
Multifaceted Role of Matrix Metalloproteinases in Neurodegenerative Diseases: Pathophysiological and Therapeutic Perspectives


 , and
, and 
Abstract
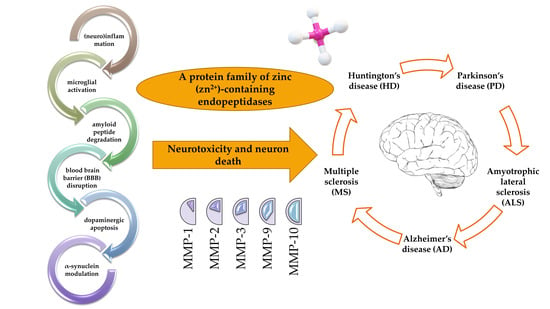
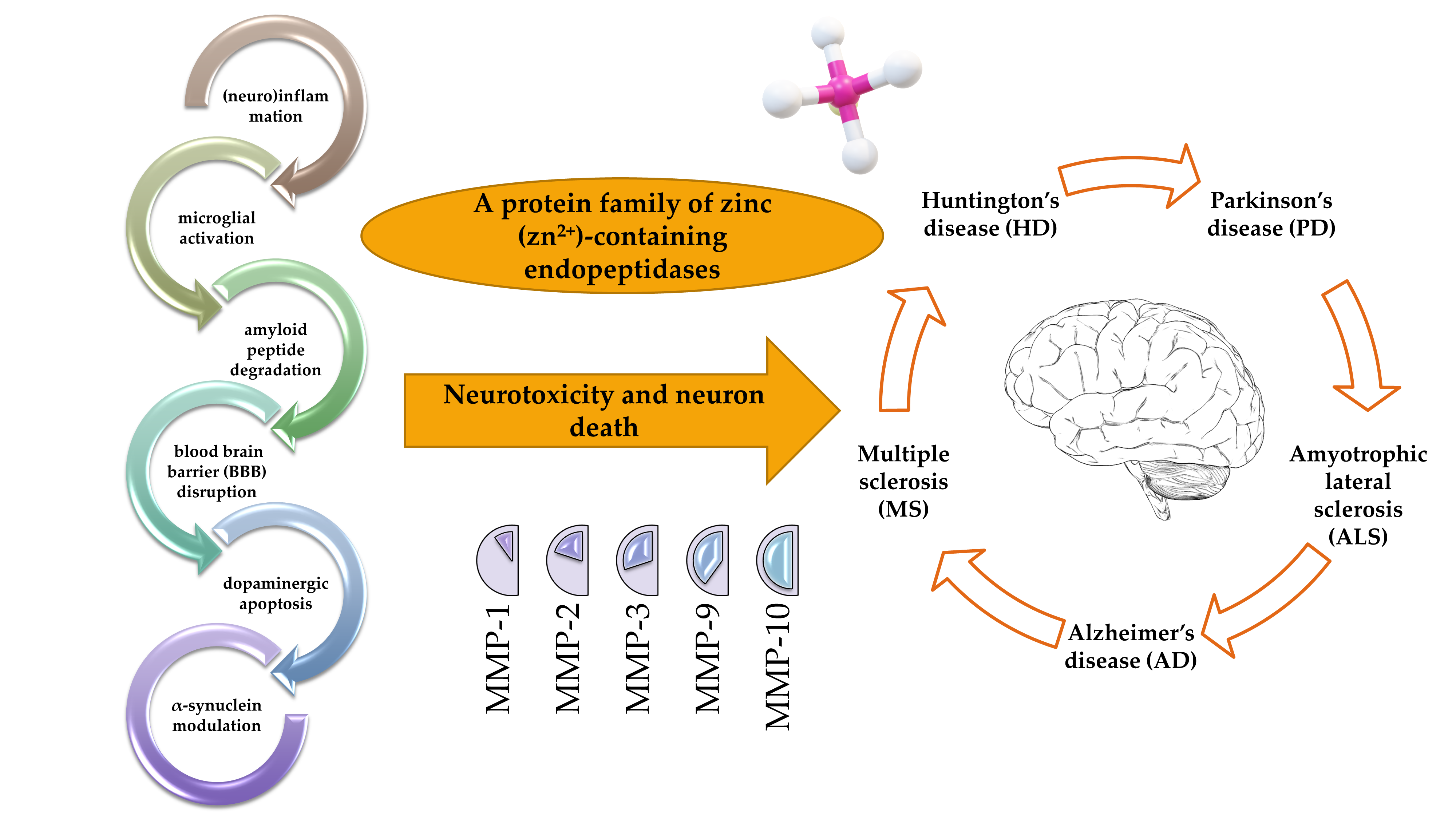
:
1. Introduction
2. An Overview of Matrix Metalloproteinases (MMPs)-Basic Structure and Function
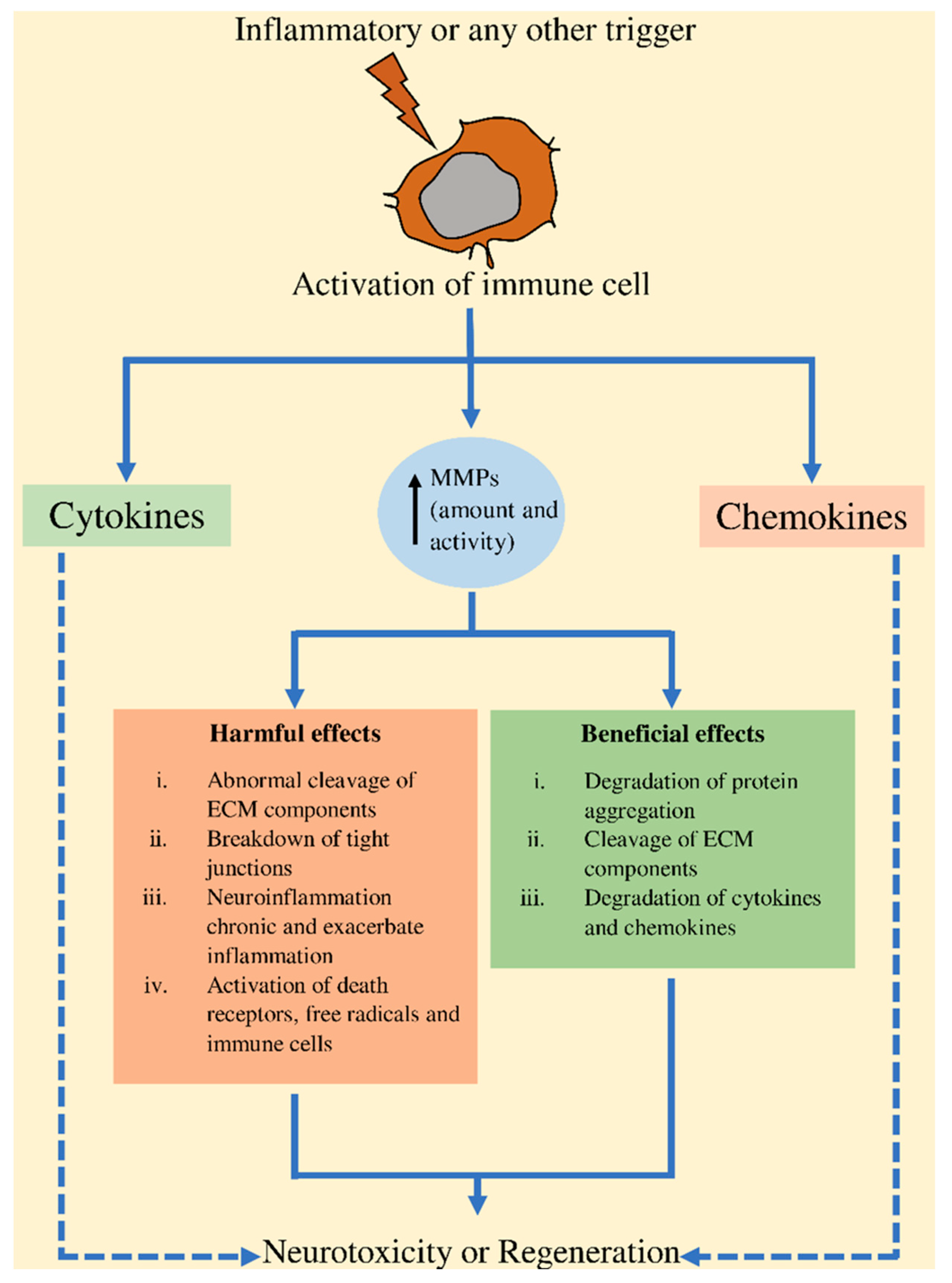
3. Involvement of MMPs in CNS
The Links between MMPs and Aquaporin-4
4. Involvement of MMPs in NDs
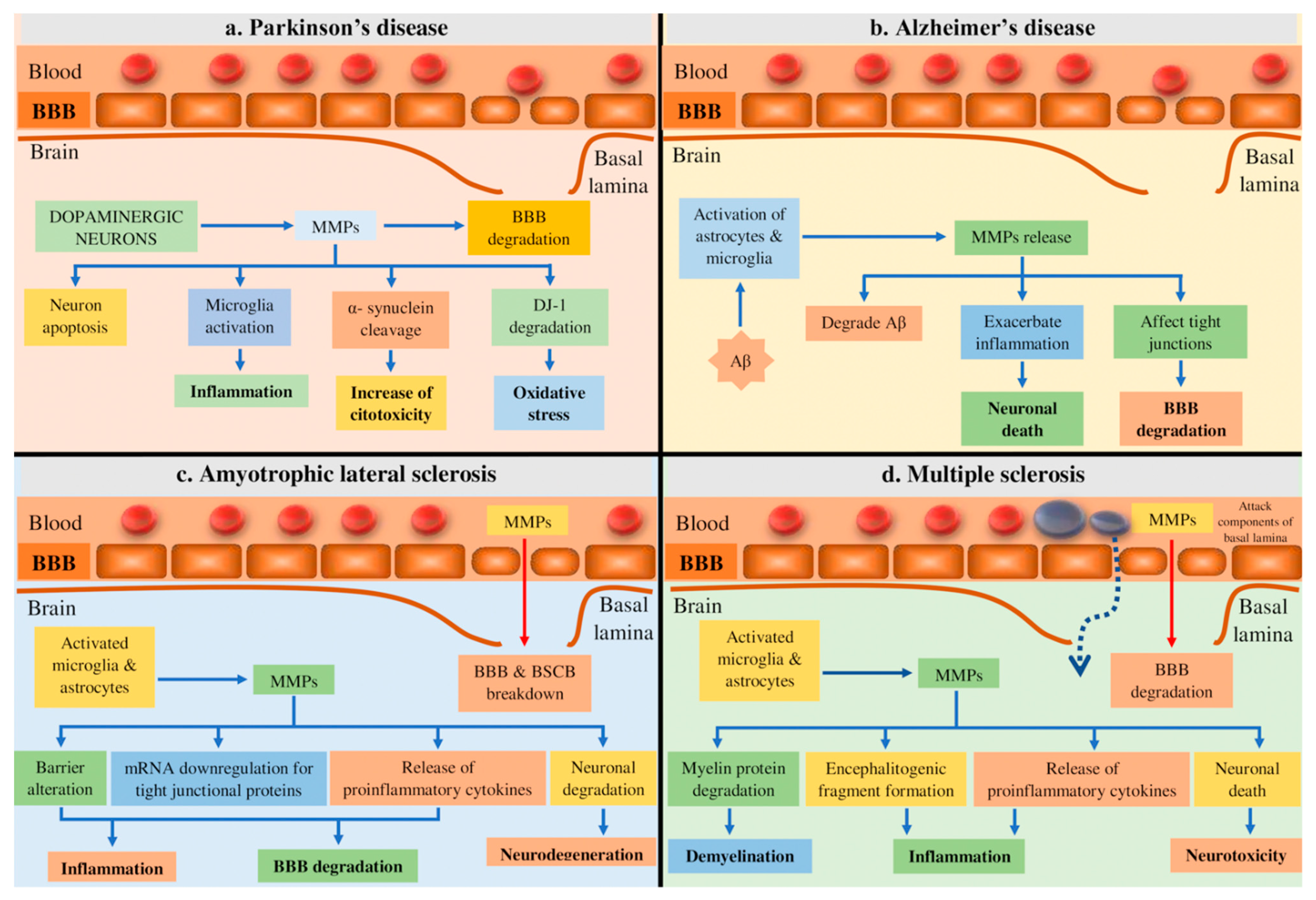
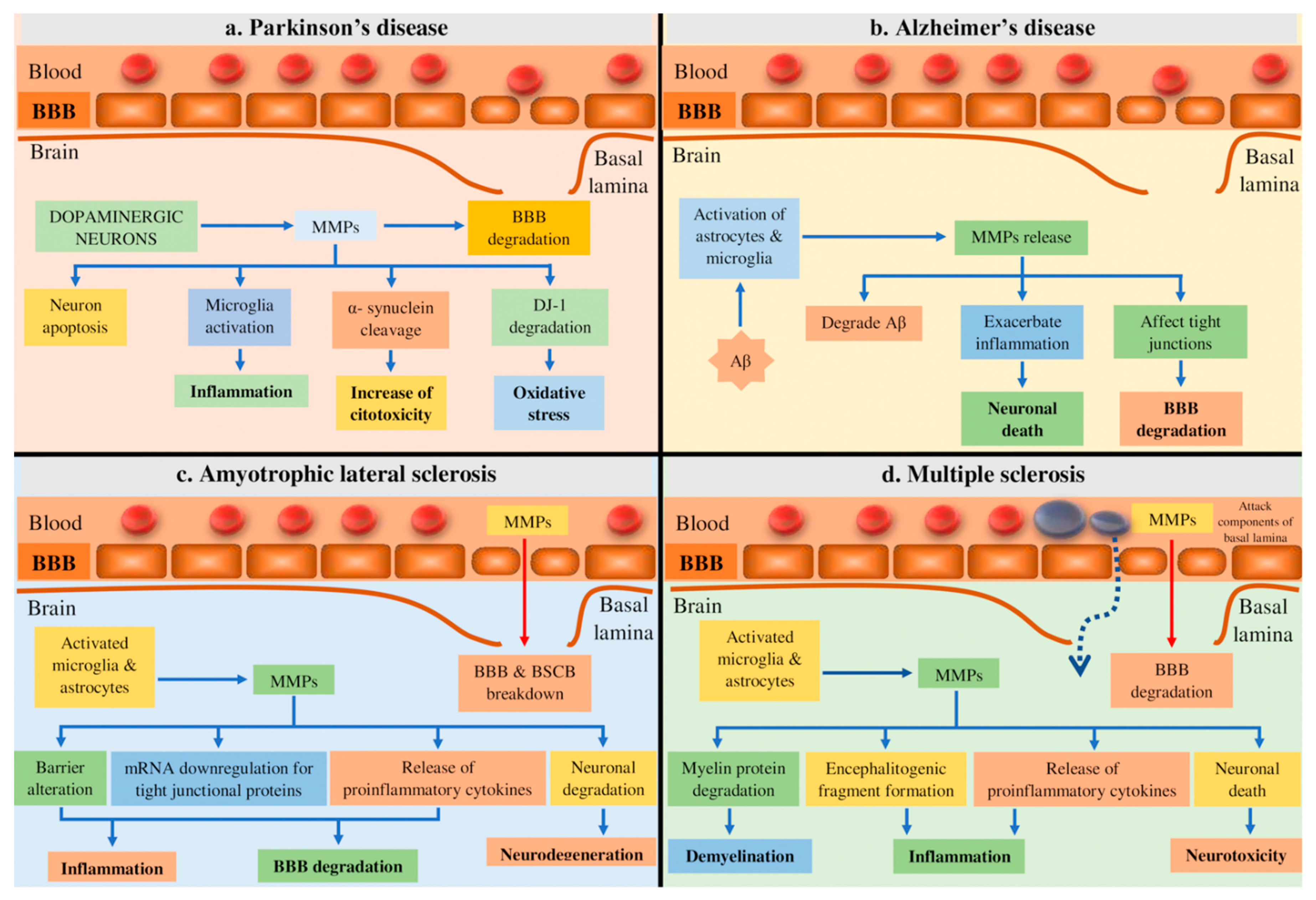
4.1. Parkinson’s Disease (PD)
4.2. Alzheimer’s Disease (AD)
4.3. Amyotrophic Lateral Sclerosis (ALS)
4.4. Multiple Sclerosis (MS)
4.5. Huntington’s Disease (HD) and Other NDs
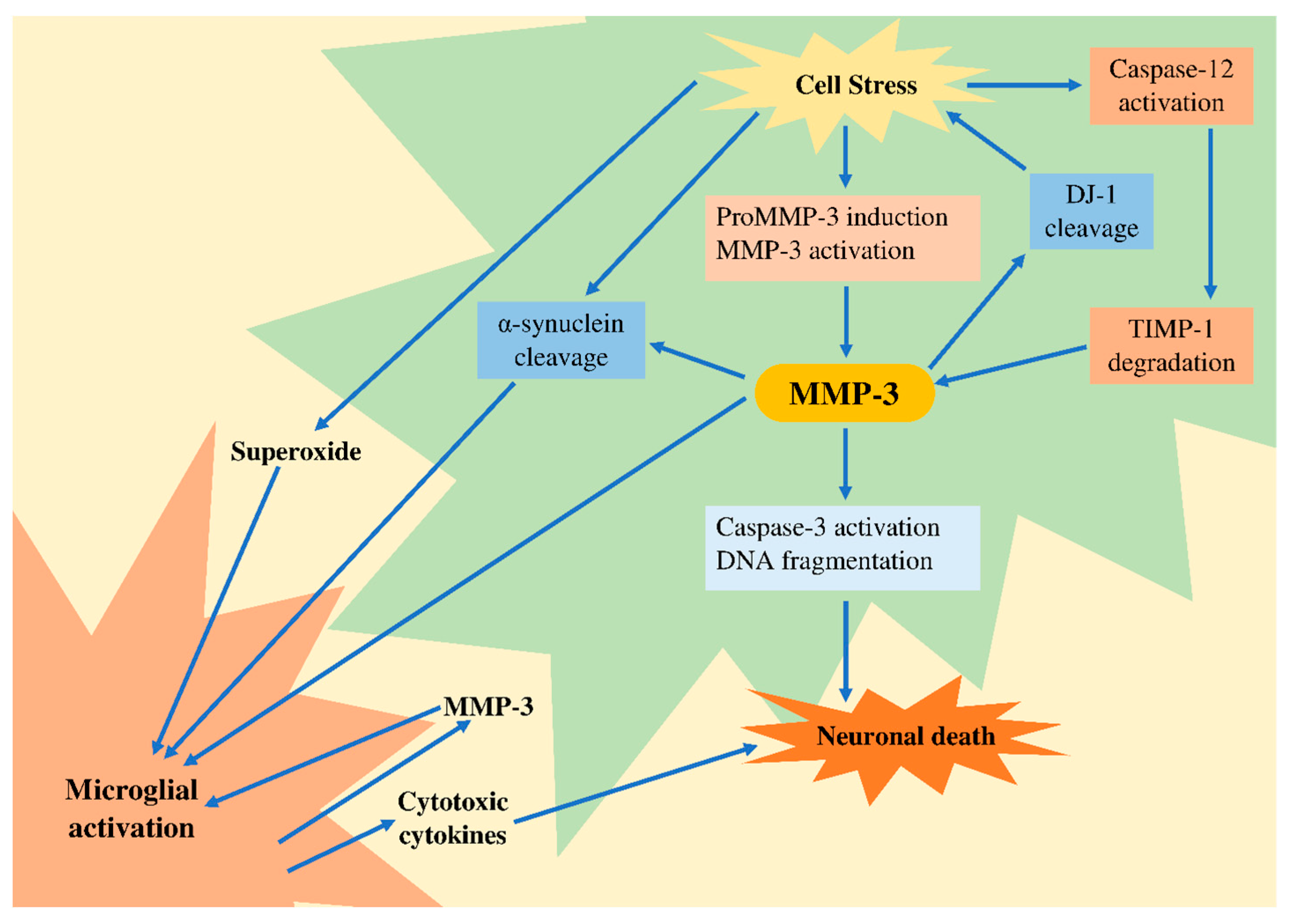
5. Potential Role of MMP-3 in Neurodegeneration
6. Therapeutic Opportunities
6.1. Alzheimer’s Disease
6.2. Parkinson’s Disease
6.3. Amyotrophic Lateral Sclerosis
6.4. Multiple Sclerosis
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lech, A.M.; Wiera, G.; Mozrzymas, J.W. Matrix metalloproteinase-3 in brain physiology and neurodegeneration. Adv. Clin. Exp. Med. 2019, 28, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.; Rochais, C.; Baranger, K.; Rivera, S.; Dallemagne, P. Matrix Metalloproteinases as New Targets in Alzheimer’s Disease: Opportunities and Challenges. J. Med. Chem. 2020, 63, 10705–10725. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Srivastava, S.K.; Chaudhuri, T.K.; Upadhyay, G. Multifaceted role of matrix metalloproteinases (MMPs). Front. Mol. Biosci. 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Brzdak, P.; Nowak, D.; Wiera, G.; Mozrzymas, J.W. Multifaceted Roles of Metzincins in CNS Physiology and Pathology: From Synaptic Plasticity and Cognition to Neurodegenerative Disorders. Front. Cell. Neurosci. 2017, 11, 178. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, G.A. Metalloproteinases and neurodegenerative diseases: Pathophysiological and therapeutic perspec-tives. Met. Med. 2015, 2, 39–50. [Google Scholar]
- Huntley, G.W. Synaptic circuit remodelling by matrix metalloproteinases in health and disease. Nat. Rev. Neurosci. 2012, 13, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Dziembowska, M.; Wlodarczyk, J. MMP9: A novel function in synaptic plasticity. Int. J. Biochem. Cell Biol. 2012, 44, 709–713. [Google Scholar] [CrossRef]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood–brain barrier: Versatile breakers and makers. Br. J. Pharm. 2016, 36, 1481–1507. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Torres, J.L.; Yescas-Gómez, P.; Torres-Romero, J.; Espinosa, O.R.; Canovas, L.L.; Tecalco-Cruz, Á.C.; Ponce-Regalado, M.D.; Alvarez-Sánchez, M.E. Matrix metalloproteinases deregulation in amyotrophic lateral sclerosis. J. Neurol. Sci. 2020, 419, 117175. [Google Scholar] [CrossRef]
- Heemels, M.-T. Neurodegenerative diseases. Nat. Cell Biol. 2016, 539, 179. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.-M.; Hwang, O. Role of matrix metalloproteinase-3 in neurodegeneration. J. Neurochem. 2010, 116, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Gentile, E.; Liuzzi, G.M. Marine pharmacology: Therapeutic targeting of matrix metalloproteinases in neuroinflammation. Drug Discov. Today 2017, 22, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.M.; Lau, L.; Yong, V.W. MMPs in the central nervous system: Where the good guys go bad. Semin. Cell Dev. Biol. 2008, 19, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.-H.; Kim, Y.-J.; Kim, Y.-G.; Joh, T.H.; Beal, M.F.; Kim, Y.-S. Role of Matrix Metalloproteinase 3-mediated α-Synuclein Cleavage in Dopaminergic Cell Death. J. Biol. Chem. 2011, 286, 14168–14177. [Google Scholar] [CrossRef] [Green Version]
- Candelario—Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metal-loproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.J.; Kim, E.-M.; Lee, J.A.; Rhim, H.; Hwang, O. Matrix metalloproteinase-3 is activated by HtrA2/Omi in dopaminergic cells: Relevance to Parkinson’s disease. Neurochem. Int. 2012, 60, 249–256. [Google Scholar] [CrossRef]
- Yong, V.W.; Zabad, R.K.; Agrawal, S.; DaSilva, A.G.; Metz, L.M. Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J. Neurol. Sci. 2007, 259, 79–84. [Google Scholar] [CrossRef]
- Yong, V.W. Metalloproteinases: Mediators of Pathology and Regeneration in the CNS. Nat. Rev. Neurosci. 2005, 6, 931–944. [Google Scholar] [CrossRef]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef]
- Ågren, M.S.; Keller, U.A.D. Matrix Metalloproteinases: How Much Can They Do? Int. J. Mol. Sci. 2020, 21, 2678. [Google Scholar] [CrossRef]
- Djurić, T.; Zivkovic, M. Overview of MMP Biology and Gene Associations in Human Diseases. Role Matrix Met. Hum. Body Pathol. 2017, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Young, D.; Das, N.; Anowai, A.; Dufour, A. Matrix Metalloproteases as Influencers of the Cells’ Social Media. Int. J. Mol. Sci. 2019, 20, 3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakiyanov, O.; Kalousová, M.; Zima, T.; Tesař, V. Matrix Metalloproteinases in Renal Diseases: A Critical Appraisal. Kidney Blood Press. Res. 2019, 44, 298–330. [Google Scholar] [CrossRef]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [Green Version]
- Radisky, E.S. Matrix metalloproteinases as drivers and therapeutic targets in breast cancer. Front. Biosci. 2015, 20, 1144–1163. [Google Scholar] [CrossRef]
- Lei, Z.; Jian, M.; Li, X.; Wei, J.; Meng, X.; Wangab, Z. Biosensors and bioassays for determination of matrix metalloproteinases: State of the art and recent advances. J. Mater. Chem. B 2019, 8, 3261–3291. [Google Scholar] [CrossRef]
- Chuang, H.-M.; Chen, Y.-S.; Harn, H.-J. The Versatile Role of Matrix Metalloproteinase for the Diverse Results of Fibrosis Treatment. Molecules 2019, 24, 4188. [Google Scholar] [CrossRef] [Green Version]
- Maskos, K. Crystal structures of MMPs in complex with physiological and pharmacological inhibitors. Biochimie 2005, 87, 249–263. [Google Scholar] [CrossRef]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef]
- Tokito, A.; Jougasaki, M. Matrix Metalloproteinases in Non-Neoplastic Disorders. Int. J. Mol. Sci. 2016, 17, 1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ra, H.-J.; Parks, W.C. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007, 26, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szóstek-Mioduchowska, A.; Słowińska, M.; Pacewicz, J.; Skarzynski, D.J.; Okuda, K. Matrix metallopeptidase expression and modulation by transforming growth factor-β1 in equine endometrosis. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page-McCaw, A.; Ewald, A.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Cauwe, B.; Steen, P.E.V.D.; Opdenakker, G. The Biochemical, Biological, and Pathological Kaleidoscope of Cell Surface Substrates Processed by Matrix Metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 113–185. [Google Scholar] [CrossRef] [Green Version]
- Blobel, C.P. Remarkable roles of proteolysis on and beyond the cell surface. Curr. Opin. Cell Biol. 2000, 12, 606–612. [Google Scholar] [CrossRef]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1803, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Radisky, E.S.; Raeeszadeh-Sarmazdeh, M.; Radisky, D.C. Therapeutic Potential of Matrix Metalloproteinase Inhibition in Breast Cancer. J. Cell. Biochem. 2017, 118, 3531–3548. [Google Scholar] [CrossRef] [Green Version]
- Yadav, L.; Puri, N.; Rastogi, V.; Satpute, P.; Ahmad, R.; Kaur, G. Matrix Metalloproteinases and Cancer—Roles in Threat and Therapy. Asian Pac. J. Cancer Prev. 2014, 15, 1085–1091. [Google Scholar] [CrossRef] [Green Version]
- Rivera, S.; García-González, L.; Khrestchatisky, M.; Baranger, K. Metalloproteinases and their tissue inhibitors in Alzheimer’s disease and other neurodegenerative disorders. Cell. Mol. Life Sci. 2019, 76, 3167–3191. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, A.M.; Spinale, F.G. Pathways of matrix metalloproteinase induction in heart failure: Bioactive molecules and transcriptional regulation. Cardiovasc. Res. 2006, 69, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akaike, T.; Sawa, T.; Miyamoto, Y.; Van Der Vliet, A.; Maeda, H. Activation of Matrix Metalloproteinases by Peroxynitrite-induced Protein S-Glutathiolation via Disulfide S-Oxide Formation. J. Biol. Chem. 2001, 276, 29596–29602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasche, Y. Matrix metalloproteinases and diseases of the central nervous system with a special emphasis on ischemic brain. Front. Biosci. 2006, 11, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arend, W.P.; Dayer, J.-M. Inhibition of the production and effects of interleukins-1 and tumor necrosis factor α in rheumatoid arthritis. Arthritis Rheum. 1995, 38, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Dual Role of Matrix Metalloproteinases (Matrixins) in Intimal Thickening and Atherosclerotic Plaque Rupture. Physiol. Rev. 2005, 85, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Lukes, A.; Mun-Bryce, S.; Lukes, M.; Rosenberg, G.A. Extracellular matrix degradation by metalloproteinases and central nervous system diseases. Mol. Neurobiol. 1999, 19, 267–284. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Dejonckheere, E.; Van Lint, P.; Demeestere, D.; Van Wonterghem, E.; Vanlaere, I.; Puimège, L.; Van Hauwermeiren, F.; De Rycke, R.; Mc Guire, C.; et al. Matrix Metalloprotease 8-Dependent Extracellular Matrix Cleavage at the Blood-CSF Barrier Contributes to Lethality during Systemic Inflammatory Diseases. J. Neurosci. 2012, 32, 9805–9816. [Google Scholar] [CrossRef]
- Gurney, K.J.; Estrada, E.Y.; Rosenberg, G.A. Blood–brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol. Dis. 2006, 23, 87–96. [Google Scholar] [CrossRef]
- Schubert-Unkmeir, A.; Konrad, C.; Slanina, H.; Czapek, F.; Hebling, S.; Frosch, M. Neisseria meningitidis Induces Brain Microvascular Endothelial Cell Detachment from the Matrix and Cleavage of Occludin: A Role for MMP-8. Plos Pathog. 2010, 6, e1000874. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Ohashi, N.; Li, W.; Eckman, C.; Nguyen, J.H. Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology 2009, 50, 1914–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brkic, M.; Balusu, S.; Van Wonterghem, E.; Gorlé, N.; Benilova, I.; Kremer, A.; Van Hove, I.; Moons, L.; De Strooper, B.; Kanazir, S.; et al. Amyloid Oligomers Disrupt Blood-CSF Barrier Integrity by Activating Matrix Metalloproteinases. J. Neurosci. 2015, 35, 12766–12778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, M.-S.; Park, J.-S.; Choi, I.-Y.; Kimf, W.-K.; Kim, H.-S. Inhibition of MMP-3 or -9 suppresses lipopolysaccharideinduced expression of proinflammatory cytokines and iNOS in microglia. J. Neurochem. 2008, 106, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, L.A.; Wetzel, M.; Rosenberg, G.A. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 2005, 50, 329–339. [Google Scholar] [CrossRef]
- Nuttall, R.K.; Silva, C.; Hader, W.; Bar-Or, A.; Patel, K.D.; Edwards, D.R.; Yong, V.W. Metalloproteinases are enriched in microglia compared with leukocytes and they regulate cytokine levels in activated microglia. Glia 2007, 55, 516–526. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Rodrigues, M.C.; Hernandez-Ontiveros, D.G.; Louis, M.K.; Willing, A.E.; Borlongan, C.V.; Sanberg, P.R. Amyotrophic lateral sclerosis: A neurovascular disease. Brain Res. 2011, 1398, 113–125. [Google Scholar] [CrossRef]
- Beroun, A.; Mitra, S.; Michaluk, P.; Pijet, B.; Stefaniuk, M.; Kaczmarek, L. MMPs in learning and memory and neuropsychiatric disorders. Cell. Mol. Life Sci. 2019, 76, 3207–3228. [Google Scholar] [CrossRef] [Green Version]
- Sanz, R.L.; Ferraro, G.B.; Kacervosky, J.; Salesse, C.; Gowing, E.; Hua, L.; Rambaldi, I.; Beaubien, F.; Holmbeck, K.; Cloutier, J.F.; et al. MT3-MMP Promotes Excitatory Synapse Formation by Promoting Nogo-66 Receptor Ectodomain Shedding. J. Neurosci. 2017, 38, 518–529. [Google Scholar] [CrossRef]
- Ould-Yahoui, A.; Sbai, O.; Baranger, K.; Bernard, A.; Gueye, Y.; Charrat, E.; Clément, B.; Gigmes, D.; Dive, V.; Girard, S.D.; et al. Role of Matrix Metalloproteinases in Migration and Neurotrophic Properties of Nasal Olfactory Stem and Ensheathing Cells. Cell Transpl. 2013, 22, 993–1010. [Google Scholar] [CrossRef]
- Muri, L.; Leppert, D.; Grandgirard, D.; Leib, S.L. MMPs and ADAMs in neurological infectious diseases and multiple sclerosis. Cell. Mol. Life Sci. 2019, 76, 3097–3116. [Google Scholar] [CrossRef]
- Chopra, S.; Overall, C.M.; Dufour, A. Matrix metalloproteinases in the CNS: Interferons get nervous. Cell. Mol. Life Sci. 2019, 76, 3083–3095. [Google Scholar] [CrossRef] [PubMed]
- Rivera, S.; Ogier, C.; Jourquin, J.; Timsit, S.; Szklarczyk, A.W.; Miller, K.; Gearing, A.J.H.; Kaczmarek, L.; Khrestchatisky, M. Gelatinase B and TIMP-1 are regulated in a cell- and time-dependent manner in association with neuronal death and glial reactivity after global forebrain ischemia. Eur. J. Neurosci. 2002, 15, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Montaner, J.; Ramiro, L.; Simats, A.; Hernández-Guillamon, M.; Delgado, P.; Bustamante, A.; Rosell, A. Matrix Metallo-proteinases and ADAMs in Stroke. Cell. Mol. Life Sci. 2019, 76, 3117–3140. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharm. Ther. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-González, L.; Pilat, D.; Baranger, K.; Rivera, S. Emerging alternative proteinases in APP metabolism and Alz-heimer’s disease pathogenesis: A focus on MT1-MMP and MT5-MMP. Front. Aging Neurosci. 2019, 11, 244. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; Macdonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799. [Google Scholar] [CrossRef] [PubMed]
- Higashida, T.; Kreipke, C.W.; Rafols, J.A.; Peng, C.; Schafer, S.; Schäfer, P.; Ding, J.Y.; Dornbos, D.; Li, X.; Guthikonda, M.; et al. The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J. Neurosurg. 2011, 114, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Zhu, P.; Yu, X.; Chen, J.; Li, J.; Yan, F.; Wang, L.; Yu, J.; Chen, G. Hydrogen sulfide attenuates brain edema in early brain injury after subarachnoid hemorrhage in rats: Possible involvement of MMP-9 induced blood-brain barrier disruption and AQP4 expression. Neurosci. Lett. 2016, 621, 88–97. [Google Scholar] [CrossRef]
- Li, M.; Na Ma, R.; Li, L.H.; Qu, Y.Z.; Gao, G.D. Astragaloside IV reduces cerebral edema post-ischemia/reperfusion correlating the suppression of MMP-9 and AQP4. Eur. J. Pharm. Ther. 2013, 715, 189–195. [Google Scholar] [CrossRef]
- Kitchen, P.; Day, R.E.; Salman, M.M.; Conner, M.T.; Bill, R.M.; Conner, A.C. Beyond water homeostasis: Diverse functional roles of mammalian aquaporins. Biochim. Biophys. Acta (BBA) Gen. Subj. 2015, 1850, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Hara-Chikuma, M.; Verkman, A.S. Physiological roles of glycerol-transporting aquaporins: The aquaglyceroporins. Cell. Mol. Life Sci. 2006, 63, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Salman, M.; Pickel, S.U.; Jennings, J.; Törnroth-Horsefield, S.; Conner, M.T.; Bill, R.M.; Conner, A.C. Water channel pore size determines exclusion properties but not solute selectivity. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salman, M.; Kitchen, P.; Woodroofe, M.N.; Brown, J.E.; Bill, R.M.; Conner, A.C.; Conner, M.T. Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)- and calmodulin-mediated mechanism. Eur. J. Neurosci. 2017, 46, 2542–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciappelloni, S.; Bouchet, D.; Dubourdieu, N.; Boué-Grabot, E.; Kellermayer, B.; Manso, C.; Marignier, R.; Oliet, S.H.; Tourdias, T.; Groc, L. Aquaporin-4 Surface Trafficking Regulates Astrocytic Process Motility and Synaptic Activity in Health and Autoimmune Disease. Cell Rep. 2019, 27, 3860–3872.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abir-Awan, M.; Kitchen, P.; Salman, M.; Conner, M.T.; Conner, A.C.; Bill, R.M. Inhibitors of Mammalian Aquaporin Water Channels. Int. J. Mol. Sci. 2019, 20, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordone, M.P.; Salman, M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; De Freitas, A.E.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Makkar, R.; Behl, T.; Bungau, S.; Zengin, G.; Mehta, V.; Kumar, A.; Uddin, M.S.; Ashraf, G.M.; Abdel-Daim, M.M.; Arora, S.; et al. Nutraceuticals in Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 4424. [Google Scholar] [CrossRef]
- Seo, J.H.; Guo, S.; Lok, J.; Navaratna, D.; Whalen, M.J.; Kim, K.-W.; Lo, E.H. Neurovascular matrix metalloproteinases and the blood-brain barrier. Curr. Pharm. Des. 2012, 18, 3645–3648. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Bungau, S.; Jhanji, R.; Kumar, A.; Mehta, V.; Zengin, G.; Brata, R.; Hassan, S.S.U.; Fratila, O. Distinctive Evidence Involved in the Role of Endocannabinoid Signalling in Parkinson’s Disease: A Perspective on Associated Therapeutic Interventions. Int. J. Mol. Sci. 2020, 21, 6235. [Google Scholar] [CrossRef]
- Kaur, G.; Behl, T.; Bungau, S.; Kumar, A.; Uddin, S.; Mehta, V.; Zengin, G.; Mathew, B.; Shah, M.A.; Arora, S. Dysregulation of the Gut-Brain Axis, Dysbiosis and Influence of Numerous Factors on Gut Microbiota Associated Parkinson’s Disease. Curr. Neuropharmacol. 2020, 19, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Salamon, A.; Zádori, D.; Szpisjak, L.; Klivényi, P.; Vécsei, L. Neuroprotection in Parkinson’s disease: Facts and hopes. J. Neural Transm. 2019, 127, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelders, G.; Baekelandt, V.; Van Der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Raymick, J.; Imam, S.Z. Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives. Int. J. Mol. Sci. 2016, 17, 904. [Google Scholar] [CrossRef] [Green Version]
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Di Paolo, T.; Tremblay, M.-È. Microglial Implication in Parkinson’s Disease: Loss of Beneficial Physiological Roles or Gain of Inflammatory Functions? Front. Cell. Neurosci. 2018, 12, 282. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yagi, S.; Yokokura, M.; Sakamoto, M. Neuroinflammation in the living brain of Parkinson’s disease. Park. Relat. Disord. 2009, 15, S200–S204. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Barcia, C.; Ros, C.M.; Annese, V.; Gómez, A.; Ros-Bernal, F.; Aguado-Llera, D.E.; Martínez-Pagán, M.E.; De Pablos, V.; Fernandez-Villalba, E.; Herrero, M.T. Erratum: IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 2012, 3, e379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. Focus Extrapyramidal Dysfunct. 2000, 2000, 277–290. [Google Scholar] [CrossRef]
- Blum-Degena, D.; Müller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Godoy, M.C.P.; Tarelli, R.; Ferrari, C.C.; Sarchi, M.I.; Pitossi, F.J. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 2008, 131, 1880–1894. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. Faseb J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, S.S.; Cho, J.J.; Choi, D.H.; Hwang, O.; Shin, D.H.; Chun, H.S.; Beal, M.F.; Joh, T.H. Matrix Metalloproteinase-3: A Novel Signaling Proteinase from Apoptotic Neuronal Cells That Activates Microglia. J. Neurosci. 2005, 25, 3701–3711. [Google Scholar] [CrossRef] [Green Version]
- McGeer, P.L.; Schwab, C.; Parent, A.; Doudet, D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann. Neurol. 2003, 54, 599–604. [Google Scholar] [CrossRef]
- Kim, Y.S.; Choi, D.H.; Block, M.L.; Lorenzl, S.; Yang, L.; Kim, Y.J.; Sugama, S.; Cho, B.P.; Hwang, O.; Browne, S.E.; et al. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. Faseb J. 2006, 21, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.-M.; Liu, B.; Hong, J.-S. Critical Role for Microglial NADPH Oxidase in Rotenone-Induced Degeneration of Dopaminergic Neurons. J. Neurosci. 2003, 23, 6181–6187. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.-K.; Ischiropoulos, H.; Przedborski, S. Blockade of Microglial Activation Is Neuroprotective in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson Disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Kim, E.-M.; Son, H.J.; Joh, T.H.; Kim, Y.S.; Kim, D.; Beal, M.F.; Hwang, O. A novel intracellular role of matrix metalloproteinase-3 during apoptosis of dopaminergic cells. J. Neurochem. 2008, 106, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Lorenzl, S.; Calingasan, N.; Yang, L.; Albers, D.S.; Shugama, S.; Gregorio, J.; Krell, H.W.; Chirichigno, J.; Joh, T.; Beal, M.F. Matrix Metalloproteinase-9 Is Elevated in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Parkinsonism in Mice. Neuromolecular Med. 2004, 5, 119–132. [Google Scholar] [CrossRef]
- Lorenzl, S.; Albers, D.S.; Narr, S.; Chirichigno, J.; Beal, M. Expression of MMP-2, MMP-9, and MMP-1 and Their Endogenous Counterregulators TIMP-1 and TIMP-2 in Postmortem Brain Tissue of Parkinson’s Disease. Exp. Neurol. 2002, 178, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Annese, V.; Herrero, M.T.; Di Pentima, M.; Gomez, A.; Lombardi, L.; Ros, C.M.; De Pablos, V.; Fernandez-Villalba, E.; De Stefano, M.E. Metalloproteinase-9 contributes to inflammatory glia activation and nigro-striatal pathway degeneration in both mouse and monkey models of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. Brain Struct. Funct. 2015, 220, 703–727. [Google Scholar] [CrossRef]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Zhao, C.; Ling, Z.; Newman, M.B.; Bhatia, A.; Carvey, P.M. TNF-α knockout and minocycline treatment attenuates blood–brain barrier leakage in MPTP-treated mice. Neurobiol. Dis. 2007, 26, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Beraud, D.; Hathaway, H.A.; Trecki, J.; Chasovskikh, S.; Johnson, D.A.; Johnson, J.A.; Federoff, H.J.; Shimoji, M.; Mhyre, T.R.; Maguire-Zeiss, K.A. Microglial Activation and Antioxidant Responses Induced by the Parkinson’s Disease Protein α-Synuclein. J. Neuroimmune Pharm. 2012, 8, 94–117. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.Y.; Park, S.M.; Lee, C.-H.; Um, J.W.; Lee, H.J.; Kim, J.; Oh, Y.J.; Lee, S.-T.; Paik, S.R.; Chung, K.C. Proteolytic Cleavage of Extracellular Secreted α-Synuclein via Matrix Metalloproteinases. J. Biol. Chem. 2005, 280, 25216–25224. [Google Scholar] [CrossRef] [Green Version]
- Levin, J.; Giese, A.; Boetzel, K.; Israel, L.; Högen, T.; Nübling, G.; Kretzschmar, H.; Lorenzl, S. Increased α-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Exp. Neurol. 2009, 215, 201–208. [Google Scholar] [CrossRef]
- Kabir, T.; Uddin, S.; Zaman, S.; Begum, Y.; Ashraf, G.M.; Bin-Jumah, M.N.; Bungau, S.G.; Mousa, S.A.; Abdel-Daim, M.M. Molecular Mechanisms of Metal Toxicity in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chadha, S.; Behl, T.; Sehgal, A.; Kumar, A.; Bungau, S. Exploring the role of mitochondrial proteins as molecular target in Alzheimer’s disease. Mitochondrion 2021, 56, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-F.; Xu, T.-H.; Yan, Y.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharm. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.L.; Figueiró, M.; Gattaz, W.F. Insights into Alzheimer disease pathogenesis from studies in transgenic animal models. Clinics 2011, 66, 45–54. [Google Scholar] [CrossRef] [Green Version]
- DeTure, M.A.; Dickson, D. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Roy, S. The physical approximation of APP and BACE-1: A key event in alzheimer’s disease pathogenesis. Dev. Neurobiol. 2018, 78, 340–347. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid Precursor Protein Trafficking, Processing, and Function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [Green Version]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Uddin, S.; Kabir, T. Oxidative stress in Alzheimer’s disease: Molecular hallmarks of underlying vulnerability. In Biological, Diagnostic and Therapeutic Advances in Alzheimer’s Disease; Ashraf, G., Alexiou, A., Eds.; Springer: Singapore, 2019; pp. 91–115. [Google Scholar] [CrossRef]
- Sinyor, B.; Mineo, J.; Ochner, C. Alzheimer’s Disease, Inflammation, and the Role of Antioxidants. J. Alzheimer’s Dis. Rep. 2020, 4, 175–183. [Google Scholar] [CrossRef]
- Feng, W.; Zhang, Y.; Wang, Z.; Xu, H.; Wu, T.; Marshall, C.; Gao, J.; Xiao, M. Microglia prevent beta-amyloid plaque formation in the early stage of an Alzheimer’s disease mouse model with suppression of glymphatic clearance. Alzheimer’s Res. Ther. 2020, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and neurotoxic effects of amyloid beta protein: Reversal by tachykinin neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory Mechanisms in Alzheimer’s Disease: Inhibition of β-Amyloid-Stimulated Proinflammatory Responses and Neurotoxicity by PPARγ Agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Cooper, C.; Liu, B.; Wilson, B.; Hong, J.-S. Microglia enhance β-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J. Neurochem. 2002, 83, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-X.; Tan, M.-S.; Yu, J.-T.; Tan, L. Matrix Metalloproteinases and Their Multiple Roles in Alzheimer’s Disease. Biomed. Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Bjerke, M.; Zetterberg, H.; Edman, Å.; Blennow, K.; Wallin, A.; Andreasson, U. Cerebrospinal Fluid Matrix Metalloproteinases and Tissue Inhibitor of Metalloproteinases in Combination with Subcortical and Cortical Biomarkers in Vascular Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 665–676. [Google Scholar] [CrossRef]
- Bruno, M.A.; Mufson, E.J.; Wuu, J.; Cuello, A.C. Increased Matrix Metalloproteinase 9 Activity in Mild Cognitive Impairment. J. Neuropathol. Exp. Neurol. 2009, 68, 1309–1318. [Google Scholar] [CrossRef]
- Lorenzl, P.D.S.; Buerger, K.; Hampel, H.; Beal, M.F. Profiles of matrix metalloproteinases and their inhibitors in plasma of patients with dementia. Int. Psychogeriatr. 2008, 20, 67–76. [Google Scholar] [CrossRef]
- Asahina, M.; Yoshiyama, Y.; Hattori, T. Expression of matrix metalloproteinase-9 and urinary-type plasminogen activator in Alzheimer’s disease brain. Clin. Neuropathol. 2001, 20, 60–63. [Google Scholar] [PubMed]
- Yan, P.; Hu, X.; Song, H.; Yin, K.; Bateman, R.J.; Cirrito, J.R.; Xiao, Q.; Hsu, F.F.; Turk, J.W.; Xu, J.; et al. Matrix Metalloproteinase-9 Degrades Amyloid-β Fibrils in Vitro and Compact Plaques in Situ. J. Biol. Chem. 2006, 281, 24566–24574. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, H.; Takuma, K.; Fukuzaki, E.; Ibi, D.; Someya, E.; Akazawa, K.-H.; Alkam, T.; Tsunekawa, H.; Mouri, A.; Noda, Y.; et al. Matrix Metalloprotease-9 Inhibition Improves Amyloid β-Mediated Cognitive Impairment and Neurotoxicity in Mice. J. Pharmacol. Exp. Ther. 2009, 331, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Kimura, K.; Haneda, M.; Ishida, Y.; Sawada, M.; Isobe, K.-I. Induction of matrix metalloproteinases (MMP3, MMP12 and MMP13) expression in the microglia by amyloid-β stimulation via the PI3K/Akt pathway. Exp. Gerontol. 2007, 42, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.-C.; Van Nostrand, W.E. Degradation of Soluble and Fibrillar Amyloid β-Protein by Matrix Metalloproteinase (MT1-MMP)in Vitro. Biochemistry 2010, 49, 1127–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nat. Cell Biol. 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Halliday, M.R.; Pomara, N.; Sagare, A.P.; Mack, W.J.; Frangione, B.; Zlokovic, B.V. Relationship Between Cyclophilin A Levels and Matrix Metalloproteinase 9 Activity in Cerebrospinal Fluid of Cognitively Normal Apolipoprotein E4 Carriers and Blood-Brain Barrier Breakdown. JAMA Neurol. 2013, 70, 1198–1200. [Google Scholar] [CrossRef] [Green Version]
- Horstmann, S.; Budig, L.; Gardner, H.; Koziol, J.; Deuschle, M.; Schilling, C.; Wagner, S. Matrix metalloproteinases in peripheral blood and cerebrospinal fluid in patients with Alzheimer’s disease. Int. Psychogeriatr. 2010, 22, 966–972. [Google Scholar] [CrossRef]
- Leake, A.; Morris, C.M.; Whateley, J. Brain matrix metalloproteinase 1 levels are elevated in Alzheimer’s disease. Neurosci. Lett. 2000, 291, 201–203. [Google Scholar] [CrossRef]
- Langenfurth, A.; Rinnenthal, J.L.; Vinnakota, K.; Prinz, V.; Carlo, A.-S.; Stadelmann, C.; Siffrin, V.; Peaschke, S.; Endres, M.; Heppner, F.; et al. Membrane-type 1 metalloproteinase is upregulated in microglia/brain macrophages in neurodegenerative and neuroinflammatory diseases. J. Neurosci. Res. 2013, 92, 275–286. [Google Scholar] [CrossRef]
- Inspector, M.; Aharon-Perez, J.; Glass-Marmor, L.; Miller, A. Matrix Metalloproteinase-9, Its Tissue Inhibitor(TIMP)-1 and CRP in Alzheimer’s Disease. Eur. Neurol. 2005, 53, 155–157. [Google Scholar] [CrossRef]
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.Ă.; Carrete, A.; Paumier, J.-M.; Py, Ă.N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer’s disease. Cell. Mol. Life Sci. 2016, 73, 217–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, M.V.D.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef] [Green Version]
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, N.; Nishiyama, A.; Kato, M.; Warita, H.; Aoki, M. Familial Amyotrophic Lateral Sclerosis. Brain Nerve 2019, 71, 1169–1181. (in Japanese). [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 1–16. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Daim, M.M.; El-Tawil, O.S.; Bungau, S.; Atanasov, A.G. Applications of Antioxidants in Metabolic Disorders and Degenerative Diseases: Mechanistic Approach. Oxidative Med. Cell. Longev. 2019, 2019, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB–mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef]
- Lim, G.P.; Backstrom, J.R.; Cullen, M.J.; Miller, C.A.; Atkinson, R.D.; Tökés, Z.A. Matrix Metalloproteinases in the Neocortex and Spinal Cord of Amyotrophic Lateral Sclerosis Patients. J. Neurochem. 2002, 67, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.I.; Appel, S.H. IgG Reactivity in the Spinal Cord and Motor Cortex in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1990, 47, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Sengillo, J.D.; Sagare, A.P.; Zhao, Z.; Ma, Q.; Zuniga, E.; Wang, Y.; Zhong, Z.; Sullivan, J.S.; Griffin, J.H.; et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1035–E1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Bowser, R.; Appel, S.H. Decreased mRNA expressıon of tıght junctıon proteıns ın lumbar spınal cords of patıents wıth als. Neurology 2009, 72, 1614–1616. [Google Scholar] [CrossRef]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Soon, C.P.; Crouch, P.J.; Turner, B.J.; McLean, C.A.; Laughton, K.M.; Atkin, J.D.; Masters, C.L.; White, A.R.; Li, Q.-X. Serum matrix metalloproteinase-9 activity is dysregulated with disease progression in the mutant SOD1 transgenic mice. Neuromuscul. Disord. 2010, 20, 260–266. [Google Scholar] [CrossRef]
- Beuche, W.; Yushchenko, M.; Mäder, M.; Maliszewska, M.; Felgenhauer, K.; Weber, F. Matrix metalloproteinase-9 is ele-vated in serum of patients with amyotrophic lateral sclerosis. NeuroReport 2000, 11, 3419–3422. [Google Scholar] [CrossRef]
- Demestre, M.; Parkin-Smith, G.; Petzold, A.; Pullen, A. The pro and the active form of matrix metalloproteinase-9 is increased in serum of patients with amyotrophic lateral sclerosis. J. Neuroimmunol. 2005, 159, 146–154. [Google Scholar] [CrossRef]
- Niebroj-Dobosz, I.; Janik, P.; Sokołowska, B.; Kwiecinski, H. Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2009, 17, 226–231. [Google Scholar] [CrossRef]
- Fang, L.; Huber-Abel, F.; Teuchert, M.; Hendrich, C.; Dorst, J.; Schattauer, D.; Zettlmeissel, H.; Wlaschek, M.; Scharffetter-Kochanek, K.; Tumani, H.; et al. Linking neuron and skin: Matrix metalloproteinases in amyotrophic lateral sclerosis (ALS). J. Neurol. Sci. 2009, 285, 62–66. [Google Scholar] [CrossRef]
- Kaplan, A.; Spiller, K.J.; Towne, C.; Kanning, K.C.; Choe, G.T.; Geber, A.; Akay, T.; Aebischer, P.; Henderson, C.E. Neuronal Matrix Metalloproteinase-9 Is a Determinant of Selective Neurodegeneration. Neuron 2014, 81, 333–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Impellizzeri, D.; Cordaro, M.; Bruschetta, G.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. N-Palmitoylethanolamine-Oxazoline as a New Therapeutic Strategy to Control Neuroinflammation: Neuroprotective Effects in Experimental Models of Spinal Cord and Brain Injury. J. Neurotrauma 2017, 34, 2609–2623. [Google Scholar] [CrossRef] [PubMed]
- Iyaswamy, A.; Krishnamoorthi, S.K.; Song, J.-X.; Yang, C.-B.; Kaliyamoorthy, V.; Zhang, H.; Sreenivasmurthy, S.G.; Malampati, S.; Wang, Z.-Y.; Zhu, Z.; et al. NeuroDefend, a novel Chinese medicine, attenuates amyloid-β and tau pathology in experimental Alzheimer’s disease models. J. Food Drug Anal. 2020, 28, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M.; Kipiani, K.; Calingasan, N.Y.; Wille, E.; Chen, J.; Heissig, B.; Rafii, S.; Lorenzl, S.; Beal, M.F. Matrix metalloproteinase-9 regulates TNF-α and FasL expression in neuronal, glial cells and its absence extends life in a transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2007, 205, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2016, 19, 1–10. [Google Scholar] [PubMed]
- Rommer, P.S.; Weber, M.S.; Illes, Z.; Zettl, U.K. Editorial: Multiple Sclerosis—From Bench to Bedside: Currents Insights Into Pathophysiological Concepts and Their Potential Impact on Patients. Front. Immunol. 2020, 11, 137. [Google Scholar] [CrossRef] [Green Version]
- Metz, L. Natural History of Multiple Sclerosis: Early Prognostic Factors. Neurol. Clin. 2011, 29, 279–292. [Google Scholar] [CrossRef]
- Vukusic, S.; Confavreux, C. Natural history of multiple sclerosis: Risk factors and prognostic indicators. Curr. Opin. Neurol. 2007, 20, 269–274. [Google Scholar] [CrossRef]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef]
- Chaudhuri, A. Multiple sclerosis is primarily a neurodegenerative disease. J. Neural Transm. 2013, 120, 1463–1466. [Google Scholar] [CrossRef]
- Könnecke, H.; Bechmann, I. The Role of Microglia and Matrix Metalloproteinases Involvement in Neuroinflammation and Gliomas. Clin. Dev. Immunol. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindberg, R.L.P.; De Groot, C.J.A.; Montagne, L.; Freitag, P.; Van Der Valk, P.; Kappos, L.; Leppert, D. The expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in lesions and normal appearing white matter of multiple sclerosis. Brain 2001, 124, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Washington, R.; Burton, J.; Todd, R.F.; Newman, W.; Dragovic, L.; Dore-Duffy, P. Expression of immunologically relevant endothelial cell activation antigens on isolated central neurvous system microvessels from patients with multiple sclerosis. Ann. Neurol. 1994, 35, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Cayrol, R.; Prat, A. Disruption of central nervous system barriers in multiple sclerosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, L.; Shah, B.; Rivers, L.; Barden, L.; Groom, A.; Chung, R.; Higazi, D.; Desmond, H.; Smith, T.; Staddon, J. Inflammation and dephosphorylation of the tight junction protein occludin in an experimental model of multiple sclerosis. Neuroscience 2007, 147, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Leppert, D.; Waubant, E.; Bürk, M.R.; Oksenberg, J.R.; Hauser, S.L. Interferon beta-1b inhibits gelatinase secretion and in vitro migration of human T cells: A possible mechanism for treatment efficacy in multiple sclerosis. Ann. Neurol. 1996, 40, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Cuzner, M.L.; Gveric, D.; Strand, C.; Loughlin, A.J.; Paemen, L.; Opdenakker, G.; Newcombe, J. The expression of tis-sue-type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: Comparison of stages in lesion evolution. J. Neuropathol. Exp. Neurol. 1996, 55, 1194–1204. [Google Scholar] [CrossRef] [Green Version]
- Cossins, J.A.; Clements, J.M.; Ford, J.; Miller, K.M.; Pigott, R.; Vos, W.; Van Der Valk, P.; De Groot, C.J.A. Enhanced expression of MMP-7 and MMP-9 in demyelinating multiple sclerosis lesions. Acta Neuropathol. 1997, 94, 590–598. [Google Scholar] [CrossRef]
- Leppert, D.; Ford, J.; Stabler, G.; Grygar, C.; Lienert, C.; Huber, S.; Miller, K.M.; Hauser, S.L.; Kappos, L. Matrix metalloproteinase-9 (gelatinase B) is selectively elevated in CSF during relapses and stable phases of multiple sclerosis. Brain 1998, 121, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.M.; Palace, J.; Stabler, G.; Ford, J.; Gearing, A.; Miller, K. Serum gelatinase B, TIMP-1 and TIMP-2 levels in multiple sclerosis. Brain 1999, 122, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Kouwenhoven, M.; Özenci, V.; Gomes, A.C.; Yarilin, D.; Giedraitis, V.; Press, R.; Link, H. Multiple sclerosis: Elevated expression of matrix metalloproteinases in blood monocytes. J. Autoimmun. 2001, 16, 463–470. [Google Scholar] [CrossRef]
- Althoff, G.E.; Wolfer, D.P.; Timmesfeld, N.; Kanzler, B.; Schrewe, H.; Pagenstecher, A. Long-Term Expression of Tissue-Inhibitor of Matrix Metalloproteinase-1 in the Murine Central Nervous System Does Not Alter the Morphological and Behavioral Phenotype but Alleviates the Course of Experimental Allergic Encephalomyelitis. Am. J. Pathol. 2010, 177, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, W.; Cannella, B.; Mazzaccaro, R.J.; Clements, J.M.; Miller, K.M.; Wucherpfenning, K.W.; Gearing, A.J.H.; Raine, C.S. Effective treatment of models of multiple sclerosis by matrix metalloproteinase inhibitors. Ann. Neurol. 1998, 44, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Masure, S.; Hurtenbach, U.; Paemen, L.; Heremans, H.; Oord, J.V.D.; Sciot, R.; Meinhardt, T.; Hämmerling, G.; Opdenakker, G.; et al. Resistance of young gelatinase B–deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J. Clin. Investig. 1999, 104, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- LaRochelle, C.; Alvarez, J.I.; Prat, A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011, 585, 3770–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilli, F.; Bertolotto, A.; Sala, A.; Hoffmann, F.; Capobianco, M.; Malucchi, S.; Glass, T.; Kappos, L.; Lindberg, R.L.P.; Leppert, D. Neutralizing antibodies against IFN- in multiple sclerosis: Antagonization of IFN- mediated suppression of MMPs. Brain 2004, 127, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Newman, T.; Woolley, S.T.; Hughes, P.M.; Sibson, N.R.; Anthony, D.C.; Perry, V. T-cell- and macrophage-mediated axon damage in the absence of a CNS-specific immune response: Involvement of metalloproteinases. Brain 2001, 124, 2203–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizuya-Oka, A.; Li, Q.; Amano, T.; Damjanovski, S.; Ueda, S.; Shi, Y.-B. Requirement for Matrix Metalloproteinase Stromelysin-3 in Cell Migration and Apoptosis during Tissue Remodeling in Xenopus laevis. J. Cell Biol. 2000, 150, 1177–1188. [Google Scholar] [CrossRef] [Green Version]
- Oh, L.Y.S.; Larsen, P.H.; Krekoski, C.A.; Edwards, D.R.; Donovan, F.; Werb, Z.; Yong, V.W. Matrix Metalloproteinase-9/Gelatinase B Is Required for Process Outgrowth by Oligodendrocytes. J. Neurosci. 1999, 19, 8464–8475. [Google Scholar] [CrossRef] [Green Version]
- Larsen, P.H.; DaSilva, A.G.; Conant, K.; Yong, V.W. Myelin Formation during Development of the CNS Is Delayed in Matrix Metalloproteinase-9 and -12 Null Mice. J. Neurosci. 2006, 26, 2207–2214. [Google Scholar] [CrossRef] [Green Version]
- Olsson, T.T.; Klementieva, O.; Gouras, G.K. Prion-like seeding and nucleation of intracellular amyloid-β. Neurobiol. Dis. 2018, 113, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.P.; Holcomb, J.; Al-Ramahi, I.; De Haro, M.; Gafni, J.; Zhang, N.; Kim, E.; Sanhueza, M.; Torcassi, C.; Kwak, S.; et al. Matrix Metalloproteinases Are Modifiers of Huntingtin Proteolysis and Toxicity in Huntington’s Disease. Neuron 2010, 67, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestroni, A.; Faull, R.L.; Strand, A.D.; Möller, T. Distinct neuroinflammatory profile in post-mortem human Huntingtonʼs disease. NeuroReport 2009, 20, 1098–1103. [Google Scholar] [CrossRef]
- Duran-Vilaregut, J.; Del Valle, J.; Manich, G.; Camins, A.; Pallàs, M.; Vilaplana, J.; Pelegrí, C. Role of matrix metalloproteinase-9 (MMP-9) in striatal blood-brain barrier disruption in a 3-nitropropionic acid model of Huntington’s disease. Neuropathol. Appl. Neurobiol. 2011, 37, 525–537. [Google Scholar] [CrossRef]
- Chang, K.; Wu, Y.-R.; Chen, Y.-C.; Chen, C.-M. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav. Immun. 2015, 44, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Lorenzl, S.; Albers, D.S.; LeWitt, P.A.; Chirichigno, J.W.; Hilgenberg, S.L.; Cudkowicz, M.E.; Beal, M.F. Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J. Neurol. Sci. 2003, 207, 71–76. [Google Scholar] [CrossRef]
- Kettlun, A.; Collados, L.; García, L.A.; Cartier, L.; Wolf, M.E.; Mosnaim, A.D.; Valenzuela, M.A. Matrix metalloproteinase profile in patients with Creuztfeldt-Jakob disease. Int. J. Clin. Pract. 2003, 57, 475–478. [Google Scholar]
- Meighan, S.E.; Meighan, P.C.; Choudhury, P.; Davis, C.J.; Olson, M.L.; Zornes, P.A.; Wright, J.W.; Harding, J.W. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J. Neurochem. 2006, 96, 1227–1241. [Google Scholar] [CrossRef]
- Li, W.; Poteet, E.; Xie, L.; Liu, R.; Wen, Y.; Yang, S.-H. Regulation of matrix metalloproteinase 2 by oligomeric amyloid β protein. Brain Res. 2011, 1387, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Daim, M.M.; Abo El-Ela, F.I.; Alshahrani, F.K.; Bin-Jumah, M.; Al-Zharani, M.; Almutairi, B.; Alyousif, M.S.; Bungau, S.; Aleya, L.; Alkahtani, S. Protective effects of thymoquinone against acrylamide-induced liver, kidney and brain oxidative damage in rats. Environ. Sci. Pollut. Res. 2020, 27, 37709–37717. [Google Scholar] [CrossRef]
- Purza, L.; Abdel-Daim, M.; Belba, A.; Iovan, C.; Bumbu, A.; Lazar, L.; Bungau, S.; Tit, D.M. Monitoring the effects of various combination of specific drug therapies at different stages of Alzheimer′s dementia. Farmacia 2019, 67, 477–481. [Google Scholar] [CrossRef]
- White, A.R.; Du, T.; Laughton, K.M.; Volitakis, I.; Sharples, R.A.; Xilinas, M.E.; Hoke, D.E.; Holsinger, R.M.D.; Evin, G.; Cherny, R.A.; et al. Degradation of the Alzheimer Disease Amyloid β-Peptide by Metal-dependent Up-regulation of Metalloprotease Activity. J. Biol. Chem. 2006, 281, 17670–17680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokołowska, B.; Jozwik, A.; Niebroj-Dobosz, I.; Janik, P.; Kwiecinski, H. Evaluation of matrix metalloproteinases in serum of patients with amyotrophic lateral sclerosis with pattern recognition methods. J. Physiol. Pharm. Off. J. Pol. Physiol. Soc. 2009, 60, 117–120. [Google Scholar]
- Deb, S.; Zhang, J.W.; Gottschall, P.E. β-Amyloid induces the production of active, matrix-degrading proteases in cultured rat astrocytes. Brain Res. 2003, 970, 205–213. [Google Scholar] [CrossRef]
- Backstrom, J.R.; Lim, G.P.; Cullen, M.J.; Tökés, Z.A. Matrix Metalloproteinase-9 (MMP-9) Is Synthesized in Neurons of the Human Hippocampus and Is Capable of Degrading the Amyloid-β Peptide (1–40). J. Neurosci. 1996, 16, 7910–7919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, A.; Sobel, R.A. Matrix Metalloproteinases in the Normal Human Central Nervous System, Microglial Nodules, and Multiple Sclerosis Lesions. J. Neuropathol. Exp. Neurol. 1996, 55, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Enghild, J.J.; Suzuki, K.; Salvesen, G. Stepwise activation mechanisms of the precursor of matrix metalloproteinase 3 (stromelysin) by proteinases and (4-aminophenyl)mercuric acetate. Biochemistry 1990, 29, 5783–5789. [Google Scholar] [CrossRef]
- Nagase, H.; Fields, C.; Fields, G. Design and characterization of a fluorogenic substrate selectively hydrolyzed by stromelysin 1 (matrix metalloproteinase-3). J. Biol. Chem. 1994, 269, 20952–20957. [Google Scholar] [CrossRef]
- McClain, J.A.; Phillips, L.L.; Fillmore, H.L. Increased MMP-3 and CTGF expression during lipopolysaccharide-induced dopaminergic neurodegeneration. Neurosci. Lett. 2009, 460, 27–31. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009, 8, 205–216. [Google Scholar] [CrossRef]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, S.; Gottschall, P.E. Increased Production of Matrix Metalloproteinases in Enriched Astrocyte and Mixed Hippocampal Cultures Treated with β-Amyloid Peptides. J. Neurochem. 2002, 66, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Asahina, M.; Hattori, T. Selective distribution of matrix metalloproteinase-3 (MMP-3) in Alzheimer’s disease brain. Acta Neuropathol. 2000, 99, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Steen, P.E.V.D.; Sang, Q.-X.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Sang, Q.-X.; Jin, Y.; Newcomer, R.; Monroe, S.; Fang, X.; Hurst, D.R.; Lee, S.; Cao, Q.; Schwartz, M. Matrix Metalloproteinase Inhibitors as Prospective Agents for the Prevention and Treatment of Cardiovascular and Neoplastic Diseases. Curr. Top. Med. Chem. 2006, 6, 289–316. [Google Scholar] [CrossRef]
- Miyazaki, K.; Hasegawa, M.; Funahashi, K.; Umeda, M. A metalloproteinase inhibitor domain in Alzheimer amyloid protein precursor. Nat. Cell Biol. 1993, 362, 839–841. [Google Scholar] [CrossRef]
- Jang, J.; Kim, K.; Yoon, J.; Park, C.B. Piezoelectric materials for ultrasound-driven dissociation of Alzheimer’s β-amyloid aggregate structure. Biomaterials 2020, 255, 120165. [Google Scholar] [CrossRef]
- Roher, A.; Kasunic, T.; Woods, A.; Cotter, R.; Ball, M.; Fridman, R. Proteolysis of Aβ Peptide from Alzheimer Disease Brain by Gelatinase, A. Biochem. Biophys. Res. Commun. 1994, 205, 1755–1761. [Google Scholar] [CrossRef]
- Yin, K.-J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.-F.; Turk, J.; et al. Matrix Metalloproteinases Expressed by Astrocytes Mediate Extracellular Amyloid-beta Peptide Catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef]
- Banning, L.C.; Ramakers, I.H.; Deckers, K.; Verhey, F.; Aalten, P. Apolipoprotein E and affective symptoms in mild cognitive impairment and Alzheimer’s disease dementia: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2019, 96, 302–315. [Google Scholar] [CrossRef]
- Hoie, E. Alzheimer’s Disease: Current Treatments and Potential New Agents. Us Pharm. 2019, 44, 20–23. [Google Scholar]
- Garcia-Alloza, M.; Prada, C.; Lattarulo, C.; Fine, S.; Borrelli, L.A.; Betensky, R.; Greenberg, S.M.; Frosch, M.P.; Bacskai, B.J. Matrix metalloproteinase inhibition reduces oxidative stress associated with cerebral amyloid angiopathyin vivoin transgenic mice. J. Neurochem. 2009, 109, 1636–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-talks among GBA Gene Mutations, GCase, and α-synuclein in GBA Associated Parkinson’s Disease with their Targeted Therapeutic Approaches: A Comprehensive Review. Transl. Neurodeg. 2021, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4+T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Madav, Y.; Wairkar, S.; Prabhakar, B. Recent therapeutic strategies targeting beta amyloid and tauopathies in Alzheimer’s disease. Brain Res. Bull. 2019, 146, 171–184. [Google Scholar] [CrossRef]
- Peress, N.; Perillo, E.D.; Zucker, S. Localization of Tissue Inhibitor of Matrix Metalloproteinases in Alzheimerʼs Disease and Normal Brain. J. Neuropathol. Exp. Neurol. 1995, 54, 16–22. [Google Scholar] [CrossRef]
- Smith, M.E.; Amaducci, L.A. Observations on the effects of protease inhibitors on the suppression of experimental allergic encephalomyelitis. Neurochem. Res. 1982, 7, 541–554. [Google Scholar] [CrossRef]
- Hewson, A.K.; Smith, T.; Leonard, J.P.; Cuzner, M.L. Suppression of experimental allergic encephalomyelitis in the Lewis rat by the matrix metalloproteinase inhibitor Ro31-9790. Inflamm. Res. 1995, 44, 345–349. [Google Scholar] [CrossRef]
- Gijbels, K.; Galardy, R.E.; Steinman, L. Reversal of experimental autoimmune encephalomyelitis with a hydroxamate inhibitor of matrix metalloproteases. J. Clin. Investig. 1994, 94, 2177–2182. [Google Scholar] [CrossRef]
- Graesser, D.; Mahooti, S.; Haas, T.; Davis, S.; Clark, R.B.; Madri, J.A. The interrelationship of alpha4 integrin and matrix metalloproteinase-2 in the pathogenesis of experimental autoimmune encephalomyelitis. Lab. Investig. 1998, 78, 1445–1458. [Google Scholar]
- Smith, M.E.; van der Maesen, K.; Somera, F.P. Macrophage and microglial responses to cytokines in vitro: Phagocytic ac-tivity, proteolytic enzyme release, and free radical production. J. Neurosci. Res. 1998, 54, 68–78. [Google Scholar] [CrossRef]
- Norga, K.; Paemen, L.; Masure, S.; Dillen, C.; Heremans, H.; Billiau, A.; Carton, H.; Cuzner, L.; Olsson, T.; Van Damme, J.; et al. Prevention of acute autoimmune encephalomyelitis and abrogation of relapses in murine models of multiple sclerosis by the protease inhibitor D-penicillamine. Inflamm. Res. 1995, 44, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Gijbels, K.; Masure, S.; Carton, H.; Opdenakker, G. Gelatinase in the cerebrospinal fluid of patients with multiple sclerosis and other inflammatory neurological disorders. J. Neuroimmunol. 1992, 41, 29–34. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Dencoff, J.E.; Correa, N.; Reiners, M.; Ford, C.C. Effect of steroids on CSF matrix metalloproteinases in multiple sclerosis: Relation to blood-brain barrier injury. Neurology 1996, 46, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.M.; Cossins, J.A.; Wells, G.M.; Corkill, D.J.; Helfrich, K.; Wood, L.; Pigott, R.; Stabler, G.; Ward, G.A.; Gearing, A.J.; et al. Matrix metalloproteinase expression during experimental autoimmune encephalomyelitis and effects of a combined matrix metalloproteinase and tumour necrosis factor-α inhibitor. J. Neuroimmunol. 1997, 74, 85–94. [Google Scholar] [CrossRef]
- Richter, F.; Zettlitz, K.A.; Seifert, O.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R.E. Monovalent TNF receptor 1-selective antibody with improved affinity and neutralizing activity. mAbs 2018, 11, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.; Butler, D.; Scallon, B.J.; O’Neill, J.K.; Turk, J.L.; Feldmann, M. Control of established experimental allergic encephalomyelitis by inhibition of tumor necrosis factor (TNF) activity within the central nervous system using monoclonal antibodies and TNF receptor-immunoglobulin fusion proteins. Eur. J. Immunol. 1994, 24, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Kanchi, P.K.; DasMahapatra, A.K. Polyproline chains destabilize the Alzheimer’s amyloid-β protofibrils: A molecular dynamics simulation study. J. Mol. Graph. Model. 2019, 93, 107456. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Huch, M. Disease modelling in human organoids. Dis. Model. Mech. 2019, 12, dmm039347. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; De Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef]
- Ma, C.; Peng, Y.; Li, H.; Chen, W. Organ-on-a-Chip: A New Paradigm for Drug Development. Trends Pharm. Sci. 2020, 42, 119–133. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MMPs Involved/ Neurodegenerative Disease | Model System | Role of MMPs | Ref. |
|---|---|---|---|
| MMP-2/PD | In vitro (PC12 cells) | Activates microglia | [97] |
| Patients | Detected in microglia and astrocytes | [99] | |
| In vitro (neuron-glia culture) | Induces DAergic neuronal death in culture of glia-neuron (mesencephalic) | [103] | |
| MMP-2/AD | In vivo (rats) | Involved in synaptic plasticity | [199] |
| In-vitro (microglial cell line) | Increased microglial expression after Aβ oligomer stimulation | [133] | |
| MMP-1/AD | In vitro (primary astrocytes) | Low MMP-9 levels and decreased MMP-2 activity after Aβ oligomer stimulation. | [139] |
| Patients | Increased MMP-1 levels in AD patients | [200,201] | |
| MMP-3/PD | In vitro (primary cultured DAergic neurons) | MMP-3 neuronal secretion | [102] |
| In vitro (primary mesencephalic cultures) | Induces NO production in microglia | [98,100] | |
| In vitro (human DAergic neuroblastoma) | α-synuclein proteolysis | [202] | |
| MMP-3/AD | Patients | Significant upregulation of MMP-3 plasma levels | [138] |
| In vivo (icv injections of Aβ oligomer) | Increased MMP-3 expression | [52] | |
| In vitro (APP-CHO cells) | Ability to degrade Aβ | [203] | |
| In vivo (icv injection of Aβ oligomer) | Enhanced permeability of BCSFB | [52] | |
| MMP-2/ALS | Patients | Increased permeability of BBB | [204] |
| Patients (serum) | To evaluate ALS progression | [161] | |
| MMP-9/AD | In vitro (astrocytes) | Detected in astrocytes when treated with fibrillar and soluble Aβ | [205] |
| In vivo (rats) | Involved in synaptic plasticity | [199] | |
| In vivo (mice) | Increased levels in hippocampus on icv injection | [132] | |
| Patients (CSF) | Activation of MMP-9/CypA in pericytes, BBB disruption | [137] | |
| In vitro (isolates from brain of patients) | Cleavage of Aβ1-40 by MMP-9 | [206] | |
| MMP-3/ALS | In vivo (G93A SOD1 mice) | Upregulation of neuronal FasL and TNF | [157] |
| In vivo (mutant SOD1 transgenic mice) | Dysregulated MMP-3 activity with ALS progression | [165] | |
| In vivo (G93A SOD1 mice) | Encourages motor cell death in neurons | [165] | |
| MMP-1/MS | Patients (monocytes) | Increased mRNA levels of MMP-1 | [182] |
| Patients (postmortem brain samples) | Weak astrocytic expression | [207] | |
| MMP-3/MS | Patients (monocytes) | Increased mRNA levels of MMP-3 | [182] |
| Patients (postmortem brain samples) | Expression in endothelial cells | [207] | |
| MMP-9/HD | In vivo (3-nitropropionic acid animal disease model) | Increased expression of MMP-9 | [195] |
| MMP-9/PD | Patients (postmortem brain tissues) | Increased expression of MMP-9 in SN | [116] |
| In vivo (MPTP induced PD in monkey and mouse model) | Primary localization of MMP-9 in neurons | [103] | |
| MMP-7/MS | Patients (monocytes) | Increased mRNA levels of MMP-7 | [182] |
| Patients (postmortem brain samples) | Secreted by blood vessels | [179] | |
| MMP-9/ALS | Patients (CSF and skin) Patients (CSF) | Elevated in CSF and skin Low CSF levels of MMP-9 | [160,161] |
| Patients (serum) | MMP-9 as marker distinguishing between healthy individuals and ALS | [204] | |
| MMP-10/HD | In vitro (striatal cell culture) | Cleaves huntingtin | [181] |
| MMP-9/MS | Patients (CSF samples) | Secreted by macrophages and T-cells, leads to damage of tissue | [160] |
| Patients (serum) | Increased serum levels together with TIMP-1 and -2 | [173] | |
| MMP-14/HD | In vitro (striatal cell culture) | MMP-14 knockdown reduces toxicity | [181] |
| MMP-12/AD | In vitro (microglial cell line) | Increase in microglia | [126] |
| MMP-23/HD | In vitro (striatal cell line) | MMP-23 knockdown reduces toxicity | [193] |
| MMP-13/AD | In vitro (microglial cell line) | Increase in microglia | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Kaur, G.; Sehgal, A.; Bhardwaj, S.; Singh, S.; Buhas, C.; Judea-Pusta, C.; Uivarosan, D.; Munteanu, M.A.; Bungau, S. Multifaceted Role of Matrix Metalloproteinases in Neurodegenerative Diseases: Pathophysiological and Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22, 1413. https://doi.org/10.3390/ijms22031413
Behl T, Kaur G, Sehgal A, Bhardwaj S, Singh S, Buhas C, Judea-Pusta C, Uivarosan D, Munteanu MA, Bungau S. Multifaceted Role of Matrix Metalloproteinases in Neurodegenerative Diseases: Pathophysiological and Therapeutic Perspectives. International Journal of Molecular Sciences. 2021; 22(3):1413. https://doi.org/10.3390/ijms22031413
Chicago/Turabian StyleBehl, Tapan, Gagandeep Kaur, Aayush Sehgal, Shaveta Bhardwaj, Sukhbir Singh, Camelia Buhas, Claudia Judea-Pusta, Diana Uivarosan, Mihai Alexandru Munteanu, and Simona Bungau. 2021. "Multifaceted Role of Matrix Metalloproteinases in Neurodegenerative Diseases: Pathophysiological and Therapeutic Perspectives" International Journal of Molecular Sciences 22, no. 3: 1413. https://doi.org/10.3390/ijms22031413
APA StyleBehl, T., Kaur, G., Sehgal, A., Bhardwaj, S., Singh, S., Buhas, C., Judea-Pusta, C., Uivarosan, D., Munteanu, M. A., & Bungau, S. (2021). Multifaceted Role of Matrix Metalloproteinases in Neurodegenerative Diseases: Pathophysiological and Therapeutic Perspectives. International Journal of Molecular Sciences, 22(3), 1413. https://doi.org/10.3390/ijms22031413