Pathological Circulating Factors in Moyamoya Disease


Abstract
:1. Introduction
2. Growth Factors
3. Circulating Progenitor Cells
4. Angiogenesis-Related Cytokines
5. Inflammatory and Immune Mediators
6. Other Factors
7. Discussion
8. Limitations
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suzuki, J.; Takaku, A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch. Neurol. 1969, 20, 288–299. [Google Scholar] [CrossRef]
- Scott, R.M.; Smith, E.R. Moyamoya disease and moyamoya syndrome. N. Engl. J. Med. 2009, 360, 1226–1237. [Google Scholar] [CrossRef] [Green Version]
- Shang, S.; Zhou, D.; Ya, J.; Li, S.; Yang, Q.; Ding, Y.; Ji, X.; Meng, R. Progress in moyamoya disease. Neurosurg. Rev. 2020, 43, 371–382. [Google Scholar] [CrossRef]
- Goto, Y.; Yonekawa, Y. Worldwide distribution of moyamoya disease. Neurol. Med. Chir. 1992, 32, 883–886. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, S.; Houkin, K. Moyamoya disease: Current concepts and future perspectives. Lancet Neurol. 2008, 7, 1056–1066. [Google Scholar] [CrossRef]
- Kamada, F.; Aoki, Y.; Narisawa, A.; Abe, Y.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2011, 56, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Freemont, P.S. The RING finger. A novel protein sequence motif related to the zinc finger. Ann. N. Y. Acad. Sci. 1993, 684, 174–192. [Google Scholar] [CrossRef]
- Elangovan, M.; Choi, E.S.; Jang, B.G.; Kim, M.S.; Yoo, Y.J. The ubiquitin-interacting motif of 26S proteasome subunit S5a induces A549 lung cancer cell death. Biochem. Biophys Res. Commun. 2007, 364, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Morito, D.; Takashima, S.; Mineharu, Y.; Kobayashi, H.; Hitomi, T.; Hashikata, H.; Matsuura, N.; Yamazaki, S.; Toyoda, A.; et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE 2011, 6, e22542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, O.Y.; Chung, J.W.; Kim, S.J.; Oh, M.J.; Kim, S.Y.; Cho, Y.H.; Cha, J.; Yeon, J.Y.; Kim, K.H.; Kim, G.M.; et al. Caveolin-1, Ring finger protein 213, and endothelial function in Moyamoya disease. Int. J. Stroke 2016, 11, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Sonobe, S.; Nishijima, Y.; Niizuma, K.; Sakata, H.; Kure, S.; Tominaga, T. Genetics and biomarkers of Moyamoya disease: Significance of RNF213 as a susceptibility gene. J. Stroke 2014, 16, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Kim, J.H.; Phi, J.H.; Kim, Y.Y.; Kim, J.E.; Wang, K.C.; Cho, B.K.; Kim, S.K. Plasma matrix metalloproteinases, cytokines and angiogenic factors in moyamoya disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 673–678. [Google Scholar] [CrossRef]
- Sakamoto, S.; Kiura, Y.; Yamasaki, F.; Shibukawa, M.; Ohba, S.; Shrestha, P.; Sugiyama, K.; Kurisu, K. Expression of vascular endothelial growth factor in dura mater of patients with moyamoya disease. Neurosurg. Rev. 2008, 31, 77–81. [Google Scholar] [CrossRef]
- Park, Y.S.; Jeon, Y.J.; Kim, H.S.; Chae, K.Y.; Oh, S.H.; Han, I.B.; Kim, H.S.; Kim, W.C.; Kim, O.J.; Kim, T.G.; et al. The role of VEGF and KDR polymorphisms in moyamoya disease and collateral revascularization. PLoS ONE 2012, 7, e47158. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wang, R.; Zhang, D.; Zhang, Y.; Zhang, Q.; Zhao, J. Expression of circulating vascular endothelial growth factor-antagonizing cytokines and vascular stabilizing factors prior to and following bypass surgery in patients with moyamoya disease. Exp. Ther. Med. 2014, 8, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Sawamura, Y.; Houkin, K.; Kamiyama, H.; Abe, H. The cerebrospinal fluid in patients with moyamoya disease (spontaneous occlusion of the circle of Willis) contains high level of basic fibroblast growth factor. Neurosci. Lett. 1993, 160, 214–216. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Houkin, K.; Takahashi, A.; Abe, H. Angiogenic factors in moyamoya disease. Stroke 1996, 27, 2160–2165. [Google Scholar] [CrossRef]
- Nanba, R.; Kuroda, S.; Ishikawa, T.; Houkin, K.; Iwasaki, Y. Increased expression of hepatocyte growth factor in cerebrospinal fluid and intracranial artery in moyamoya disease. Stroke 2004, 35, 2837–2842. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Aoyagi, M.; Tajima, S.; Wachi, H.; Fukai, N.; Matsushima, Y.; Yamamoto, K. Increase in elastin gene expression and protein synthesis in arterial smooth muscle cells derived from patients with Moyamoya disease. Stroke 1997, 28, 1733–1738. [Google Scholar] [CrossRef]
- Kim, J.H.; Jung, J.H.; Phi, J.H.; Kang, H.S.; Kim, J.E.; Chae, J.H.; Kim, S.J.; Kim, Y.H.; Kim, Y.Y.; Cho, B.K.; et al. Decreased level and defective function of circulating endothelial progenitor cells in children with moyamoya disease. J. Neurosci. Res. 2010, 88, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Rafat, N.; Beck, G.; Pena-Tapia, P.G.; Schmiedek, P.; Vajkoczy, P. Increased levels of circulating endothelial progenitor cells in patients with Moyamoya disease. Stroke 2009, 40, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, T.; Taguchi, A.; Matsuyama, T.; Shimizu, Y.; Kikuchi-Taura, A.; Soma, T.; Stern, D.M.; Yoshikawa, H.; Kasahara, Y.; Moriwaki, H.; et al. Increase in circulating CD34-positive cells in patients with angiographic evidence of moyamoya-like vessels. J. Cereb. Blood Flow Metab. 2008, 28, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Son, S.M.; Mook-Jung, I.; Moon, Y.J.; Lee, J.Y.; Wang, K.C.; Kang, H.S.; Phi, J.H.; Choi, S.A.; Chong, S.; et al. Mitochondrial abnormalities related to the dysfunction of circulating endothelial colony-forming cells in moyamoya disease. J. Neurosurg. 2018, 129, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.H.; Chu, K.; Lee, S.T.; Park, H.K.; Kim, D.H.; Kim, J.H.; Bahn, J.J.; Song, E.C.; Kim, M.; Lee, S.K.; et al. Circulating endothelial progenitor cells as a pathogenetic marker of moyamoya disease. J. Cereb. Blood Flow Metab. 2008, 28, 1795–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Moon, Y.J.; Lee, H.O.; Park, A.K.; Choi, S.A.; Wang, K.C.; Han, J.W.; Joung, J.G.; Kang, H.S.; Kim, J.E.; et al. Deregulation of retinaldehyde dehydrogenase 2 leads to defective angiogenic function of endothelial colony-forming cells in pediatric Moyamoya disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1670–1677. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Moon, Y.J.; Kim, Y.Y.; Park, W.Y.; Park, A.K.; Wang, K.C.; Kim, J.E.; Phi, J.H.; Lee, J.Y.; Kim, S.K. Smooth-muscle progenitor cells isolated from patients with moyamoya disease: Novel experimental cell model. J. Neurosurg. 2014, 120, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Hojo, M.; Hoshimaru, M.; Miyamoto, S.; Taki, W.; Nagata, I.; Asahi, M.; Matsuura, N.; Ishizaki, R.; Kikuchi, H.; Hashimoto, N. Role of transforming growth factor-beta1 in the pathogenesis of moyamoya disease. J. Neurosurg. 1998, 89, 623–629. [Google Scholar] [CrossRef]
- Weng, L.; Cao, X.; Han, L.; Zhao, H.; Qiu, S.; Yan, Y.; Wang, X.; Chen, X.; Zheng, W.; Xu, X.; et al. Association of increased Treg and Th17 with pathogenesis of moyamoya disease. Sci. Rep. 2017, 7, 3071. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Yoo, J.I.; Cho, B.K.; Hong, S.J.; Kim, Y.K.; Moon, J.A.; Kim, J.H.; Chung, Y.N.; Wang, K.C. Elevation of CRABP-I in the cerebrospinal fluid of patients with Moyamoya disease. Stroke 2003, 34, 2835–2841. [Google Scholar] [CrossRef] [Green Version]
- Blecharz-Lang, K.G.; Prinz, V.; Burek, M.; Frey, D.; Schenkel, T.; Krug, S.M.; Fromm, M.; Vajkoczy, P. Gelatinolytic activity of autocrine matrix metalloproteinase-9 leads to endothelial de-arrangement in Moyamoya disease. J. Cereb. Blood Flow Metab. 2018, 38, 1940–1953. [Google Scholar] [CrossRef]
- Fujimura, M.; Watanabe, M.; Narisawa, A.; Shimizu, H.; Tominaga, T. Increased expression of serum Matrix Metalloproteinase-9 in patients with Moyamoya disease. Surg. Neurol. 2009, 72, 476–480. [Google Scholar] [CrossRef]
- Takagi, Y.; Kikuta, K.; Nozaki, K.; Fujimoto, M.; Hayashi, J.; Imamura, H.; Hashimoto, N. Expression of hypoxia-inducing factor-1 alpha and endoglin in intimal hyperplasia of the middle cerebral artery of patients with Moyamoya disease. Neurosurgery 2007, 60, 338–345. [Google Scholar] [CrossRef]
- Lin, R.; Xie, Z.; Zhang, J.; Xu, H.; Su, H.; Tan, X.; Tian, D.; Su, M. Clinical and immunopathological features of Moyamoya disease. PLoS ONE 2012, 7, e36386. [Google Scholar] [CrossRef]
- Fujimura, M.; Fujimura, T.; Kakizaki, A.; Sato-Maeda, M.; Niizuma, K.; Tomata, Y.; Aiba, S.; Tominaga, T. Increased serum production of soluble CD163 and CXCL5 in patients with moyamoya disease: Involvement of intrinsic immune reaction in its pathogenesis. Brain Res. 2018, 1679, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Kim, D.H.; Oh, M.J.; Cho, Y.H.; Kim, E.H.; Moon, G.J.; Ki, C.S.; Cha, J.; Kim, K.H.; Jeon, P.; et al. Cav-1 (Caveolin-1) and arterial remodeling in adult Moyamoya disease. Stroke 2018, 49, 2597–2604. [Google Scholar] [CrossRef] [PubMed]
- Noda, A.; Suzuki, Y.; Takayasu, M.; Watanabe, K.; Takagi, T.; Hara, M.; Yoshida, J. Elevation of nitric oxide metabolites in the cerebrospinal fluid of patients with moyamoya disease. Acta Neurochir. 2000, 142, 1275–1279. [Google Scholar] [CrossRef]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Jaipersad, A.S.; Lip, G.Y.; Silverman, S.; Shantsila, E. The role of monocytes in angiogenesis and atherosclerosis. J. Am. Coll. Cardiol. 2014, 63, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Aoyagi, M.; Fukai, N.; Matsushima, Y.; Yamamoto, K. Differences in cellular responses to mitogens in arterial smooth muscle cells derived from patients with moyamoya disease. Stroke 1998, 29, 1188–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishita, R.; Sakaki, M.; Yamamoto, K.; Iguchi, S.; Aoki, M.; Yamasaki, K.; Matsumoto, K.; Nakamura, T.; Lawn, R.; Ogihara, T.; et al. Impairment of collateral formation in lipoprotein(a) transgenic mice: Therapeutic angiogenesis induced by human hepatocyte growth factor gene. Circulation 2002, 105, 1491–1496. [Google Scholar] [CrossRef] [Green Version]
- Houkin, K.; Ito, M.; Sugiyama, T.; Shichinohe, H.; Nakayama, N.; Kazumata, K.; Kuroda, S. Review of past research and current concepts on the etiology of moyamoya disease. Neurol. Med. Chir. 2012, 52, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Paschalaki, K.E.; Randi, A.M. Recent advances in endothelial colony forming cells toward their use in clinical translation. Front. Med. 2018, 5, 295. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, K.; Date, I. Moyamoya disease. Brain Nerve 2008, 60, 37–42. [Google Scholar]
- Boylan, J.F.; Gudas, L.J. The level of CRABP-I expression influences the amounts and types of all-trans-retinoic acid metabolites in F9 teratocarcinoma stem cells. J. Biol. Chem. 1992, 267, 21486–21491. [Google Scholar] [CrossRef]
- Mikami, T.; Suzuki, H.; Komatsu, K.; Mikuni, N. Influence of Inflammatory disease on the pathophysiology of Moyamoya disease and quasi-Moyamoya disease. Neurol. Med. Chir. 2019, 59, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Martinive, P.; DeWever, J.; Batova, Z.; Daneau, G.; Pelat, M.; Ghisdal, P.; Gregoire, V.; Dessy, C.; Balligand, J.L.; et al. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ. Res. 2004, 95, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.S.; Min, K.T.; Kim, T.G.; Lee, Y.H.; Cheong, H.J.; Yeom, I.S.; Choi, J.U.; Kim, D.S.; Kim, N.K. Age-specific eNOS polymorphisms in moyamoya disease. Childs Nerv. Syst. 2011, 27, 1919–1926. [Google Scholar] [CrossRef]
- Dai, D.; Lu, Q.; Huang, Q.; Yang, P.; Hong, B.; Xu, Y.; Zhao, W.; Liu, J.; Li, Q. Serum miRNA signature in Moyamoya disease. PLoS ONE 2014, 9, e102382. [Google Scholar] [CrossRef]
- Uchino, H.; Ito, M.; Kazumata, K.; Hama, Y.; Hamauchi, S.; Terasaka, S.; Sasaki, H.; Houkin, K. Circulating miRNome profiling in Moyamoya disease-discordant monozygotic twins and endothelial microRNA expression analysis using iPS cell line. BMC Med. Genom. 2018, 11, 72. [Google Scholar] [CrossRef]
- Lee, M.J.; Fallen, S.; Zhou, Y.; Baxter, D.; Scherler, K.; Kuo, M.F.; Wang, K. The impact of Moyamoya disease and RNF213 mutations on the spectrum of plasma protein and MicroRNA. J. Clin. Med. 2019, 8, 1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
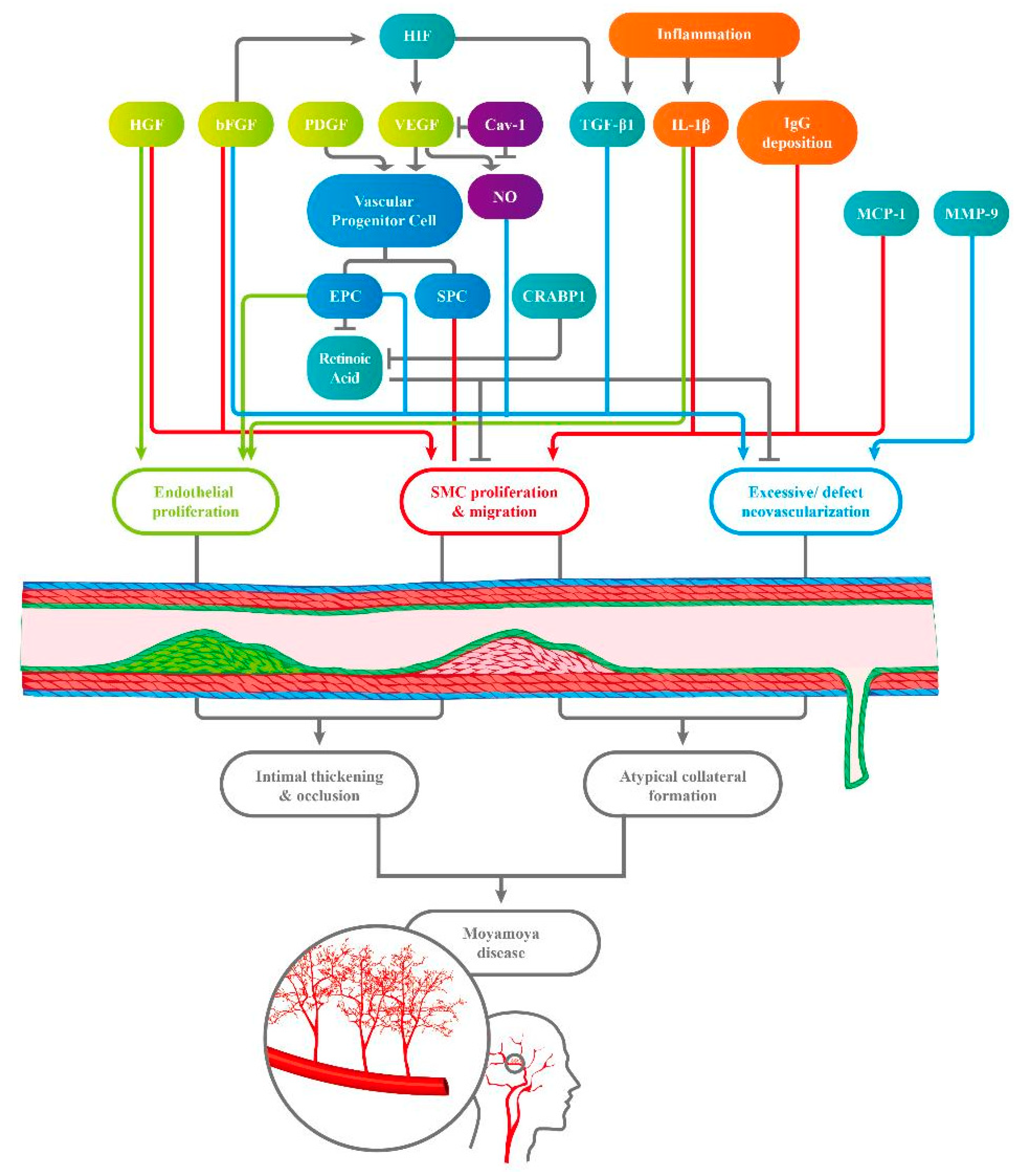

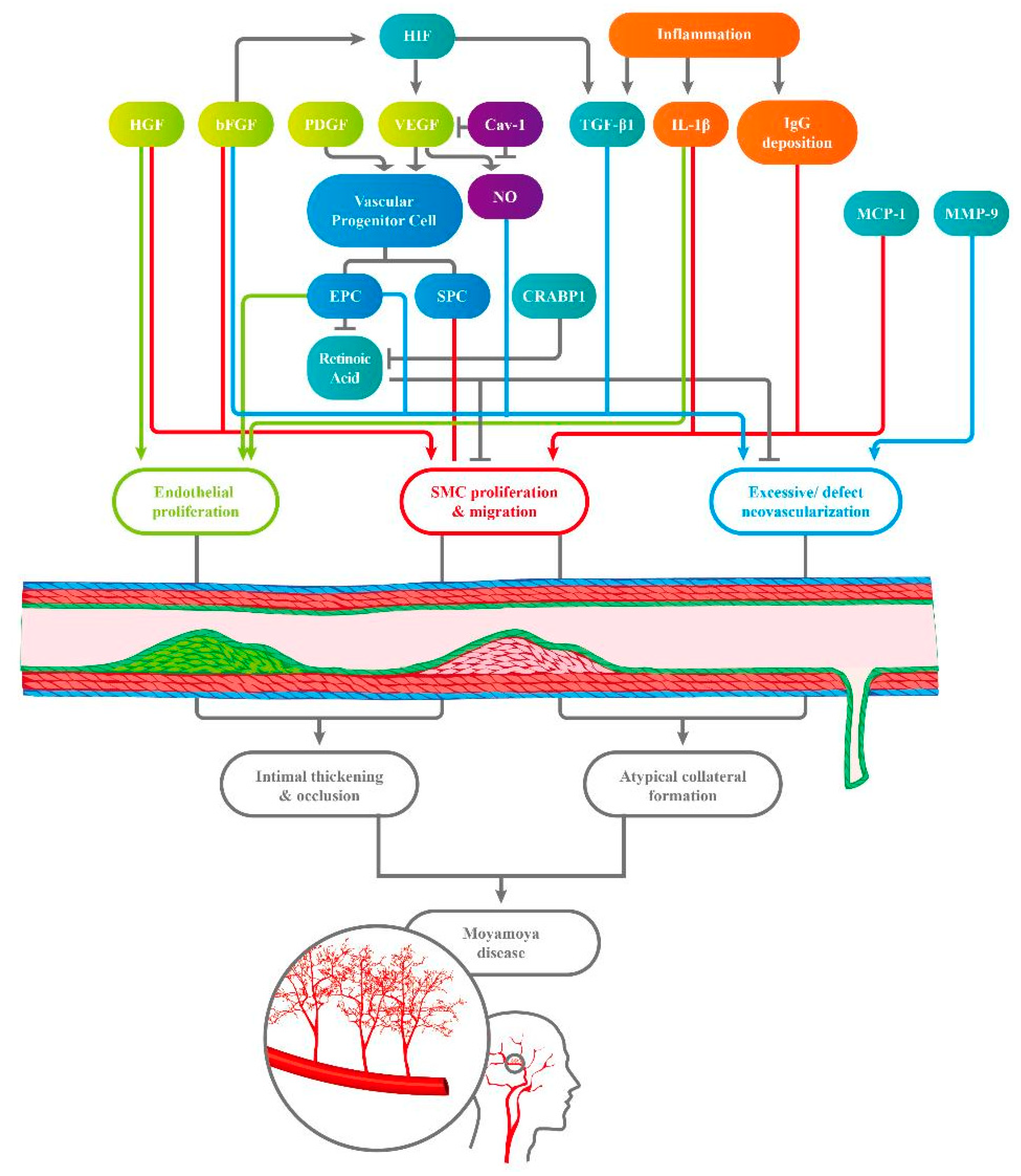{kind=link}
| Substance | Sampled Population/Sampled Objects | Abnormal Findings in MMD Patients | Hypothetic Pathogenetic Pathway | First Author | |
|---|---|---|---|---|---|
| Growth Factor | VEGF | MMD patients/ plasma | VEGF ↑ | VEGF ↑ → recruitment of vascular progenitor cells and collateral vessel formation | Kang et al. [12] |
| MMD patients/ Dura mater | VEGF ↑ | VEGF ↑ → migration, proliferation, and neovascularization in vascular cells | Sakamoto et al. [13] | ||
| Pediatric MMD/ gene | VEGF−634CC ↓ → better post-operative collateral vessel formation | VEGF −634G allele → poor collateral vessel formation (in pediatric MMD) | Park et al. [14] | ||
| MMD patients/ serum | sVEGF1-R and sVEGF2-R ↓ → better collateral vessel formation | 1. VEGF2-R → initial steps in angiogenesis 2. VEGF1-R and VEGF2-R↓ → pathological angiogenesis↓ | He et al. [15] | ||
| bFGF | MMD patients/ CSF | (bFGF) ↑ | bFGF ↑ → pathogenesis in MMD | Takahashi et al. [16] | |
| MMD patients/ CSF | bFGF ↑ (after neovascularization) | bFGF ↑ → proliferation of mesoderm, neuroectoderm-derived cells, and SMC ↑ → stenosis and occlusion of ICA | Yoshimoto et al. [17] | ||
| HGF | MMD patients/ CSF | (HGF) ↑ (in CSF, the intima and the media of the carotid fork) | HGF (strong angiogenic inducer) ↑ → proliferation of endothelial cells↑ and migration of SMCs ↑ | Nanba et al. [18] | |
| PDGF-BB | MMD patients/ plasma | PDGF-BB↑ PDGF receptors↓ (on SMCs) | PDGF-BB ↑ → vascular progenitor cells differentiating into a SMC lineage → intima hyperplasia | Kang et al. [12] | |
| MMD patients/ SMC strains | PDGF-AA & PDGF-BB → SMC migration ↑ | PDGF ↑ → intima thickening ↑ | Yamamoto et al. [19] | ||
| Circulating progenitor Cells | ECFCs/ EPCs | Pediatric MMD/ Peripheral blood | tube formation-type ECFCs ↓ senescent-like phenotype ECFCs ↑ | EPCs ↓ → delayed repairing in ischemia vessels and abnormal differentiating activities → ineffective vasculogenesis | Kim et al. [20] |
| MMD patients/ Peripheral blood | ECFCs ↑ | EPCs ↑ → activity of resting endothelial cells ↑ → arteriogenesis and angiogenesis ↑ | Rafat et al. [21] | ||
| Patients with ICA or MCA stenosis or occlusion/ peripheral blood | Circulating CD34 + cells ↑ | Circulating ECFCs ↑ → pathological neo-vascularization in ischemia brain | Yoshihara et al. [22] | ||
| MMD patients/ peripheral blood | The mitochondria in ECFCs display morphological and functional abnormalities. | ECFCs with abnormal function → delayed repair of the defect vessels → vessel occlusion | Choi et al. [23] | ||
| Adult MMD/ CFU and outgrowth cell | 1. CFU↓ (in advanced MMD) 2. EOC↑ (in early MMD) | Inefficient subtype of EPCs → vascular occlusion and angiogenesis | Jung et al. [24] | ||
| Pediatric MMD/ ECFCs in vitro and in vivo | RALDH2 gene in ECFCs↓ → capillary formation↓ | RALDH2 ↓ → RA ↓ → defective tube formation activity | Lee et al. [25] | ||
| SPCs | MMD patients/ peripheral blood | Mutation in ACTA2 gene → proliferation in SMCs ↑ | SMCs ↑ → occlusive disease | Guo et al. [26] | |
| MMD patients/ peripheral blood | SMCs make an irregular arrangement and thickened tubules | Multiple genes express differentially → SMCs shows irregular morphology | Kang et al. [27] | ||
| Angiogenesis-related cytokines | TGF-β1 | MMD patients/ SMCs from superficial temporal artery and serum | TGF-β1 ↑ (in SMCs and in serum) | TGF-β1 ↑ → connective tissues genes ↑ → promoting neovascularization | Hojo et al. [28] |
| MMD patients/ SMCs | TGF-β1 ↑ → elastin mRNA ↑ | TGF-β1 ↑ (produced by MMD SMCs and in inflammatory stimulation) → elastin synthesis ↑ | Yamamoto et al. [19] | ||
| MMD patients/ Peripheral blood | Fr III Treg cells ↑ (lack of suppressive functions) | TGF-β↑ (induced by circulating Treg) → VEGF ↑ | Weng et al. [29] | ||
| CRABP-1 | Pediatric MMD/ CSF | CRABP-1 ↑ | CRABP-1 ↑→ retinoid acid ↓ → SMCs migration & proliferation ↑ (induced by growth factors) | Kim et al. [30] | |
| MMPs | MMD patients/ Serum | autocrine activities of MMP-9 ↑ | MMP-9 ↑→ gelatinase↑ → collagen IV↓ and vascular remodeling → angiogenesis ↑ | Blecharz-Lang et al. [31] | |
| MMD patients/ Peripheral blood | Plasma MMP-9 ↑ | MMP-9↑ → intimal hyperplasia and excessive collateral vessel formation | Kang et al. [12] | ||
| MMD patients/ Serum | Serum MMP ↑ | MMP-9↑ → pathological angiogenesis and defect vascular structure → hemorrhage in MMD | Fujimura et al. [32] | ||
| HIF-1α | MMD patients/ MCA sample | HIF-1α ↑ (in the intima and endothelium) | HIF-1α ↑ → transcription of other growth factors and cytokines ↑ → intima proliferation ↑ | Takagi et al. [33] | |
| MCP-1 | MMD patients/ Serum | MCP-1↑ | MCP-1 ↑ → the migration of stromal cells↑ → pathological collateral vessel formation | Kang et al. [12] | |
| Inflammatory mediator | IgG Immune complex | MMD patients/ Intracranial vessels | IgG and S100A4 protein ↑ (in vascular wall) | IgG immune complex deposition ↑ → damage in internal elastic intima → S10A4 + SMC migrates into intima → stenosis and occlusion in vessels. | Lin et al. [34] |
| M2 macrophage | MMD patients/ Serum | CD163 + M2-polarized macrophage and CXCL5 levels ↑ | M2 macrophage ↑ → autoimmune activities ↑ | Fujimura et al. [35] | |
| IL-1β | MMD patients/ Peripheral blood | IL-1β ↑ | IL-1β ↑ → macrophage, endothelial cells and SMCs ↑ → vascular permeability ↑&endothelial dysfunction ↑ | Kang et al. [12] | |
| Others | Caveolin-1 | MMD patients/ serum | Serum Cav-1 ↓ | Cav-1↑ → typical angiogenesis ↓ and apoptosis in SMC↑ → impaired angiogenesis | Chung et al. [36] |
| MMD patients/ ? | Caveolin-1 ↓ (especially in RNF213 variant carriers) | Cav-1 is a crucial mediator in MMD | Bang et al. [10] | ||
| NO metabolites | MMD patients/ CSF | 1. NO metabolites levels ↑ (in CSF) 2. NO metabolites ↓ after the bypass surgery | Defect collateral vessel formation → the NO metabolites ↑ | Noda et al. [37] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Y.-C.; Wei, L.-F.; Hu, C.-J.; Tu, Y.-K. Pathological Circulating Factors in Moyamoya Disease. Int. J. Mol. Sci. 2021, 22, 1696. https://doi.org/10.3390/ijms22041696
Fang Y-C, Wei L-F, Hu C-J, Tu Y-K. Pathological Circulating Factors in Moyamoya Disease. International Journal of Molecular Sciences. 2021; 22(4):1696. https://doi.org/10.3390/ijms22041696
Chicago/Turabian StyleFang, Yao-Ching, Ling-Fei Wei, Chaur-Jong Hu, and Yong-Kwang Tu. 2021. "Pathological Circulating Factors in Moyamoya Disease" International Journal of Molecular Sciences 22, no. 4: 1696. https://doi.org/10.3390/ijms22041696
APA StyleFang, Y. -C., Wei, L. -F., Hu, C. -J., & Tu, Y. -K. (2021). Pathological Circulating Factors in Moyamoya Disease. International Journal of Molecular Sciences, 22(4), 1696. https://doi.org/10.3390/ijms22041696