
Multicellular 3D Models to Study Tumour-Stroma Interactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Rise and Fall of 2D Cell Cultures
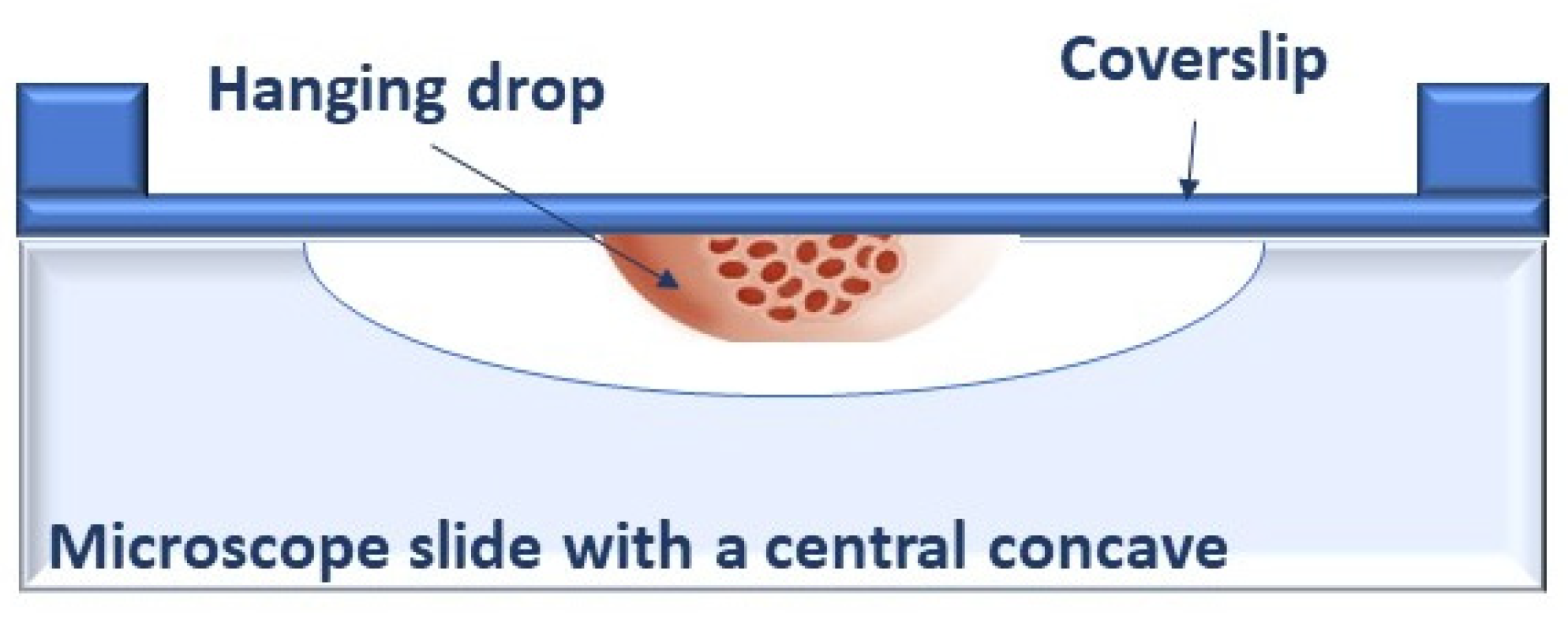
2. Back to the Past: Restarting from 3D Spheroids
3. Cell–ECM Interactions in 3D Spheroids
3.1. Matrix Stiffness
3.2. Matrix Remodelling
3.3. Matrix and Immune Cells
4. Cell–Cell Communications
4.1. Cancer-Associated Fibroblasts (CAFs)
4.2. Immune Cells
4.3. Endothelial Cells
5. Beyond Multicellular Spheroids
5.1. Engineered Metastatic Niches
5.2. Cancer-on-Chips
6. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nurse, P. The Incredible Life and Times of Biological Cells. Science 2000, 289, 1711–1716. [Google Scholar] [CrossRef] [Green Version]
- Ringer, S. A Further Contribution Regarding the Influence of the Different Constituents of the Blood on the Contraction of the Heart. J. Physiol 1883, 4, 29–42. [Google Scholar] [CrossRef]
- Harrison, R.G.; Greenman, M.J.; Mall, F.P.; Jackson, C.M. Observations of the Living Developing Nerve Fiber. Anat. Rec. 1907, 1, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Foty, R. A Simple Hanging Drop Cell Culture Protocol for Generation of 3D Spheroids. J. Vis. Exp. 2011, e2720. [Google Scholar] [CrossRef]
- Carrel, A.; Burrows, M.T. Cultivation of adult tissues and organs outside of the body. J. Am. Med. Assoc. 1910, 55, 1379–1381. [Google Scholar] [CrossRef] [Green Version]
- Carrel, A. On the permanent life of tissues outside of the organism. J. Exp Med. 1912, 15, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Earle, W.R.; Schilling, E.L.; Stark, T.H.; Straus, N.P.; Brown, M.F.; Shelton, E. Production of Malignancy in Vitro. IV. The Mouse Fibroblast Cultures and Changes Seen in the Living Cells. JNCI J. Natl. Cancer Inst. 1943, 4, 165–212. [Google Scholar] [CrossRef]
- Scherer, W.F.; Syverton, J.T.; Gey, G.O. Studies on the propagation in vitro of poliomyelitis viruses: IV. viral multiplication in a stable strain of human malignant epithelial cells (strain hela) derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 1953, 97, 695–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, J.R. HeLa Cells 50 Years on: The Good, the Bad and the Ugly. Nat. Rev. Cancer 2002, 2, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, X.; Guo, Q.; Yang, J.; Yang, Y.; Wei, S.; Su, X. HeLa-CCL2 Cell Heterogeneity Studied by Single-Cell DNA and RNA Sequencing. PLoS ONE 2019, 14, e0225466. [Google Scholar] [CrossRef] [Green Version]
- Nelson-Rees, W.A.; Daniels, D.W.; Flandermeyer, R.R. Cross-Contamination of Cells in Culture. Science 1981, 212, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Frattini, A.; Fabbri, M.; Valli, R.; De Paoli, E.; Montalbano, G.; Gribaldo, L.; Pasquali, F.; Maserati, E. High Variability of Genomic Instability and Gene Expression Profiling in Different HeLa Clones. Sci. Rep. 2015, 5, 15377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and Transcriptional Evolution Alters Cancer Cell Line Drug Response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Hinohara, K.; Polyak, K. Intratumoral Heterogeneity: More than Just Mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Hynds, R.E.; Vladimirou, E.; Janes, S.M. The Secret Lives of Cancer Cell Lines. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Breslin, S.; O’Driscoll, L. The Relevance of Using 3D Cell Cultures, in Addition to 2D Monolayer Cultures, When Evaluating Breast Cancer Drug Sensitivity and Resistance. Oncotarget 2016, 7, 45745–45756. [Google Scholar] [CrossRef] [Green Version]
- Däster, S.; Amatruda, N.; Calabrese, D.; Ivanek, R.; Turrini, E.; Droeser, R.A.; Zajac, P.; Fimognari, C.; Spagnoli, G.C.; Iezzi, G.; et al. Induction of Hypoxia and Necrosis in Multicellular Tumor Spheroids Is Associated with Resistance to Chemotherapy Treatment. Oncotarget 2016, 8, 1725–1736. [Google Scholar] [CrossRef] [Green Version]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of Cancer Cells in 2D vs 3D Culture Reveals Differences in AKT–MTOR–S6K Signaling and Drug Responses. J. Cell Sci. 2017, 130, 203–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, W.; Tong, X.; Yang, C.; Zhang, F.; Bao, C.; Chen, H.; Liu, L.; Li, M.; Ye, F.; Fan, Q.; et al. Behaviors of Glioblastoma Cells in in Vitro Microenvironments. Sci. Rep. 2019, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Melissaridou, S.; Wiechec, E.; Magan, M.; Jain, M.V.; Chung, M.K.; Farnebo, L.; Roberg, K. The Effect of 2D and 3D Cell Cultures on Treatment Response, EMT Profile and Stem Cell Features in Head and Neck Cancer. Cancer Cell Int. 2019, 19, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Zoetemelk, M.; Rausch, M.; Colin, D.J.; Dormond, O.; Nowak-Sliwinska, P. Short-Term 3D Culture Systems of Various Complexity for Treatment Optimization of Colorectal Carcinoma. Sci. Rep. 2019, 9, 7103. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Corrigendum: Estimation of Clinical Trial Success Rates and Related Parameters. Biostatistics 2019, 20, 366. [Google Scholar] [CrossRef] [PubMed]
- Moscona, A. The development in vitro of chimeric aggregates of dissociated embryonic chick and mouse cells. Proc. Natl. Acad. Sci. USA 1957, 43, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inch, W.R.; McCredie, J.A.; Sutherland, R.M. Growth of Nodular Carcinomas in Rodents Compared with Multi-Cell Spheroids in Tissue Culture. Growth 1970, 34, 271–282. [Google Scholar] [PubMed]
- Haycock, J.W. 3D Cell Culture: A Review of Current Approaches and Techniques. Methods Mol. Biol. 2011, 695, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, K.; Haeger, J.-D.; Heger, J.; Pastuschek, J.; Photini, S.M.; Yan, Y.; Lupp, A.; Pfarrer, C.; Mrowka, R.; Schleußner, E.; et al. Generation of Multicellular Breast Cancer Tumor Spheroids: Comparison of Different Protocols. J. Mammary Gland. Biol. Neoplasia 2016, 21, 89–98. [Google Scholar] [CrossRef]
- Schmidt, M.; Scholz, C.-J.; Polednik, C.; Roller, J. Spheroid-Based 3-Dimensional Culture Models: Gene Expression and Functionality in Head and Neck Cancer. Oncol. Rep. 2016, 35, 2431–2440. [Google Scholar] [CrossRef] [Green Version]
- Stadler, M.; Scherzer, M.; Walter, S.; Holzner, S.; Pudelko, K.; Riedl, A.; Unger, C.; Kramer, N.; Weil, B.; Neesen, J.; et al. Exclusion from Spheroid Formation Identifies Loss of Essential Cell-Cell Adhesion Molecules in Colon Cancer Cells. Sci. Rep. 2018, 8, 1151. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, M. Identification of the Region of Alpha-Catenin That Plays an Essential Role in Cadherin-Mediated Cell Adhesion. J. Biol. Chem. 1998, 273, 29524–29529. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Thompson, C.B. Metabolic Regulation of Cell Growth and Proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D Tumor Spheroids: An Overview on the Tools and Techniques Used for Their Analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D Tumor Spheroids as in Vitro Models to Mimic in Vivo Human Solid Tumors Resistance to Therapeutic Drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, L.; Rigal, L.; Fehrenbach, J.; Severac, C.; Ducommun, B.; Lobjois, V. Characterization of the Physical Properties of Tumor-Derived Spheroids Reveals Critical Insights for Pre-Clinical Studies. Sci. Rep. 2019, 9, 6597. [Google Scholar] [CrossRef]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Herrmann, D.; Conway, J.R.W.; Vennin, C.; Magenau, A.; Hughes, W.E.; Morton, J.P.; Timpson, P. Three-Dimensional Cancer Models Mimic Cell-Matrix Interactions in the Tumour Microenvironment. Carcinogenesis 2014, 35, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Holle, A.W.; Young, J.L.; Spatz, J.P. In Vitro Cancer Cell-ECM Interactions Inform in Vivo Cancer Treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Hartanto, Y.; Zhang, H. Advances in Multicellular Spheroids Formation. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef]
- Bissell, M.J.; Hall, H.G.; Parry, G. How Does the Extracellular Matrix Direct Gene Expression? J. Theor. Biol. 1982, 99, 31–68. [Google Scholar] [CrossRef]
- Eliceiri Brian, P. Integrin and Growth Factor Receptor Crosstalk. Circ. Res. 2001, 89, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-Associated Fibroblasts: An Emerging Target of Anti-Cancer Immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Brito, I.; Lammerding, J. Beyond Tissue Stiffness and Bioadhesivity: Advanced Biomaterials to Model Tumor Microenvironments and Drug Resistance. Trends Cancer 2018, 4, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular Matrix (ECM) Stiffness and Degradation as Cancer Drivers. J. Cell Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A Tense Situation: Forcing Tumour Progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen Density Promotes Mammary Tumor Initiation and Progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; del Río Hernández, A. Matrix Stiffness Induces Epithelial–Mesenchymal Transition and Promotes Chemoresistance in Pancreatic Cancer Cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Emon, M.A.B.; Staudacher, J.J.; Thomas, A.L.; Zessner-Spitzenberg, J.; Mancinelli, G.; Krett, N.; Saif, M.T.; Jung, B. Increased Stiffness of the Tumor Microenvironment in Colon Cancer Stimulates Cancer Associated Fibroblast-Mediated Prometastatic Activin A Signaling. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Wang, S.E.; Xiang, B.; Zent, R.; Quaranta, V.; Pozzi, A.; Arteaga, C.L. Transforming Growth Factor β Induces Clustering of HER2 and Integrins by Activating Src-Focal Adhesion Kinase and Receptor Association to the Cytoskeleton. Cancer Res. 2009, 69, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Wolf, K. Plasticity of Cell Migration: A Multiscale Tuning Model. J. Cell Biol. 2010, 188, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, M.G.; Cappellini, E.; Vicentini, L.M. Silencing of Eps8 Blocks Migration and Invasion in Human Glioblastoma Cell Lines. Exp Cell Res. 2012, 318, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.H.; Trapani, L.M.; Sieminski, A.L.; MacKellar, D.; Gong, H.; Kamm, R.D.; Wells, A.; Lauffenburger, D.A.; Matsudaira, P. Migration of Tumor Cells in 3D Matrices Is Governed by Matrix Stiffness along with Cell-Matrix Adhesion and Proteolysis. Proc. Natl. Acad. Sci. USA 2006, 103, 10889–10894. [Google Scholar] [CrossRef] [Green Version]
- Ehrbar, M.; Sala, A.; Lienemann, P.; Ranga, A.; Mosiewicz, K.; Bittermann, A.; Rizzi, S.C.; Weber, F.E.; Lutolf, M.P. Elucidating the Role of Matrix Stiffness in 3D Cell Migration and Remodeling. Biophys. J. 2011, 100, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.; Lynch, C.D.; Sheetz, M.P. Differential Matrix Rigidity Response in Breast Cancer Cell Lines Correlates with the Tissue Tropism. PLoS ONE 2009, 4, e6361. [Google Scholar] [CrossRef]
- Jain, A.; Betancur, M.; Patel, G.D.; Valmikinathan, C.M.; Mukhatyar, V.J.; Vakharia, A.; Pai, S.B.; Brahma, B.; MacDonald, T.J.; Bellamkonda, R.V. Guiding Intracortical Brain Tumour Cells to an Extracortical Cytotoxic Hydrogel Using Aligned Polymeric Nanofibres. Nat. Mater. 2014, 13, 308–316. [Google Scholar] [CrossRef]
- Huang, D.; Kidoaki, S. Stiffness-Optimized Drug-Loaded Matrix for Selective Capture and Elimination of Cancer Cells. J. Drug Deliv. Sci. Technol. 2020, 55, 101414. [Google Scholar] [CrossRef]
- Joyce, M.H.; Lu, C.; James, E.R.; Hegab, R.; Allen, S.C.; Suggs, L.J.; Brock, A. Phenotypic Basis for Matrix Stiffness-Dependent Chemoresistance of Breast Cancer Cells to Doxorubicin. Front. Oncol. 2018, 8, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, X.; Yang, S.-T. A Dual Fluorescent 3-D Multicellular Coculture of Breast Cancer MCF-7 and Fibroblast NIH-3T3 Cells for High Throughput Cancer Drug Screening. Biochem. Eng. J. 2019, 148, 152–161. [Google Scholar] [CrossRef]
- Juurikka, K.; Butler, G.S.; Salo, T.; Nyberg, P.; Åström, P. The Role of MMP8 in Cancer: A Systematic Review. Int. J. Mol. Sci. 2019, 20, 4506. [Google Scholar] [CrossRef] [Green Version]
- Balbín, M.; Fueyo, A.; Tester, A.M.; Pendás, A.M.; Pitiot, A.S.; Astudillo, A.; Overall, C.M.; Shapiro, S.D.; López-Otín, C. Loss of Collagenase-2 Confers Increased Skin Tumor Susceptibility to Male Mice. Nat. Genet. 2003, 35, 252–257. [Google Scholar] [CrossRef]
- Martino, M.M.; Hubbell, J.A. The 12th–14th Type III Repeats of Fibronectin Function as a Highly Promiscuous Growth Factor-Binding Domain. FASEB J. 2010, 24, 4711–4721. [Google Scholar] [CrossRef] [PubMed]
- Sottile, J. Regulation of Angiogenesis by Extracellular Matrix. Biochim. Biophys. Acta 2004, 1654, 13–22. [Google Scholar] [CrossRef]
- Cattaneo, M.G.; Pola, S.; Francescato, P.; Chillemi, F.; Vicentini, L.M. Human Endostatin-Derived Synthetic Peptides Possess Potent Antiangiogenic Properties in vitro and in vivo. Exp. Cell Res. 2003, 283, 230–236. [Google Scholar] [CrossRef]
- Chillemi, F.; Francescato, P.; Ragg, E.; Cattaneo, M.G.; Pola, S.; Vicentini, L. Studies on the Structure-Activity Relationship of Endostatin: Synthesis of Human Endostatin Peptides Exhibiting Potent Antiangiogenic Activities. J. Med. Chem. 2003, 46, 4165–4172. [Google Scholar] [CrossRef]
- Ouyang, X.; Ghani, A.; Mehal, W.Z. Inflammasome Biology in Fibrogenesis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 979–988. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Varol, C. Tumorigenic Interplay Between Macrophages and Collagenous Matrix in the Tumor Microenvironment. Methods Mol. Biol. 2019, 1944, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Afik, R.; Zigmond, E.; Vugman, M.; Klepfish, M.; Shimshoni, E.; Pasmanik-Chor, M.; Shenoy, A.; Bassat, E.; Halpern, Z.; Geiger, T.; et al. Tumor Macrophages Are Pivotal Constructors of Tumor Collagenous Matrix. J. Exp Med. 2016, 213, 2315–2331. [Google Scholar] [CrossRef]
- Sangaletti, S.; Carlo, E.D.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-Derived SPARC Bridges Tumor Cell-Extracellular Matrix Interactions toward Metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socovich, A.M.; Naba, A. The Cancer Matrisome: From Comprehensive Characterization to Biomarker Discovery. Semin. Cell Dev. Biol. 2019, 89, 157–166. [Google Scholar] [CrossRef]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering Cell–Cell Interactions and Communication from Gene Expression. Nat. Rev. Genet. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-Led Collective Invasion of Carcinoma Cells with Differing Roles for RhoGTPases in Leading and Following Cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Sugimoto, M.; Kitagawa, Y.; Yamada, M.; Yajima, Y.; Utoh, R.; Seki, M. Micropassage-Embedding Composite Hydrogel Fibers Enable Quantitative Evaluation of Cancer Cell Invasion under 3D Coculture Conditions. Lab. Chip 2018, 18, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Attieh, Y.; Clark, A.G.; Grass, C.; Richon, S.; Pocard, M.; Mariani, P.; Elkhatib, N.; Betz, T.; Gurchenkov, B.; Vignjevic, D.M. Cancer-Associated Fibroblasts Lead Tumor Invasion through Integrin-Β3–Dependent Fibronectin Assembly. J. Cell Biol. 2017, 216, 3509–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, T.; Weber, A.; Neuwirt, H.; Grünbacher, G.; Ploner, C.; Klocker, H.; Sampson, N.; Eder, I.E. Cancer-Associated Fibroblastsmodify the Response of Prostate Cancer Cells to Androgen and Anti-Androgens in Three-Dimensional Spheroid Culture. Int. J. Mol. Sci. 2016, 17, 1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steer, A.; Cordes, N.; Jendrossek, V.; Klein, D. Impact of Cancer-Associated Fibroblast on the Radiation-Response of Solid Xenograft Tumors. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular Characterization of the Tumor Microenvironment in Breast Cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Sheng, J.; Gao, D.; Li, F.; Durrans, A.; Ryu, S.; Lee, S.B.; Narula, N.; Rafii, S.; Elemento, O.; et al. Transcriptome Analysis of Individual Stromal Cell Populations Identifies Stroma-Tumor Crosstalk in Mouse Lung Cancer Model. Cell Rep. 2015, 10, 1187–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, T.-L.; Sheng, J.; Leung, C.S.; Li, F.; Kim, J.; Ho, S.Y.; Matzuk, M.M.; Lu, K.H.; Wong, S.T.C.; Mok, S.C. Systematic Identification of Druggable Epithelial-Stromal Crosstalk Signaling Networks in Ovarian Cancer. J. Natl. Cancer Inst. 2019, 111, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and Cancer: From Mechanisms to Therapeutic Implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P. The Interaction of Anticancer Therapies with Tumor-Associated Macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Tevis, K.M.; Cecchi, R.J.; Colson, Y.L.; Grinstaff, M.W. Mimicking the Tumor Microenvironment to Regulate Macrophage Phenotype and Assessing Chemotherapeutic Efficacy in Embedded Cancer Cell/Macrophage Spheroid Models. Acta Biomater. 2017, 50, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiet, R.; Van Goethem, E.; Cougoule, C.; Balor, S.; Valette, A.; Al Saati, T.; Lowell, C.A.; Le Cabec, V.; Maridonneau-Parini, I. The Process of Macrophage Migration Promotes Matrix Metalloproteinase-Independent Invasion by Tumor Cells. J. Immunol. 2011, 187, 3806–3814. [Google Scholar] [CrossRef] [Green Version]
- Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 689. [Google Scholar] [CrossRef] [Green Version]
- Akins, E.A.; Aghi, M.K.; Kumar, S. Incorporating Tumor-Associated Macrophages into Engineered Models of Glioma. iScience 2020, 23. [Google Scholar] [CrossRef]
- Jain, N.; Moeller, J.; Vogel, V. Mechanobiology of Macrophages: How Physical Factors Coregulate Macrophage Plasticity and Phagocytosis. Annu. Rev. Biomed. Eng. 2019, 21, 267–297. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.M.H.; Kuczek, D.E.; Kalvisa, A.; Siersbæk, M.S.; Thorseth, M.-L.; Johansen, A.Z.; Carretta, M.; Grøntved, L.; Vang, O.; Madsen, D.H. Collagen Density Modulates the Immunosuppressive Functions of Macrophages. J. Immunol. 2020, 205, 1461–1472. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Rauniyar, K.; Jha, S.K.; Jeltsch, M. Biology of Vascular Endothelial Growth Factor C in the Morphogenesis of Lymphatic Vessels. Front. Bioeng. Biotechnol. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Nagl, L.; Horvath, L.; Pircher, A.; Wolf, D. Tumor Endothelial Cells (TECs) as Potential Immune Directors of the Tumor Microenvironment–New Findings and Future Perspectives. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Georganaki, M.; van Hooren, L.; Dimberg, A. Vascular Targeting to Increase the Efficiency of Immune Checkpoint Blockade in Cancer. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Maishi, N.; Annan, D.A.; Kikuchi, H.; Hida, Y.; Hida, K. Tumor Endothelial Heterogeneity in Cancer Progression. Cancers 2019, 11, 1511. [Google Scholar] [CrossRef] [Green Version]
- Sökeland, G.; Schumacher, U. The Functional Role of Integrins during Intra- and Extravasation within the Metastatic Cascade. Mol. Cancer 2019, 18, 12. [Google Scholar] [CrossRef]
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M.; et al. Heterogeneity of Tumor Endothelial Cells. Am. J. Pathol. 2012, 180, 1294–1307. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-C.; Pan, M.-R.; Hung, W.-C. Two Birds, One Stone: Double Hits on Tumor Growth and Lymphangiogenesis by Targeting Vascular Endothelial Growth Factor Receptor 3. Cells 2019, 8, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roudsari, L.C.; Jeffs, S.E.; Witt, A.S.; Gill, B.J.; West, J.L. A 3D Poly(Ethylene Glycol)-Based Tumor Angiogenesis Model to Study the Influence of Vascular Cells on Lung Tumor Cell Behavior. Sci. Rep. 2016, 6, 32726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, D.; Fiorelli, R.; Barrientos, E.S.; Melendez, E.L.; Sanai, N.; Mehta, S.; Nikkhah, M. A Three-Dimensional (3D) Organotypic Microfluidic Model for Glioma Stem Cells-Vascular Interactions. Biomaterials 2019, 198, 63–77. [Google Scholar] [CrossRef]
- Meng, F.; Meyer, C.M.; Joung, D.; Vallera, D.A.; McAlpine, M.C.; Panoskaltsis-Mortari, A. 3D Bioprinted In Vitro Metastatic Models via Reconstruction of Tumor Microenvironments. Adv. Mater. 2019, 31, 1806899. [Google Scholar] [CrossRef]
- Kim, S.-H.; Song, Y.; Seo, H.R. GSK-3β Regulates the Endothelial-to-Mesenchymal Transition via Reciprocal Crosstalk between NSCLC Cells and HUVECs in Multicellular Tumor Spheroid Models. J. Exp. Clin. Cancer Res. 2019, 38, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcu, R.; Choi, Y.J.; Xue, J.; Fortin, C.L.; Wang, Y.; Nagao, R.J.; Xu, J.; MacDonald, J.W.; Bammler, T.K.; Murry, C.E.; et al. Human Organ-Specific Endothelial Cell Heterogeneity. iScience 2018, 4, 20–35. [Google Scholar] [CrossRef] [Green Version]
- Medina-Leyte, D.J.; Domínguez-Pérez, M.; Mercado, I.; Villarreal-Molina, M.T.; Jacobo-Albavera, L. Use of Human Umbilical Vein Endothelial Cells (HUVEC) as a Model to Study Cardiovascular Disease: A Review. Appl. Sci. 2020, 10, 938. [Google Scholar] [CrossRef] [Green Version]
- Seano, G.; Jain, R.K. Vessel Co-Option in Glioblastoma: Emerging Insights and Opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Garcia, H.; Alvarado-Estrada, K.; Schiapparelli, P.; Quinones-Hinojosa, A.; Trifiletti, D.M. Engineering Three-Dimensional Tumor Models to Study Glioma Cancer Stem Cells and Tumor Microenvironment. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma Stem-like Cells Give Rise to Tumour Endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial Cells Promote 3D Invasion of GBM by IL-8-Dependent Induction of Cancer Stem Cell Properties. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Yang, W.; Warrington, N.M.; Taylor, S.J.; Whitmire, P.; Carrasco, E.; Singleton, K.W.; Wu, N.; Lathia, J.D.; Berens, M.E.; Kim, A.H.; et al. Sex Differences in GBM Revealed by Analysis of Patient Imaging, Transcriptome, and Survival Data. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Addis, R.; Campesi, I.; Fois, M.; Capobianco, G.; Dessole, S.; Fenu, G.; Montella, A.; Cattaneo, M.G.; Vicentini, L.M.; Franconi, F. Human Umbilical Endothelial Cells (HUVECs) Have a Sex: Characterisation of the Phenotype of Male and Female Cells. Biol. Sex. Differ. 2014, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, M.G.; Vanetti, C.; Decimo, I.; Di Chio, M.; Martano, G.; Garrone, G.; Bifari, F.; Vicentini, L.M. Sex-Specific ENOS Activity and Function in Human Endothelial Cells. Sci. Rep. 2017, 7, 9612. [Google Scholar] [CrossRef]
- Huxley, V.H.; Kemp, S.S.; Schramm, C.; Sieveking, S.; Bingaman, S.; Yu, Y.; Zaniletti, I.; Stockard, K.; Wang, J. Sex Differences Influencing Micro- and Macrovascular Endothelial Phenotype in vitro. J. Physiol. 2018, 596, 3929–3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, E.; Lorenz, M.; Völker, U.; Stangl, K.; Hammer, E.; Stangl, V. Sex-Specific Differences in the Intracellular Proteome of Human Endothelial Cells from Dizygotic Twins. J. Proteom. 2019, 201, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Blaschke, B.; Benn, A.; Hammer, E.; Witt, E.; Kirwan, J.; Fritsche-Guenther, R.; Gloaguen, Y.; Bartsch, C.; Vietzke, A.; et al. Sex-Specific Metabolic and Functional Differences in Human Umbilical Vein Endothelial Cells from Twin Pairs. Atherosclerosis 2019, 291, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, M.G.; Banfi, C.; Brioschi, M.; Lattuada, D.; Vicentini, L.M. Sex-Dependent Differences in the Secretome of Human Endothelial Cells. Biol. Sex Differ. 2021, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.H.; Orbach, S.M.; Bushnell, G.G.; Oakes, R.S.; Jeruss, J.S.; Shea, L.D. Engineered Niches to Analyze Mechanisms of Metastasis and Guide Precision Medicine. Cancer Res. 2020, 80, 3786–3794. [Google Scholar] [CrossRef]
- Matei, I.; Rampersaud, S.; Lyden, D. Engineered Niches Model the Onset of Metastasis. Nat. Biomed. Eng. 2018, 2, 885–887. [Google Scholar] [CrossRef]
- Seib, F.P.; Berry, J.E.; Shiozawa, Y.; Taichman, R.S.; Kaplan, D.L. Tissue Engineering a Surrogate Niche for Metastatic Cancer Cells. Biomaterials 2015, 51, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-Chips: Into the next Decade. Nat. Rev. Drug Discov. 2020, 1–17. [Google Scholar] [CrossRef]
- Sleeboom, J.J.F.; Amirabadi, H.E.; Nair, P.; Sahlgren, C.M.; Toonder, J.M.J. den Metastasis in Context: Modeling the Tumor Microenvironment with Cancer-on-a-Chip Approaches. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling Cancer in Microfluidic Human Organs-on-Chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef]
- Huang, J.-W.; Pan, H.-J.; Yao, W.-Y.; Tsao, Y.-W.; Liao, W.-Y.; Wu, C.-W.; Tung, Y.-C.; Lee, C.-H. Interaction between Lung Cancer Cell and Myofibroblast Influenced by Cyclic Tensile Strain. Lab. Chip 2013, 13, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, Z.; Fan, H.; Song, J.; Liu, Y.; Gao, Z.; Wang, Q. Cancer-Associated Fibroblasts Promote Non-Small Cell Lung Cancer Cell Invasion by Upregulation of Glucose-Regulated Protein 78 (GRP78) Expression in an Integrated Bionic Microfluidic Device. Oncotarget 2016, 7, 25593–25603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowski-Daspit, A.S.; Tien, J.; Nelson, C.M. Interstitial Fluid Pressure Regulates Collective Invasion in Engineered Human Breast Tumors via Snail, Vimentin, and E-Cadherin. Integr. Biol. 2016, 8, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.D.; Searson, P.C. Live-Cell Imaging of Invasion and Intravasation in an Artificial Microvessel Platform. Cancer Res. 2014, 74, 4937–4945. [Google Scholar] [CrossRef] [Green Version]
- Polacheck, W.J.; Charest, J.L.; Kamm, R.D. Interstitial Flow Influences Direction of Tumor Cell Migration through Competing Mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 11115–11120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalchman, J.; Fujioka, S.; Chung, S.; Kikkawa, Y.; Mitaka, T.; Kamm, R.D.; Tanishita, K.; Sudo, R. A Three-Dimensional Microfluidic Tumor Cell Migration Assay to Screen the Effect of Anti-Migratory Drugs and Interstitial Flow. Microfluid. Nanofluid 2013, 14, 969–981. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.L.; Tung, C.-K.; Zheng, A.; Kim, B.J.; Wu, M. Interstitial Flows Promote Amoeboid over Mesenchymal Motility of Breast Cancer Cells Revealed by a Three Dimensional Microfluidic Model. Integr. Biol. 2015, 7, 1402–1411. [Google Scholar] [CrossRef]
- Sung, K.E.; Yang, N.; Pehlke, C.; Keely, P.J.; Eliceiri, K.W.; Friedl, A.; Beebe, D.J. Transition to Invasion in Breast Cancer: A Microfluidic in Vitro Model Enables Examination of Spatial and Temporal Effects. Integr. Biol. 2011, 3, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Hassell, B.A.; Goyal, G.; Lee, E.; Sontheimer-Phelps, A.; Levy, O.; Chen, C.S.; Ingber, D.E. Human Organ Chip Models Recapitulate Orthotopic Lung Cancer Growth, Therapeutic Responses, and Tumor Dormancy In Vitro. Cell Rep. 2017, 21, 508–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zervantonakis, I.K.; Hughes-Alford, S.K.; Charest, J.L.; Condeelis, J.S.; Gertler, F.B.; Kamm, R.D. Three-Dimensional Microfluidic Model for Tumor Cell Intravasation and Endothelial Barrier Function. Proc. Natl. Acad. Sci. USA 2012, 109, 13515–13520. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.P.; Lu, J.; Seon, H.; Lee, A.P.; Flanagan, L.A.; Kim, H.-Y.; Putnam, A.J.; Jeon, N.L. Engineering Microscale Cellular Niches for Three-Dimensional Multicellular Co-Cultures. Lab. Chip 2009, 9, 1740–1748. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wang, F.; Zhang, J.; Sun, X.; Yan, Y.; Wang, Y.; Ouyang, J.; Zhang, J.; Honore, T.; Ge, J.; et al. Study on Development of Composite Hydrogels With Tunable Structures and Properties for Tumor-on-a-Chip Research. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Kim, G.-M.; Le, K.H.T.; Giannitelli, S.M.; Lee, Y.J.; Rainer, A.; Trombetta, M. Electrospinning of PCL/PVP Blends for Tissue Engineering Scaffolds. J. Mater. Sci. Mater. Med. 2013, 24, 1425–1442. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T. Organoids for Drug Discovery and Personalized Medicine. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 447–462. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colombo, E.; Cattaneo, M.G. Multicellular 3D Models to Study Tumour-Stroma Interactions. Int. J. Mol. Sci. 2021, 22, 1633. https://doi.org/10.3390/ijms22041633
Colombo E, Cattaneo MG. Multicellular 3D Models to Study Tumour-Stroma Interactions. International Journal of Molecular Sciences. 2021; 22(4):1633. https://doi.org/10.3390/ijms22041633
Chicago/Turabian StyleColombo, Elisabetta, and Maria Grazia Cattaneo. 2021. "Multicellular 3D Models to Study Tumour-Stroma Interactions" International Journal of Molecular Sciences 22, no. 4: 1633. https://doi.org/10.3390/ijms22041633
APA StyleColombo, E., & Cattaneo, M. G. (2021). Multicellular 3D Models to Study Tumour-Stroma Interactions. International Journal of Molecular Sciences, 22(4), 1633. https://doi.org/10.3390/ijms22041633