, and 16 as a white powder.
3′-O-Acetyl-7,8-di-chloro-7-deaza-2′-deoxy-guanosine (14): Yield was 40% for three steps. 1H-NMR (500 MHZ, DMSO-d6) δ (ppm): 10.98 (bs, 1H), 6.46 (bs, 2H), 6.36–6.29 (m, 1H), 5.31–5.29 (m, 1H), 4.92 (t, 1H, J = 6.4 Hz), 3.96–3.93 (m, 1H), 3.66–3.62 (m, 1H), 3.60–3.54 (m, 1H), 3.27–3.23 (m, 1H), 2.28–2.24 (m, 1H), 2.07 (s, 3H). HR-ESI-MS (m/z) calcd. for C13H14N4O5Cl2Na [M + Na]+: 399.0234, 401.0204, 403.0175, Found: 399.0252, 401.0225, 403.0199.
3′-O-Acetyl-7,8-di-bromo-7-deaza-2′-deoxy-guanosine (15): Yield was 59% for three steps. 1H-NMR (500 MHZ, DMSO-d6) δ (ppm): 10.75 (bs, 1H), 6.40 (bs, 2H), 6.30–6.27 (m, 1H), 5.33–5.31 (m, 1H), 4.94 (t, 1H, J = 6.3 Hz), 3.97–3.96 (m, 1H), 3.68–3.66 (m, 1H), 3.61–3.57 (m, 1H), 3.38–3.36 (m, 1H), 2.27–2.23 (m, 1H), 2.07 (s, 3H). HR-ESI-MS (m/z) calcd. for C13H15N4O5Br2 [M + H]+: 464.9404, 466.9383, 468.9363, Found: 464.9437, 466.9420, 468.9398.
3′-O-Acetyl-7,8-di-iodo-7-deaza-2′-deoxy-guanosine (16): Yield was 18% for three steps. 1H-NMR (500MHZ, DMSO) δ (ppm): 10.67 (s, 1H), 6.29–6.27 (m, 3H), 5.35–5.32 (m, 1H), 5.02 (t, 1H, J = 6.3 Hz), 3.98–3.96 (m, 1H), 3.72–3.67(m, 1H), 3.63–3.58 (m, 1H), 3.44–3.38 (m, 1H), 2.23–2.19 (m, 1H), 2.07 (s, 3H). HR-ESI-MS (m/z) calcd. for C13H14N4O5Cl2Na [M + Na]+: 582.8946, found: 582.8971.
3.1.3. General Procedure for the Synthesis of 5′-Monophosphate Compounds
The corresponding 3′-OAc compounds (14, 15, 16, and 17 [28]) (0.021 mmol) and 2-cyanoethylphosphate (0.11 mmol) were dissolved in pyridine (0.58 mL), under an argon atmosphere at room temperature, and DCC (0.15 mmol) were then added to the reaction mixture. The resulting mixture was stirred at room temperature for 48 h. The reaction mixture was concentrated under reduced pressure, and then the residue was dissolved in 28% ammonia solution and stirred for 6 h at 40 °C. The reaction mixture was filtered off, and the filtrate was washed with diethyl ether, then aqueous layer was concentrated under reduced pressure. The resulting residue was purified by reversed-phase HPLC to give corresponding monophosphate compounds as a white material after lyophilization. (HPLC conditions: A: 0.1 M TEAA, B: MeCN; Flow rate: 1.0 mL/min; Shiseido MG1 (4.6 × 250 mm); UV detector: 254 nm; 35 °C).
7,8-di-Chloro-7-deaza-2′-deoxy-guanosine monophosphate (5): Yield was 31% for two steps. 1H-NMR (500 MHZ, D2O) δ (ppm) 6.48 (t, 1H, J = 9.6 Hz), 4.87–4.84 (m, 1H), 4.09–4.01 (m, 2H), 3.96–3.92 (m, 1H), 3.26–3.21 (m, 1H), 2.37–2.32 (m, 1H). 31P-NMR (202 MHz, D2O) δ (ppm) 3.79. HR-ESI-MS (m/z) calcd. for C11H12N4O7Cl2P [M − H]−: 412.9826, 414.9797, 416.9767, found: 412.9826, 414.9805, 416.9781.
7,8-di-Bromo-7-deaza-2′-deoxy-guanosine monophosphate (6): Yield was 27% for two steps. 1H-NMR (500 MHZ, D2O) δ (ppm) 6.54–6.51 (m, 1H), 4.87–4.84 (m, 1H), 4.17–4.11 (m, 2H), 4.00–3.98 (m, 1H), 3.34–3.28 (m, 1H), 2.36–2.31 (m, 1H). 31P-NMR (202 MHz, D2O) δ (ppm) 1.68. HR-ESI-MS (m/z) calcd. for C11H12N4O7Br2P [M − H]−: 500.8864, 502.8795, 504.8775, found: 500.8864, 502.8831, 504.8819.
7,8-di-Iodo-7-deaza-2′-deoxy-guanosine monophosphate (7): Yield was 9% for two steps. 1H-NMR (500 MHZ, D2O) δ (ppm) 6.48 (t, 1H, J = 8.4 Hz), 4.87–4.84 (m, 1H), 4.17–4.09 (m, 2H), 3.99–3.94 (m, 1H), 3.36–3.30 (m, 1H), 2.34–2.28 (m, 1H). 31P-NMR (202 MHz, D2O) δ (ppm) 3.98. HR-ESI-MS (m/z) calcd. for C11H12N4O7I2P [M − H]−: 596.8538, found: 596.8579.
8-Iodo-7-deaza-2′-deoxy-guanosine monophosphate (8): Yield was 30% for two steps. 1H-NMR (500 MHZ, D2O) δ (ppm) 7.05 (s, 1H), 6.28 (t, 1H, J = 7.0 Hz), 4.88–4.85 (m, 1H), 4.22–4.16 (m, 1H), 4.15–4.13 (m, 1H), 4.01–3.97 (m, 1H), 3.41–3.35 (m, 1H), 2.38–2.33 (m, 1H). 31P-NMR (202 MHZ, D2O) δ (ppm) 3.42. HR-ESI-MS (m/z) calcd. for C11H13N4O7IP [M − H]−: 470.9572, found: 470.9591.
3.1.4. General Procedure for the Synthesis of 5′-Triphosphate Compounds
The solution of 2-chloro-4-H-1,3,2-benzodioxaphosphorin-4-one (0.26 mmol) in DMF (0.35 mL) and tributyl amine (0.5 mL) were added dropwise to the solution of tributyl ammonium pyrophosphate (0.26 mmol) in DMF (0.35 mL) and stirred for 30 min under an argon atmosphere at room temperature. This reaction mixture was added to the solution of the corresponding diol compounds (1, 2, and 3) (0.13 mmol) in DMF (0.1 mL). After stirring for 2 h, the oxidation reagent (4 mL, 1.0% I2 in pyridine/H2O = 98/2) was added to the reaction mixture and stirred for 10 min, and then the reaction was quenched by 5.0% NaHSO3 solution (4 mL). After ethanol precipitation, the resulting residue was purified by reversed-phase HPLC to give corresponding monophosphate compounds as a white material after lyophilization. (HPLC conditions: A: 0.1 M TEAA, B: MeCN; Flow rate: 1.0 mL/min; Shiseido MG1 (4.6 × 250 mm); UV detector: 254 nm; 35 °C).
7,8-di-Chloro-7-deaza-2′-deoxy-guanosine triphosphate (9): Yield was 30%. 1H-NMR (500 MHZ, D2O) δ (ppm) 6.50 (t, 1H, J = 7.5 Hz), 4.87–4.84 (m, 1H), 4.33–4.31 (m, 1H), 4.22–4.17 (m, 2H), 3.32–3.28 (m, 1H), 2.36–2.31 (m, 1H). 31P-NMR (202 MHz, D2O) δ (ppm) −10.9, −11.3, −23.3. HR-ESI-MS (m/z) calcd. for C11H14N4O13Cl2P3 [M − H]−: 572.9195, 574.9123, 576.9093, found: 572.9149, 574.9122, 576.9090.
7,8-di-Bromo-7-deaza-2′-deoxy-guanosine triphosphate (10): Yield was 14%. 1H-NMR (500 MHZ, D2O) δ (ppm) 6.52 (t, 1H, J = 7.5 Hz), 4.92–4.90 (m, 1H), 4.39–4.37 (m, 1H), 4.23–4.17 (m, 2H), 3.38–3.31 (m, 1H), 2.35–2.30 (m, 1H). 31P-NMR (202 MHz, D2O) δ (ppm) −9.4, −11.1, −23.0. HR-ESI-MS (m/z) calcd. for C11H14N4O13Br2P3 [M − H]−: 660.8142, 662.8122, 664.8102, found: 660.8171, 662.8152, 664.8132.
7,8-di-Iodo-7-deaza-2′-deoxy-guanosine triphosphate (11): Yield was 14%. 1H-NMR (500 MHZ, D2O) δ (ppm) 6.48 (t, 1H, J = 7.5 Hz), 4.94–4.92 (m, 1H), 4.40–4.36 (m, 1H), 4.24–4.15 (m, 2H), 3.38–3.30 (m, 1H), 2.31–2.26 (m, 1H). 31P-NMR (202 MHz, D2O) δ (ppm) −9.9, −11.1, −23.0 HR-ESI-MS (m/z) calcd. for C11H14N4O13I2P3 [M − H]−: 756.7865, found: 756.7902.
3.2. Hydrolysis of 7,8-Dihalogenated-7-deaza-dGTP, 8-CF3-7-deaza-dGTP, and 8-oxo-dGTP by MTH1
The reaction mixture was pre-incubated at 37 °C for 10 min before the addition of MTH1 to a final volume of 100 L (final concentration: 20 mM Tris-HCl, pH 7.5, 4 mM MgCl2, 40 mM NaCl, 80 g/mL bovine serum albumin (BSA), 8 mM dithiothreitol (DTT), 10% glycerol; 0.5 pmol MTH1; 50 μM 7,8-dihalogenated-7-deaza-dGTP (7, 10, and 13), 8-CF3-7-deaza-dGTP (4), and 8-oxo-dGTP). The resulting solutions were incubated at 37 °C for various times and quenched with 100 μL of stop solution (5 mM EDTA). All samples were loaded on ice before the analysis with HPLC (HPLC conditions: A: 0.1 M TEAA, B: CH3CN; Flow rate: 1.0 mL/min; Shiseido MG1 (4.6 × 250 mm); UV detector: 254 nm; column oven: 35 °C).
3.3. Inhibitory Effects of New 7-Deaza-dG Derivatives on the Hydrolytic Activity of MTH1
Twenty microliters of MTH1 buffer (100 mM Tris-HCl, pH 7.5, 20 mM MgCl2, 200 mM NaCl, 400 g/mL BSA, 40 mM DTT, and 50% glycerol), 10 μL (500 μM) of 8-oxo-dGTP, 55 μL of MilliQ, and 10 μL of various concentrations of substituted 7-deaza-dG derivatives were pre-incubated at 37 °C for 10 min (concentrations used: 7-deaza-dG, 7,8-dihalogenated-7-deaza-dG were 0, 0.5, 1, 2.5, 5, 10, 25, 50, 100, and 250 μM in 1% DMSO; 8-CF3-7-deazadGMP (3) was 0, 0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, 10, and 25 μM; 7,8-dihalogenated-7-deaza-dGMP and –dGTP were 0, 0.0125, 0.025, 0.05, 0.1, 0.25, 0.5, 1, and 2.5 μM). Five microliters of MTH1 (0.1 pmol/μL in MTH1 buffer) was added, and the reaction mixture was incubated at 37 °C for 10 min. The resulting solutions were quenched with 100 μL of stop solution (5 mM EDTA). All samples were loaded on ice before the analysis with HPLC (HPLC conditions: A: 0.1 M TEAA, B: MeCN; Flow rate: 1.0 mL/min; Shiseido MG1 (4.6 × 250 mm); UV detector: 254 nm; 35 °C). The relative hydrolysis ratio values (ratio with inhibitor/ratio without inhibitor) were plotted at each inhibitor concentration. IC50 values were assessed using images.
3.4. Molecular Docking
The three-dimensional (3D) crystal structure of MTH1 complexed with 8-oxo-dGTP (PDB ID: 5FSI) was downloaded from the Protein Data Bank (PDB). AutoDockTools 1.5.6 (ADT, http://autodock.scripps.edu/) was used to remove the water and oxo-dGTP molecules, add hydrogen atoms, merge the non-polar hydrogen atoms, check and repair missing atoms, and assign Gasteiger charges and the AD4 atom type for the 5FSI crystal structure. The area of the protein involved in the docking calculation was also set with a ADT of 60 × 60 × 60 Å3 grid size and the center x, y, and z coordinates were set at 14.1, 19.6, and 9.2, respectively, to cover the whole protein (blind docking). The MTH1 structure prepared was then saved in the .pdbqt format. The structure of 8-oxo-dGTP was extracted from the 5FSI crystal structure and used as the standard in the docking study. ADT was then used to prepare the docking input file of the 8-oxo-dGTP structure, which included the detection of torsion angles, setting the number of torsions to the maximum, the assignment of Gasteiger charges, and conversion into the .pdbqt format for flexible ligand docking using AutoDock Vina. The processed oxodGTP structure was redocked back into its crystal structure using AutoDock Vina with an optimized exhaustiveness at 400, employing 5 different seedings of docking approaches with an output of 20 final conformations for each docking (total docking output conformations = 100). The root-mean-square deviation (RMSD) values between the resulting redocked conformations and the original conformation in the crystal structure of the ligand were then calculated using Discovery Studio 4.5 (DS) software. These redocked conformations with RMSD values less than 5 Å, when compared to the 8-oxo-dGTP moiety in the 5FSI crystal structure, were grouped and defined as “Percentage of 8-oxo-dGTP-like Binding”. A conformation with the lowest aforementioned RMSD value was selected as the best conformation for the binding interaction analysis through a 2D-interaction diagram generated by DS. The value of the lowest binding energy corresponding to the best conformation, which represents its binding affinity, was also extracted from the docking result. The best performing triphosphate derivatives in vitro, 8-dihalogenated-7-deaza-dGTPs (7, 10, and 13), were then selected and prepared by modifying 8-oxo-dGTP using DS for subsequent molecular docking to predict their binding properties.
4. Conclusions
We herein designed and successfully synthesized various 7-substituted and 7,8-dihalogenated 7-deaza-dG derivatives. These derivatives were designed as molecules that bind more strongly to the 8-oxo-dGTP repair enzyme, MTH1, based on the findings of our previous studies. We found that 7,8-dihalogenated 7-deaza-dGTPs (7, 10, and 13) were not hydrolyzed by MTH1. The triphosphate part was not hydrolyzed, which is consistent with our previous findings and is a very important property. 7,8-Dichloro-7-deaza-dG derivatives (5, 6, and 7), 7,8-dibromo-7-deaza-dG derivatives (8, 9, and 10), and 7,8-diiodo-7-deaza-dG derivatives (11, 12, and 13) exerted markedly stronger inhibitory effects on the hydrolytic activity of MTH1 than previously reported corresponding 8-iodo-7-deaza-dG derivatives (14, 15, and 16). The present results confirmed that the involvement of phosphate groups was also very important for binding to MHT1. This result was supported by the Ki value. Furthermore, the inhibition mode was competitive inhibition with the substrate, according to steady-state kinetic parameters. The binding mode was also predicted by the computational molecular docking simulation, and these 7,8-dihalogenated-7-deaza-dGTP derivatives were able to bind at an active site of MTH1 and showed similar binding shapes with the substrate, 8-oxo-dGTP. The present results are the first example of the development of novel nucleotide analogues for strong binding to nucleotide repair enzymes. In the future, these compounds will be modified with membrane-permeable molecules and developed into chemical tools for the inhibition of repair enzyme activity, selectivity, and elucidation of detailed enzyme functions or their distribution in cells.
Supplementary Materials
Supplementary Materials, NMR, Mass spectrum and ADME prediction results, may be found at https://www.mdpi.com/1422-0067/22/3/1274/s1. Figure S1: Predicted ADME parameter of compound 9, Figure S2: Predicted ADME parameter of compound 10, Figure S3: Predicted ADME parameter of compound 11, Figure S4: Predicted ADME parameter of compound 12.
Author Contributions
H.S. performed the experiments, analyzed the data, and contributed to the writing of the manuscript. R.I. performed the experiments. C.H.H. performed the computational method. S.S. reviewed and edited the manuscript. Y.T. contributed to project administration, data analysis, and the writing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Grants-in-Aid for Scientific Research (B), Japan, grant number JP19H03351 for Y.T. and Fukuoka Foundation for Sound Health Cancer Research Fund for Y.T.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| MTH1 | human mutt homolog 1, also called NUDT1 |
| 7-deaza-dG | 7-deaza-2′-deoxyguanosine |
| 8-oxo-dGTP | 8-oxo-2′-deoxyguanosine-5′-triphosphate |
| 8-oxo-dGMP | 8-oxo-2′-deoxyguanosine-5′-monophosphate |
| ROS | reactive oxygen species |
| IC50 | half maximal inhibitory concentration |
| Ki | inhibition constant |
| NCS | N-chlorosuccinimide |
| NBC | N-bromosuccinimide |
| NIS | N-iodosuccinimide |
| POCl3 | phosphoryl chloride |
| TBSCl | tert-butylchlorodimethylsilane |
| DMAP | 4-dimethylaminopyridine |
| Et3N-3HF | triethylamine trihydrofluoride |
| TEA | triethylamine |
| DCC | N,N’-dicyclohexylcarbodiimide |
| DMF | N,N-dimethylformamide |
| HPLC | high performance liquid chromatography |
| Tris-HCl | tris(hydroxymethyl)aminomethane hydrochloride |
| BSA | bovine serum albumin |
| DTT | dithiothreitol |
| KM | michaelis constant |
| kcat | turnover number |
| Asp | aspartic acid |
| Trp | tryptophan |
| Lys | lysine |
References
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.A.; Crapo, J.D. Biology of disease: Free radicals and tissue injury. Lab. Investig. 1982, 47, 412–426. [Google Scholar] [PubMed]
- Topal, M.D.; Baker, M.S. DNA precursor pool: A significant target for N-methyl-N-nitrosourea in C3H/10T1/2 clone 8 cells. Proc. Natl Acad. Sci. USA 1982, 79, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Steenken, S.; Jovanovic, S.V. One-electron redox potentials of purines and pyrimidines. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Burrows, C.J.; Muller, J.G. Oxidative nucleobase modifications leading to strand scission. Chem. Rev. 1998, 98, 1109–1152. [Google Scholar] [CrossRef]
- Candeias, L.P.; Steenken, S. Reaction of HO· with guanine derivatives in aqueous solution: Formation of two different redox-active OH-adduct radicals and their unimolecular transformation reactions. Properties of G(-H). Chem. Eur. J. 2000, 6, 475–484. [Google Scholar] [CrossRef]
- Plum, G.E.; Grollman, A.P.; Johnson, F.; Breslauer, K.J. Influence of the oxidatively damaged adduct 8-oxodeoxyguanosine on the conformation, energetics, and thermodynamic stability of a DNA duplex. Biochemistry 1995, 34, 16148–16160. [Google Scholar] [CrossRef]
- Gannett, P.M.; Sura, T.P.; Virginia, W. Base pairing of 8-oxoguanosine and 8-oxo-2’-deoxyguanosine with 2’-deoxyadenosine, 2’-deoxycytosine, 2’-deoxyguanosine, and thymidine. Chem. Res. Toxicol. 1993, 6, 690–700. [Google Scholar] [CrossRef]
- McAuley-Hecht, K.E.; Leonard, G.A.; Gibson, N.J.; Thomson, J.B.; Watson, W.P.; Hunter, W.N.; Brown, T. Crystal structure of a DNA duplex containing 8-hydroxydeoxyguanine-adenine base pairs. Biochemistry 1994, 33, 10266–10270. [Google Scholar] [CrossRef]
- Culp, S.J.; Cho, B.P.; Kadlubar, F.F.; Evans, F.E. Structural and conformational analyses of 8-hydroxy-2’-deoxyguanosine. Chem. Res. Toxicol. 1989, 2, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.H.; Goddard, W.A.; Noyes, K.T.; Sowers, L.C.; Hwang, S.; Chung, D.S. First principles calculations of the tautomers and pKa values of 8-oxoguanine: implications for mutagenicity and repair. Chem. Res. Toxicol. 2002, 15, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Takeshita, M.; Grollaman, A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Hsu, G.W.; Ober, M.; Carell, T.; Beese, L.S. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature 2004, 431, 217–221. [Google Scholar] [CrossRef]
- Nakabeppu, Y. Molecular genetics and structural biology of human MutT homolog, MTH1. Mutat. Res. 2001, 477, 59–70. [Google Scholar] [CrossRef]
- Sakai, Y.; Furuichi, M.; Takahashi, M.; Mishima, M.; Iwai, S.; Shirakawa, M.; Nakabeppu, Y. A molecular basis for the selective recognition of 2-hydroxy-dATPand 8-oxo-dGTP by human MTH1. J. Biol. Chem. 2002, 277, 8579–8587. [Google Scholar] [CrossRef]
- Kamiya, H.; Yakushiji, H.; Dugue, L.; Tanimoto, M.; Pochet, S.; Nakabeppu, Y.; Harashima, H. Probing the substrate recognition mechanism of the human MTH1 protein by nucleotide analogs. J. Mol. Biol. 2004, 336, 843–850. [Google Scholar] [CrossRef]
- Gad, H.; Koolmeister, T.; Jemth, A.S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Huber, K.V.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.S.; Göktürk, C.; Sanjiv, K.; Strömberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef]
- Yin, Y.; Chen, F. Targeting human MutT homolog 1 (MTH1) for cancer eradication: Current progress and perspectives. Acta Pharm. Sin. B 2020, 10, 2259–2271. [Google Scholar] [CrossRef]
- Berglund, U.W.; Sanjiv, K.; Gad, H.; Kalderén, C.; Koolmeister, T.; Pham, T.; Gokturk, C.; Jafari, R.; Maddalo, G.; Seashore-Ludlow, B.; et al. Validation and development of MTH1 inhibitors for treatment of cancer. Ann. Oncol. 2016, 27, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Moukengue, B.; Brown, H.K.; Charrier, C.; Battaglia, S.; Baud’huin, M.; Quillard, T.; Pham, T.M.; Pateras, I.S.; Gorgoulis, V.G.; Helleday, T.; et al. TH1579, MTH1 inhibitor, delays tumour growth and inhibits metastases development in osteosarcoma mode. EBioMedicine 2020, 53, 102704. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Sanjiv, K.; Gad, H.; Pham, T.; Gokturk, C.; Rasti, A.; Zhao, Z.; He, K.; Feng, M.; Zang, Y.; et al. Karonudib is a promising anticancer therapy in hepatocellular carcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Petrocchi, A.; Leo, E.; Reyna, N.J.; Hamilton, M.M.; Shi, X.; Parker, C.A.; Mseeh, F.; Bardenhagen, J.P.; Leonard, P.; Cross, J.B.; et al. Identification of potent and selective MTH1 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1503–1507. [Google Scholar] [CrossRef]
- Kettle, J.G.; Alwan, H.; Bista, M.; Breed, J.; Davies, N.L.; Eckersley, K.; Fillery, S.; Foote, K.M.; Goodwin, L.; Jones, D.R.; et al. Potent and selective inhibitors of MTH1 probe its role in cancer cell survival. J. Med. Chem. 2016, 59, 2346–2361. [Google Scholar] [CrossRef]
- Kawamura, T.; Kawatani, M.; Muroi, M.; Kondoh, Y.; Futamura, Y.; Aono, H.; Tanaka, M.; Honda, K.; Osada, H. Proteomic profiling of small-molecule inhibitors reveals dispensability of MTH1 for cancer cell survival. Sci. Rep. 2016, 6, 26521. [Google Scholar] [CrossRef]
- Yin, Y.; Taniguchi, Y.; Sasaki, S. Synthesis of 8-halogenated-7-deaza-2’-deoxyguanosine as an 8-oxo-2’-deoxyguanosine analogue and evaluation of its base pairing properties. Tetrahedron 2014, 70, 2040–2047. [Google Scholar] [CrossRef]
- Yin, Y.; Sasaki, S.; Taniguchi, Y. Inhibitory effect of 8-halogenated 7-deaza-2′-deoxyguanosine triphosphates on human 8-oxo-2′-deoxyguanosine triphosphatase, hMTH1, activities. ChemBioChem 2016, 17, 566–569. [Google Scholar] [CrossRef]
- Yin, Y.; Sasaki, S.; Taniguchi, Y. Effects of 8-halo-7-deaza-2’-deoxyguanosine triphosphate on DNA synthesis by DNA polymerase and cell proliferation. Bioorg. Med. Chem. 2016, 24, 3856–3861. [Google Scholar] [CrossRef]
- Thavoncharoensub, N.; Maruyama, K.; Heh, C.H.; Leong, K.H.; Shi, H.; Shigematsu, Y.; Sasaki, S.; Taniguchi, Y. Synthesis of γ-N-modified 8-oxo-2′-deoxyguanosine triphosphate and its characterization. Nucleosides Nucleotides Nucleic Acids 2019, 38, 578–589. [Google Scholar] [CrossRef]
- Taktakishvili, M.; Nair, V. A new method for the phosphorylation of nucleosides. Tetrahedron Lett. 2000, 41, 7173–7176. [Google Scholar] [CrossRef]
- Caton-Williams, J.; Lin, L.; Smith, M.; Huang, Z. Convenient synthesis of nucleoside 5′-triphosphates for RNA transcription. Chem. Commun. 2011, 47, 8142–8144. [Google Scholar] [CrossRef] [PubMed]
- Caton-Williams, J.; Hoxhaj, R.; Fiaz, B.; Huang, Z. Use of a novel 5′-regioselective phosphitylating reagent for one-pot synthesis of nucleoside 5′-triphosphates from unprotected nucleosides. Curr. Protoc. Nucleic Acid Chem. 2013, 52, 1.30.1–1.30.21. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).